

Abstract
While strong familial evidence supports a substantial genetic contribution to the etiology of autism spectrum disorders (ASD), specific genetic abnormalities have been identified in only a small minority of all cases. In order to comprehensively delineate the genetic components of autism including the identification of rare and common variants, overall sample sizes an order of magnitude larger than those currently under study are critically needed. This will require rapid and scalable subject assessment paradigms that obviate clinic-based time-intensive behavioral phenotyping, which is a rate-limiting step. Here, we test the accuracy of a web-based approach to autism phenotyping implemented within the Interactive Autism Network (IAN). Families who were registered with the IAN and resided near one of the three study sites were eligible for the study. One hundred seven children ascertained from this pool who were verbal, age 4–17 years, and had Social Communication Questionnaire (SCQ) scores ≥12 (a profile that characterizes a majority of ASD -affected children in IAN) underwent a clinical confirmation battery. One hundred five of the 107 children were ASD positive (98%) by clinician’s best estimate. One hundred four of these individuals (99%) were ASD positive by developmental history using the Autism Diagnostic Interview-Revised (ADI-R) and 97 (93%) were positive for ASD by developmental history and direct observational assessment (Autism Diagnostic Observational Schedule or expert clinician observation). These data support the reliability and feasibility of the IAN-implemented parent-report paradigms for the ascertainment of clinical ASD for large-scale genetic research.
Keywords: autism, ASD, sample size, genetic studies, rapid phenotyping paradigm
INTRODUCTION
Autism spectrum disorder (ASD) is a highly heterogeneous group of neurodevelopmental disorders encompassing Autistic Disorder, Asperger’s syndrome and pervasive developmental disorder—not otherwise specified (PDD-NOS). Children affected with ASD show abnormalities in communication and social behaviors and often manifest repetitive behaviors [APA, 2000]. Usually the symptoms can be observed by 18 months of age and the diagnosis predominantly occurs before age 4 years [Howlin and Asgharian, 1999; Mandell et al., 2005]. The prevalence of diagnosed ASD has increased significantly in the past decade for unclear reasons, and the current estimate is around 1 in 110 children [CDC, 2009]. Boys are affected much more often than girls at a ratio of approximately 4:1 [CDC, 2009].
Efforts to conduct large-scale investigations of autism have been historically constrained by the cost of conducting time-intensive developmental history and observational assessments which have become a research standard in the field of autism, nearly universally required by funding agencies and scientific journals. The recent development of rapid phenotyping methods, and advances in our understanding of the genetic and factoral structure of autism have raised serious questions about the relative value of traditional diagnostic assessments for autism, especially when weighed against their considerable costs and the scientific urgency of moving forward with larger-scale research efforts to advance understanding of the causes and possible treatments for these conditions.
Here, we present the current status of autism genetic research, and describe the critical role of a rapid phenotyping paradigm for developing a feasible and cost-effective approach to the next phase of gene discovery in autism. Over the past 20 years, the genetic basis of autism has been well established, as evidenced by higher concordance rate of ASD observed in monozygotic twins (~90%) than in dizygotic twins (~10%) as well as a high sibling recurrence risk [Bailey et al., 1995]. These observations have spurred considerable interest in defining the genetic basis of autism while some successes in associating autism with specific chromosomal anomalies have occurred, the vast majority of autism remains idiopathic, and conversely we expect that most gene variants contributing to autism risk have yet to be observed. Numerous linkage studies have implicated multiple genomic regions of interest, but the identification of specific genes that contribute to risk within these intervals remains largely elusive [Ashley-Koch et al., 1999; Barrett et al., 1999; Philippe et al., 1999; IMGSAC, 2001; Auranen et al., 2002; Yonan et al., 2003; Stone et al., 2004; Cantor et al., 2005; Szatmari et al., 2007; Weiss et al., 2009]. Recently, characterization of rare structural variation (e.g., copy number variation or CNV) has contributed to the progress towards the identification of genetic risk variants [Jacquemont et al., 2006; Kumar et al., 2008; Marshall et al., 2008; Weiss et al., 2008]. However, even these strong genetic findings demonstrate the clinical heterogeneity affiliated with specific genetic lesions [Bijlsma et al., 2009]. Genome-wide association analyses highlight variants of unknown function between CDH10 and CDH9 at 5p14.1 [Ma et al., 2009; Wang et al., 2009] with small effect size, and near SEMA5A on chromosome 5p15 with protective effect [Weiss et al., 2009]. Rare mutations or variants in A2BP1, NRXN1B, NRXN1B, NRSXN1A, NLGN3, and NLGN4 have been associated with autism [Jamain et al., 2003; Laumonnier et al., 2004; Yan et al., 2005; Feng et al., 2006; Martin et al., 2007; Yan et al., 2008]. Due to the now observed very high genetic heterogeneity of ASD, the discovery and validation sample sizes for genetic investigation remain unsatisfactory for identifying the majority of the genetic contribution to autism.
Most of the sample collection efforts for genetic studies so far have employed recruitment procedures that require substantial time investment per family and expensive evaluator time for individual phenotype assessment. This approach naturally limits the pacing of collection of biomaterials due to funding and evaluator time constraints, as well as logistical issues for the subjects. The largest single effort to date was implemented by the Autism Genetic Resource Exchange (AGRE), which lessened geographical constraints by deploying trained evaluators and blood draw specialists to homes throughout the US to recruit a large set of affected sibling pair families for genetic analysis [Geschwind et al., 2001]. A number of collaborative efforts have been formed and organized into a cohesive analytical group (i.e., Autism Genome Project Consortium). With these efforts, around 3,300 families with at least one affected child with autism are available for genetic analysis in North America and Europe as reflected in the largest genetic study published to date [Weiss et al., 2009]. These sample sets are proving to be largely inadequate to identify common variant associations. Further, since many of the genetic causes of ASD traits are proving to be, in each instance, rare variants, the field will require very large sample sizes to distinguish causative mutations/alleles from benign or non-causal rare variants present in the population.
Thus, we need to explore how to cost-effectively recruit tens of thousands of affected individuals and their families to enable the appropriate large-scale collection of affected individuals for genetic. Most genetic studies performed to date, especially those that aggregate data from multiple sites, rely on clinical evaluation, Autism Diagnostic Interview (ADI-R) or Autism Diagnostic Observation Schedule (ADOS) in an effort to standardize phenotypic criteria across various clinical sites. However, it is not at all clear that these currently employed time-consuming methods provide the most efficient approach to phenotyping for genetic studies of ASD. Although the use of these gold standard diagnostic instruments is often extended to address the qualitative and quantitative measurement of autistic symptomatology, item level reliability is challenging. The clinic-based or rater-based phenotypic approaches used to date need to be compared and contrasted to what could be achieved by allocating a similar level of resources for rapid, web-implemented, and much lower cost per sample phenotyping that permits the collection of at least an order of magnitude more samples for the same cost and time.
Here we assess the phenotyping approach implemented within the Interactive Autism Network (IAN), which is the largest online registry of families with an affected child with ASD in the US with approximately 10,000 autism families registered and 100–300 new families registering each month. High-quality and reliable web-based entry approaches have resulted in apparently high-quality phenotypic data, and broad access to the Internet makes the registry directly available to vast numbers of families. Specifically, IAN has implemented parent report of Social Communication Questionnaire (SCQ). Here, we sought to assess the overall accuracy of the parent provided autism diagnosis for children who are verbal per parent report and are between the ages of 4 and 17 by sampling from the IAN registrants for clinic-based evaluation with ADI-R, ADOS, and clinician best estimate. This is a critical step to use IAN for a large-scale recruitment of samples for genetic analysis. With this validation of the IAN-based parental reports, it is possible to integrate a US-wide system for the collection of blood or saliva for the generation of high-quality genomic DNA for genetic analysis in a feasible, scalable, and cost-effective fashion, greatly expanding the set of genomic DNAs available for genetic analysis [Rylander-Rudqvist et al., 2006].
MATERIALS AND METHODS
Recruitment
In order to sample IAN registrants who could participate in the evaluation of IAN phenotyping, a recruitment email was sent to 692 families already registered in IAN who met the following criteria: (i) answered “yes” to SCQ question number 1, “Is she/he now able to talk using short phrases or sentences?”; (ii) had SCQ score ≥12; (iii) reported to have only one child affected with ASD in the family (simplex); and (iv) identified that they were living within 120 miles from University of California, Los Angeles (UCLA), 50 miles from Kennedy Krieger Institute, Baltimore (KKI) and 125 miles from Washington University, St. Louis (WUSTL). The SCQ-lifetime version that reflects lifetime experience was used. It was indicated in the recruitment email that the families enrolling in the study will be requested to (i) bring previous medical documentation(s) showing ASD diagnosis of their affected child, (ii) deliver a teacher’s packet to the affected child’s teacher, and (iii) have an appointment at the clinic to be assessed using the current “gold standard” diagnostic instruments: ADI-R, ADOS and Vineland Adaptive Behavior Scales, Second Edition (Vineland-II). Each individual was also asked to (iv) donate blood and/or saliva during their clinic visit, but not required. The recruitment letter also included the information about the incentives (clinic visit $50, blood donation $25, and saliva donation $25) and the travel reimbursement (up to $50). The families interested in participating in the study were asked to contact each site by email or phone for further information and setting up the appointments. The study was approved by the Institutional Review Board (IRB) at each site. In total, 109 individuals were recruited to UCLA, KKI or WUSTL. There were a few cases (N = 12 at UCLA, N = 3 at KKI, and N = 2 at WUSTL) where the families were not initially registered in IAN yet but had learned of the study and subsequently enrolled in IAN to participate.
Measures
At each site, the families were consented and underwent three ASD evaluations: ADI-R, Vineland-II and an observational confirmation procedure (ADOS at UCLA and KKI and expert-clinician-observation or ADOS at WUSTL). By combining the results from three evaluations, clinicians made best estimate (BE) diagnosis from DSM-IV. ADI-R was administered to the parent or caretaker in clinic or over the phone. When the subject met the threshold algorithm score in each of the three functional domains (Language/Communication: 8 for verbal and 7 for non-verbal, Reciprocal Social Interactions: 10, Restricted, Repetitive, and Stereotyped Behaviors and Interests: 3) and exhibited evidence of onset of the disorder by 36 months of age, a classification of “autism” was given. If the subject underscored in any one of the three functional domains or did not exhibit evidence of onset of the disorder by 36 months of age, a classification of “not autism” was given. ADI-R is structured to best exemplify DSM-IV and International Classification of Disease-10 (ICD-10) criteria for autism and has been demonstrated to have high sensitivity and moderate specificity with BE [Lord et al., 1994]. There are no standard cutoffs for ASD, PDD-NOS, and Asperger’s syndrome on ADI-R.
Structured clinical observations were undertaken on all participants. Two sites used the ADOS, a structured clinical assessment in widespread use for the diagnosis of autism and ASD. The ADOS was performed in clinic environment by trained examiners at UCLA and KKI with a 30- to 45-min observation period of social and communication behaviors related to the diagnosis of autism or other PDD. One of the four modules was administered according to the language level and chronological age of the individual being assessed. In each module, a subject had to exceed the cutoff for either autism or ASD (slightly lower cutoff than autism) in “social reciprocity” and “communication” domain as well as combined. Substantial agreement in categorical assignment of diagnosis is exhibited between ADI-R and ADOS [Lord et al., 1999]. WUSTL used clinician-observation, which ostensibly assessed the same social, language and behavior domains as the ADOS on a subset of the subjects. These subjects were observed and videotaped in the context of 15-min interactions with trained, master’s-level clinicians who engaged the subjects in conversation and symbolic interactive play activity in order to elicit social-communicative behavior. The videotapes were reviewed by a Board Certified Child and Adolescent Psychiatrist with extensive experience in the diagnosis and treatment of autism spectrum conditions (co-author, JNC) for evidence of DSM-IV criteria in fulfillment of a diagnosis of an autism spectrum disorder. A case was deemed “confirmed” if there was direct evidence from the videotaped observation of social, communicative or stereotypic behaviors (a) sufficient to support a diagnosis of an ASD; and (b) the qualifying behaviors were deemed unlikely to be attributable to a non-ASD psychiatric diagnosis. Inter -rater agreement on case designation (confirmation vs. non-confirmation of ASD) with both a clinical child psychologist and a doctoral-level school psychologist was calculated in an independent sample using identical review methods and exceeded 90% (manuscript in preparation).
Vineland-II is designed to assess daily functioning of an individual, both normal and handicapped, whose age ranges from preschool to early adulthood. Various dimensions of adaptive behavior are captured through three domains of measurement—Communication, Daily Living Skills, and Socialization Skills. Standard scores of 100 are calculated from population norms for the instrument. Vineland-II has been proven useful in characterizing individuals with autism spectrum disorders who show distinctive low scores in Socialization [Carter et al., 1998; Gillham et al., 2000; Tomanik et al., 2007]. For each domain, standard scores and age equivalent adaptive behavior scores were generated and the overall adaptive behavior composite score created by combining the scores across domains.
Teacher’s packet including the teacher Social Responsiveness Scale (SRS) form, $15 gift card ($20 at KKI), pre-stamped envelope and instructions were given to the families for delivery preferably to the school teacher who had known the proband for more than 3 months or a therapist or extracurricular teacher, who observed the child in social situations. Parents who home-schooled their children were not allowed to do the teacher SRS. The SRS is a 65-item quantitative rating scale that can be completed by teachers or parents to measure the severity of autism symptoms occurring in natural social settings. The norms for the SRS and its psychometric properties in clinical and general population are described in the instruction manual, which also provides the guidelines on how to convert the raw scores into t-scores that adjusts for modest discrepancies observed between different raters and male and female subjects [Constantino and Gruber, 2005].
All of the clinical data were entered into Internet System for Assessing Autistic Children (ISAAC) database (http://www.autismtools.org).
Data Analysis
t-Test implemented in “Simple Interactive Statistical Analysis” tool available online (http://www.quantitativeskills.com/sisa/statistics/ t-test.htm) was used to calculate the P-values for the difference of the mean age and SCQ score. The P-value for the difference of distribution of the gender and parent reported diagnosis in IAN was calculated by fisher exact test. The 95% confidence interval was calculated for each concordance rate using a Bayesian Calculator provided by http://www.causascientia.org/math_stat/Proportion-CI.html. The exact intervals were calculated because this was an extreme case where the number of subjects found not to be ASD was very small.
RESULTS
Recruitment and Clinic Evaluation
A total of 109 individuals were recruited into the clinical evaluation at one of the three sites between 18 May and 22 December, 2009. Thirty-one were evaluated at UCLA, 42 at KKI and 36 at WUSTL, and all completed ADI-R, ADOS (or equivalent) and/or Vineland-II evaluation. Table I summarizes the affected individuals enrolled at each site and in total. Of the 109 subjects, 107 completed the ADI-R, 82 completed the ADOS, and 27 subjects were assessed by expert clinician review of videotaped observation, 108 completed the Vineland-II, and 107 subjects completed all three assessments.
TABLE I.
Study Summary
| UCLA (n=31) | KKI (n=42) | WUSTL (n=36) | Total (n=109) | |
|---|---|---|---|---|
| Male/female | 27:4 | 30:12 | 32:4 | 89:20 (4.45:1) |
| SCQ scores ≥12 | 31 (100%) | 42 (100%) | 36 (100%) | 109 (100%) |
| Verbal by SCQ (Q1) | 31 (100%) | 42 (100%) | 36 (100%) | 109 (100%) |
| Mean age (SD) | 10.0 (4.3) | 9.2 (3.8) | 9.6 (3.4) | 9.6 (3.8) |
| Medical record doc collected | 28 (90%) | 42 (100%) | 36 (100%) | 106 (97%) |
| Teacher SRS report collected | 20 (65%) | 41 (98%) | 33 (92%) | 94 (88%) |
| ADI-R completed | 31 (100%) | 41 (98%) | 35 (97%) | 107 (98%) |
| ADOS completed | 31 (100%) | 42 (100%) | 9 (25%)a | 82 (75%) |
| Vineland-II completed | 31 (100%) | 41 (98%) | 36 (100%) | 108 (99%) |
SD, standard deviation.
All subjects not administered with ADOS were assessed by expert clinician review of videotaped observation.
The study population was compared to the pool of all potential subjects registered in IAN who meet our basic inclusion criteria (simplex family, SCQ ≥12, SCQ verbal and age 4–17) to determine if there was a bias in the sampling. No significant differences were found between the two groups in terms of gender distribution (Fig. 1a, P-value: 0.6), reported diagnosis distribution (Fig. 1b, P-value: 0.8), mean age (Fig. 1c, P-value: 0.4), and mean SCQ score (Fig. 1d, P-value: 0.6).
FIG. 1.
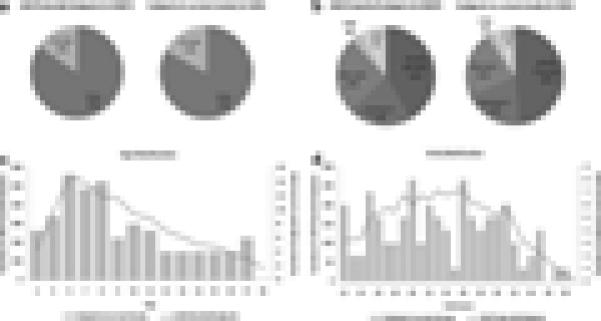
Comparison of the sample stats between the enrolled families and all samples in IAN with the same criteria. a: Gender distribution; (b) diagnosis distribution: the distribution of diagnosis that the parents have reported to IAN. If the current diagnosis was different than the first diagnosis, the current diagnosis was counted. If neither the first diagnosis nor the current diagnosis was reported, registration diagnosis was counted; (c) age distribution: for the subjects enrolled in current study, age was determined on the date of examination (DOE) and for the IAN potential subjects, it was determined as of 31 August, 2009; (d) SCQ score distribution.
Data Analysis
Of 107 individuals who completed all three clinical assessments, 99% (n = 106) met criteria for autism on the basis of ADI-R (Table II). The one subject who did not meet the ADI-R cutoff for autism did not score sufficiently highly within the ‘social’ domain (cutoff 10) for autism and had score of 8 in this domain, but otherwise met criteria for autism. 89.7% (n = 96) met criteria for Autism or ASD on the basis of ADOS or equivalent clinician assessment. Of the 11 that did not meet criteria using the ADOS scoring algorithm, one subject was inconclusive on ADOS and resulted in BE of “selective mutism and social anxiety” in addition to autism. For that individual, the ADI-R was positive. There was one subject diagnosed with “ASD-by-history” as on clinician assessment: the subject was found to not be sufficiently symptomatic at the time of the evaluation but consistent with ASD by the ADI -R and clearly not attributable to any other disorder. Four subjects passed the cutoff for ASD in the “communication” or “social” domain or both but did not pass the total cutoff on the ADOS, three subjects were one point away from the “communication” domain cutoff for ASD but passed the social and total cutoffs for ASD and one subject did not meet the cutoffs for any. However, all eight of them were found to be on the autism spectrum by clinician’s best estimate. The remaining one subject was found not to be on the spectrum by either ADOS or BE even though the ADI-R classified the subject as “autism.” The clinicians concluded that the subject most likely has childhood schizophrenia, which is very difficult to distinguish from autism and might be expected to score positive on the ADI-R given overlapping symptomatology. In total, 88.8% (n = 95, 95% CI: 81.2–94.1%) were on the spectrum based on both ADI-R (classified as “autism”) and ADOS (classified as “autism” or “ASD”), 100% (n = 107, 95% CI: 97.2–100%) by either ADI-R or ADOS and 98.1% (n = 105, 95% CI: 93.4–99.8%) by BE (73% Autistic Disorder, 18% PDD-NOS, 3% Asperger’s syndrome, and 4% not specified ASD). The lower concordance rate for the ADOS could be explained by the fact that ADOS does not reflect past behavior that may be compensated for at the time of the relatively brief ADOS evaluation. Thus, the ADI-R may be more consistent with SCQ and parental report as well as prior clinician diagnosis.
TABLE II.
Percentage of Subjects Concordant for ASD Diagnosis as a Function of Individual Components of Rapid Phenotyping Paradigm
| Total number | ADI-R Pos (%) | ADOS Pos (%) | Both Pos (%) | Either Pos (%) | Clinician BE Pos (%) | |
|---|---|---|---|---|---|---|
| Parent report of ASD DX SCQ ≥12 |
107 | 99.1 | 89.7 | 88.8 | 100 | 98.1 |
| Medical documentation of ASD | 104 | 99.0 | 89.4 | 88.5 | 100 | 98.1 |
| Teacher-report SRS ≥65 | 51 | 100 | 92.2 | 92.2 | 100 | 100 |
Of 107 individuals who completed all three clinical assessments, 86% (n = 92) had their teacher SRS returned. Two of the reports were insufficiently completed and were thus excluded. The mean t-score for the 90 subject evaluations was 66.3 (SD: 10.0). A total of 51 subjects (56.6%) had t-score ≥65, and 39 did not meet threshold criteria by SRS. Of the 39 who did not meet threshold criteria, 100% met diagnostic criteria by ADOS or ADI-R. Thus, the teacher SRS is not sufficiently sensitive for affected autistic individuals. However, of these 51 subjects, 47 (92.1%, 95% CI: 81.1–97.8%) were determined to be on the spectrum by ADOS, and 100% were on the spectrum by ADI-R and BE (95% CI: 94.3–100%) indicating high specificity if teacher SRS is positive. There was one proband who was diagnosed “autism” by both ADI-R and ADOS but determined not to be autistic by BE. The clinicians concluded that negative behaviors and oppositionality of the proband contributed to the elevated scores on both assessments. This particular proband has acquired multiple teachers’ SRS reports and all four of them had t-score total <65. Thus, there was much lower concordance of teacher completed SRS with the clinic-based assessments.
Vineland-II assessment was performed to further characterize the study sample, and indicates that the study sample has typical score distributions as other clinic-based autism samples. A summary of the Vineland-II assessment standard and adaptive behavior scores for each domain and the composite score for this population are shown in Table III.
TABLE III.
Vineland-II Summary: Standard Scores and Adaptive Level for Each Domain
| Mean | SD | |
|---|---|---|
| Communication | ||
| Standard scores | 80.9 | 14.7 |
| Adaptive level | 3.9 | 0.8 |
| Daily living | ||
| Standard scores | 79.0 | 14.6 |
| Adaptive level | 4.0 | 0.8 |
| Socialization | ||
| Standard scores | 71.6 | 15.6 |
| Adaptive level | 4.3 | 0.8 |
| Composite | ||
| Standard scores | 75.6 | 13.7 |
| Adaptive level | 4.0 | 1.0 |
DISCUSSION
Here, we have assessed the accuracy of fully implemented web-based parent report within a disease-specific patient registry (IAN). We observe that even in the small sampling performed within this study of a large potential sample set, the rapid phenotyping paradigm has a very high concordance rate with the clinic-based assessments, which are considered the reasonable standards for affectation status for inclusion in genetic studies. The high concordance rates observed imply that parent reports, as implemented in the parent-centric IAN registry, are highly reliable and provide a 95% confidence of a 93.4% concordance rate with ADI-R or ADOS based clinician’s diagnosis in verbal children. These observations are generally supportive of other reports of the self-selected populations of families with a child affected by autism. For instance, the distribution of scores on the SRS of affected children enrolled in IAN is similar to the data provided from clinically defined populations with ASD [Constantino et al., 2007].
There were a few subjects (n = 11, 10.2%) who were negative by ADOS, but all of them were positive for ASD based on ADI-R and clinician’s BE. This is not unexpected as ADOS is a 30-min snapshot during which symptoms displayed in other settings (e.g., social settings like school where children are sometimes at their worst) may be unobserved. As seen in one of the subjects here, an additional scenario involves children who, at or before age 4, were manifesting autistic symptomatology severe enough to warrant a clinical diagnosis (by developmental history), but who may have improved over time and did not meet severity criteria by research observation at the time of this study. Conversely, the ADOS also has the risk of over-interpreting behaviors elicited in the anomalous context of a 1-on-1 assessment by an unknown examiner. Because of these shortcomings of ADOS, phenotyping for genetic studies rely primarily on ADI-R supplemented by ADOS [Sebat et al., 2007; Szatmari et al., 2007; Strom et al., 2009; Wang et al., 2009; Weiss et al., 2009]. The high concordance rate observed in the presented data is important to document, as IAN is a potentially major source of recruitment into specific autism studies.
There are two caveats to the generality of the observations made here. First, all of the probands were identified through participation in IAN with web-based registration. The simple fact of the need for web-based registration will bias the sample collected, but in many ways this is similar to the bias of individual participation in biomedical research which does not randomly sample the whole population [Rothman and Greenland, 1998]. One risk of the web-implemented approach is that there may be a potential discrete cause of ASD in a given child, and yet not be known by the parents. In this study, all of the probands in the current study had idiopathic autism, which is of course true of the vast majority of individuals diagnosed with autism currently, implying that this will be a rare issue. Diagnostic misspecification, including individuals who have an alternate diagnosis, is only modestly problematic for the identification of autism risk alleles given the already observed genetic heterogeneity of ASD. Even if 10% of the ASD cases were misdiagnosed, we would need to sample a population approximately 20% larger to compensate for the loss of genetic power [Edwards et al. 2005]. Second, we assessed individuals who were at least minimally verbal as screened through online questionnaire to the parents to analyze the more common idiopathic autism group registered in IAN and because the quantitative measures employed in our rapid phenotyping paradigm have not yet been validated for their precision when used for children with comorbid intellectual disability, a majority of whom are rendered non-verbal by virtue of this comorbidity (non-verbal children comprise a 25% minority of all ASD-affected children in the IAN register). Thus, our conclusions here may be restricted to minimally verbal affected children and the concordance rate may not be as high for harder to assess non-verbal children.
Although teacher-report SRS had very high positive predictive value, reliance of the teacher-report SRS would result in the exclusion of potentially half of the documented ASD-affected children and improvement in accuracy over parent-report alone did not reach statistical significance in this study. The major factor was a lack of response from the teacher-SRS, but there was also a decreased sensitivity relative to parent-SRS. The very high positive predictive value of teacher-report SRS and relatively low sensitivity suggests that the threshold score used for case identification may have been set too high in severity. This is currently being more closely examined in an analysis of SRS data from over 1,200 children in the IAN register (manuscript in review)—it is likely that setting a slightly lower threshold will result in optimization of sensitivity, specificity, and positive predictive value. We also observe that the collection of the teacher SRS was challenging for some families and might therefore limit the feasibility of large-scale implementation that is easily scalable.
It is important to emphasize that we view this IAN/web-based approach as complementary to and synergistic with more comprehensively phenotyped sample sets, such as those being accumulated by AGRE, the Simons Simplex Collection, and various academic collaborative research projects. While we demonstrate here that the core phenotypic assessment of the autism phenotype as reported by parents results in autism study samples generally consistent with sample sets used in published autism genetic studies that relied on trained rater assessments, there are certainly many important avenues of research that cannot be directly addressed by this sampling strategy. For instance, the analysis of complex developmental syndromes characterized by both mental retardation and autistic traits would not be feasible in this context, nor would longitudinal studies that assess developmental trajectory, changes in brain imaging over time, or novel neuropsychological endophenotypes. However, given the tremendous genetic heterogeneity of ASD and unknown environmental influences makes the need for very large sample size collection compelling. This web-based recruitment permits the characterization of individuals with specific genetic etiologies. For instance, after the individuals who have a specific genetic etiology for autism are identified, IAN has established approved means to re-contact the specific families in order to perform additional phenotyping. We view the implemented web-based processes as a powerful community tool to rapidly identify willing research participants, implement rapid and facile DNA collection strategies distributed across the country, and subsequently affiliate generated genetic data with individual IAN participants.
ACKNOWLEDGMENTS
We gratefully acknowledge the families and teachers who have participated in this study. We thank Angela Matevosyan, Ana Quintana, Karin Best, PhD, Pegeen Cronin, PhD, Tamar Apelian, PsyD, Allison Kahal, PsyD, Svetlana Ravinovich, PsyD, Linda Quirmbach, PhD, Grace Gengoux, PhD and Joni Zuckerbrow-Miller at UCLA CART (Center for Autism Research and Treatment); Sarah S. Marvin, Eleeshabah Yahudah, Rachel Friedman, Connie Anderson, PhD and Shai Scheller at IAN; Patricia Rao, PhD, Jessica Jacques, MA and Deborah Crawford, MS, CCC-SLP at KKI CARD (Center for Autism and Related Disorders) and Fellana R. Randall, Anna Abbacchi, MS and Teddi Gray at Washington University in St. Louis for their support in recruitment and clinic evaluation. We also thank Vlad Kustanovich and Dusan Bosnjakovic at AGRE for facilitating the data entry into ISAAC. IAN, the Interactive Autism Network, is a web project of Kennedy Krieger Institute sponsored by the Autism Speaks Foundation, Inc. This study was funded by Autism Speaks as part of the High Risk High Impact (HRHI) Project: Sponsor Award No. 5667 “Pilot Project to Assess Web-based Family Recruitment for Autism Genetics Studies” and was partially supported by R01MH081754.
Footnotes
Conflict of interest: John N. Constantino receives royalties from Western Psychological Services for the commercial distribution of the Social Responsiveness Scale.
The authors have no financial disclosures.
REFERENCES
- American Psychiatric Association . Text Revision (DSM-IV-TR) 4th Edition American Psychiatric Association; Washington, DC: 2000. Diagnostic and Statistical Manual of Mental Disorders. [Google Scholar]
- Ashley-Koch A, Wolpert CM, Menold MM, Zaeem L, Basu S, Donnelly SL, Ravan SA, Powell CM, Qumsiyeh MB, Aylsworth AS, et al. Genetic studies of autistic disorder andchromosome 7. Genomics. 1999;61(3):227–236. doi: 10.1006/geno.1999.5968. [DOI] [PubMed] [Google Scholar]
- Auranen M, Vanhala R, Varilo T, Ayers K, Kempas E, Ylisaukko-Oja T, Sinsheimer JS, Peltonen L, Jarvela I. A genomewide screen for autism-spectrum disorders: Evidence for a major susceptibility locus on chromosome 3q25-27. Am J Hum Genet. 2002;71(4):777–790. doi: 10.1086/342720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, Rutter M. Autism as a strongly genetic disorder: Evidence from a British twin study. Psychol Med. 1995;25(1):63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- Barrett S, Beck JC, Bernier R, Bisson E, Braun TA, Casavant TL, Childress D, Folstein SE, Garcia M, Gardiner MB, et al. An autosomal genomic screen for autism. Collaborative linkage study of autism. Am J Med Genet. 1999;88(6):609–615. doi: 10.1002/(sici)1096-8628(19991215)88:6<609::aid-ajmg7>3.3.co;2-c. [DOI] [PubMed] [Google Scholar]
- Bijlsma EK, Gijsbers AC, Schuurs-Hoeijmakers JH, van Haeringen A, Fransen van de Putte DE, Anderlid BM, Lundin J, Lapunzina P, Perez Jurado LA, Delle Chiaie B, et al. Extending the phenotype of recurrent rearrangements of 16p11.2: Deletions in mentally retarded patients without autism and in normal individuals. Eur J Med Genet. 2009;52(2–3):77–87. doi: 10.1016/j.ejmg.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Cantor RM, Kono N, Duvall JA, Alvarez-Retuerto A, Stone JL, Alarcon M, Nelson SF, Geschwind DH. Replication of autism linkage: Finemapping peak at 17q21. Am J Hum Genet. 2005;76(6):1050–1056. doi: 10.1086/430278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AS, Volkmar FR, Sparrow SS, Wang JJ, Lord C, Dawson G, Fombonne E, Loveland K, Mesibov G, Schopler E. The Vineland Adaptive Behavior Scales: Supplementary norms for individuals with autism. J Autism Dev Disord. 1998;28(4):287–302. doi: 10.1023/a:1026056518470. [DOI] [PubMed] [Google Scholar]
- Autism and Developmental Disabilities Monitoring Network Surveillance Year 2006 Principal Investigators. Centers for Disease Control and Prevention (CDC) Prevalence of autism spectrum disorders—Autism and Developmental Disabilities Monitoring Network, United States, 2006. MMWR Surveill Summ. 2009;58(10):1–20. [PubMed] [Google Scholar]
- Constantino JN, Gruber CP. The Social Responsiveness Scale Manual. Western Psychological Services; Los Angeles: 2005. [Google Scholar]
- Constantino JN, Lavesser PD, Zhang Y, Abbacchi AM, Gray T, Todd RD. Rapid quantitative assessment of autistic social impairment by classroom teachers. J Am Acad Child Adolesc Psychiatry. 2007;46(12):1668–1676. doi: 10.1097/chi.0b013e318157cb23. [DOI] [PubMed] [Google Scholar]
- Edwards BJ, Haynes C, Levenstien MA, Finch SJ, Gordon D. Power and sample size calculations in the presence of phenotype errors for case/control genetic association studies. BMC Genet. 2005;6:18. doi: 10.1186/1471-2156-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Schroer R, Yan J, Song W, Yang C, Bockholt A, Cook EH, Jr, Skinner C, Schwartz CE, Sommer SS. High frequency of neurexin 1beta signal peptide structural variants in patients with autism. Neurosci Lett. 2006;409(1):10–13. doi: 10.1016/j.neulet.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Geschwind DH, Sowinski J, Lord C, Iversen P, Shestack J, Jones P, Ducat L, Spence SJ. The autism genetic resource exchange: A resource for the study of autism and related neuropsychiatric conditions. Am J Hum Genet. 2001;69(2):463–466. doi: 10.1086/321292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillham JE, Carter AS, Volkmar FR, Sparrow SS. Toward a developmental operational definition of autism. J Autism Dev Disord. 2000;30(4):269–278. doi: 10.1023/a:1005571115268. [DOI] [PubMed] [Google Scholar]
- Howlin P, Asgharian A. The diagnosis of autism and Asperger syndrome: Findings from a survey of 770 families. Dev Med Child Neurol. 1999;41(12):834–839. doi: 10.1017/s0012162299001656. [DOI] [PubMed] [Google Scholar]
- IMGSAC (International Molecular Genetic, Study of Autism Consortium) A genomewide screen for autism: Strong evidence for linkage to chromosomes 2q, 7q, and 16p. Am J Hum Genet. 2001;3:570–581. doi: 10.1086/323264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont ML, Sanlaville D, Redon R, Raoul O, Cormier-Daire V, Lyonnet S, Amiel J, Le Merrer M, Heron D, de Blois MC, et al. Array -based comparative genomic hybridisation identifies high frequency of cryptic chromosomal rearrangements in patients with syndromic autism spectrum disorders. J Med Genet. 2006;43(11):843–849. doi: 10.1136/jmg.2006.043166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamain S, Quach H, Betancur C, Rastam M, Colineaux C, Gillberg IC, Soderstrom H, Giros B, Leboyer M, Gillberg C, et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat Genet. 2003;34(1):27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA, Gilliam TC, Nowak NJ, Cook EH, Jr, Dobyns WB, et al. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 2008;17(4):628–638. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- Laumonnier F, Bonnet-Brilhault F, Gomot M, Blanc R, David A, Moizard MP, Raynaud M, Ronce N, Lemonnier E, Calvas P, et al. X-linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am J Hum Genet. 2004;74(3):552–557. doi: 10.1086/382137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord C, Rutter M, Le Couteur A. Autism Diagnostic Interview-Revised: A revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord. 1994;24(5):659–685. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- Lord C, Rutter M, Dilavore PC, Risi S. Autism Diagnostic Observation Schedule-WPS. Western Psychological Services; Los Angeles: 1999. [Google Scholar]
- Ma D, Salyakina D, Jaworski JM, Konidari I, Whitehead PL, Andersen AN, Hoffman JD, Slifer SH, Hedges DJ, Cukier HN, et al. A genome-wide association study of autism reveals a common novel risk locus at 5p14.1. Ann Hum Genet. 2009;73(Pt3):263–273. doi: 10.1111/j.1469-1809.2009.00523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell DS, Novak MM, Zubritsky CD. Factors associated with age of diagnosis among children with autism spectrum disorders. Pediatrics. 2005;116(6):1480–1486. doi: 10.1542/peds.2005-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, Shago M, Moessner R, Pinto D, Ren Y, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82(2):477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin CL, Duvall JA, Ilkin Y, Simon JS, Arreaza MG, Wilkes K, Alvarez-Retuerto A, Whichello A, Powell CM, Rao K, et al. Cytogenetic and molecular characterization of A2BP1/FOX1 as a candidate gene for autism. Am J Med Genet Part B. 2007;144B(7):869–876. doi: 10.1002/ajmg.b.30530. [DOI] [PubMed] [Google Scholar]
- Philippe A, Martinez M, Guilloud-Bataille M, Gillberg C, Rastam M, Sponheim E, Coleman M, Zappella M, Aschauer H, Van Maldergem L, et al. Genome-wide scan for autism susceptibility genes. Paris Autism Research International Sibpair Study. Hum Mol Genet. 1999;8(5):805–812. doi: 10.1093/hmg/8.5.805. [DOI] [PubMed] [Google Scholar]
- Rothman KJ, Greenland S, editors. Modern epidemiology. Lippincott Williams & Wilkins; Philadelphia, PA: 1998. Precision and validity in epidemiologic studies; pp. 115–134. [Google Scholar]
- Rylander-Rudqvist T, Hakansson N, Tybring G, Wolk A. Quality and quantity of saliva DNA obtained from the self-administrated oragene method-a pilot study on the cohort of Swedish men. Cancer Epidemiol Biomarkers Prev. 2006;15(9):1742–1745. doi: 10.1158/1055-9965.EPI-05-0706. [DOI] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316(5823):445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone JL, Merriman B, Cantor RM, Yonan AL, Gilliam TC, Geschwind DH, Nelson SF. Evidence for sex-specific risk alleles in autism spectrum disorder. Am J Hum Genet. 2004;75(6):1117–1123. doi: 10.1086/426034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom SP, Stone JL, Ten Bosch, JR, Merriman B, Cantor RM, Geschwind DH, Nelson SF. High-density SNP association study of the 17q21 chromosomal region linked to autism identifies CACNA1G as a novel candidate gene. Mol Psychiatry. 2009 doi: 10.1038/mp.2009.41. DOI: 10.1038/mp.2009.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu XQ, Vincent JB, Skaug JL, Thompson AP, Senman L, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007;39(3):319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomanik SS, Pearson DA, Loveland KA, Lane DM, Bryant Shaw J. Improving the reliability of autism diagnoses: Examining the utility of adaptive behavior. J Autism Dev Disord. 2007;37(5):921–928. doi: 10.1007/s10803-006-0227-6. [DOI] [PubMed] [Google Scholar]
- Wang K, Zhang H, Ma D, Bucan M, Glessner JT, Abrahams BS, Salyakina D, Imielinski M, Bradfield JP, Sleiman PM, et al. Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459(7246):528–533. doi: 10.1038/nature07999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LA, Arking DE, Daly MJ, Chakravarti A. A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009;461(7265):802–808. doi: 10.1038/nature08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358(7):667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- Yan J, Noltner K, Feng J, Li W, Schroer R, Skinner C, Zeng W, Schwartz CE, Sommer SS. Neurexin 1alpha structural variants associated with autism. Neurosci Lett. 2008;438(3):368–370. doi: 10.1016/j.neulet.2008.04.074. [DOI] [PubMed] [Google Scholar]
- Yan J, Oliveira G, Coutinho A, Yang C, Feng J, Katz C, Sram J, Bockholt A, Jones IR, Craddock N, et al. Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol Psychiatry. 2005;10(4):329–332. doi: 10.1038/sj.mp.4001629. [DOI] [PubMed] [Google Scholar]
- Yonan AL, Alarcon M, Cheng R, Magnusson PK, Spence SJ, Palmer AA, Grunn A, Juo SH, Terwilliger JD, Liu J, et al. A genomewide screen of 345 families for autism-susceptibility loci. Am J Hum Genet. 2003;73(4):886–897. doi: 10.1086/378778. [DOI] [PMC free article] [PubMed] [Google Scholar]