

Abstract
Hereditary frontotemporal dementia associated with mutations in the microtubule-associated protein tau gene (MAPT) is a protean disorder. Three neuropathologic subtypes can be recognized, based on the presence of inclusions made of tau isoforms with three and four repeats, predominantly three repeats and mostly four repeats. This is relevant for establishing a correlation between structural magnetic resonance imaging and positron emission tomography using tracers specific for aggregated tau. Longitudinal studies will be essential to determine the evolution of anatomical alterations from the asymptomatic stage to the various phases of disease following the onset of symptoms.
Keywords: FTDP-17 MAPT, tau aggregation, neurofibrillary tangle, Pick body, tau, [F18]-T807
Introduction
Inherited forms of frontotemporal dementia (FTD) have been known for many years [1–4], but as the clinical and pathological features are heterogeneous, the nomenclature has been variable, with disorders being called familial Pick disease, familial progressive subcortical gliosis, familial presenile dementia with tangles, autosomal-dominant parkinsonism and dementia with pallido-ponto-nigral degeneration. The major clinical manifestations include behavioural disturbances, aphasia, cognitive impairment and parkinsonism. Individuals from 13 families, with FTD and genetic linkage to chromosome 17q[21–22], were presented at a Consensus Conference at the University of Michigan in 1996 [5]. It was agreed that the unifying name should take into account the clinical features, as well as the genetic linkage, rather than the neuropathology, which was incomplete. Tau inclusions had been described in affected individuals from only four of the 13 families. Thus, the concept of FTD and Parkinsonism linked to Chromosome 17 (FTDP-17) was born. The disorder in one family had been named ‘multiple system tauopathy with presenile dementia’ (MSTD) [6]. As a result, the term ‘tauopathy’ was also introduced, and it is often used to refer to disorders in which tau protein deposition is the predominant feature.
In June 1998, mutations in the microtubule-associated protein tau gene (MAPT) were reported in affected individuals from nine of the 13 families [7–9]. They all suffered from a dementia syndrome, whereas some also had parkinsonism. The central neuropathologic feature was the presence of filamentous hyperphosphorylated tau protein in neurons or in both neurons and glia. The remaining four families had mutations in the Granulin gene (GRN), which is 1.54 megabase pairs centromeric to MAPT [10],[11]. Thus, FTDP-17 has been divided into FTDP-17 MAPT and FTDP-17 GRN [12].
FTD associated with MAPT mutations is a disorder that affects multiple domains including behaviour, language, memory and motor function. It often begins with psychiatric symptoms and can mimic Pick disease, primary progressive aphasia, Alzheimer disease (AD), progressive supranuclear palsy (PSP) or corticobasal degeneration (CBD). Neuropathology and neuroimaging reveal diverse pictures, consistent with variability of the clinical phenotype. It is important for clinicians, neuropathologists and imaging researchers to be aware that MAPT mutations can cause such a protean disorder. Their discovery established that tau dysfunction alone can cause neurodegeneration of multiple neuronal systems and dementia.
Epidemiology
To date, 53 pathogenic MAPT mutations have been reported in approximately 150 families [13] from Asia, Australia, Europe, and both North and South America. Molecular genetic analyses have demonstrated that some families share a common founder [14].
FTDP-17 MAPT affects men and women equally. The average age at symptom onset is 49 years, with a range from the early 20s to late 70s, similar to sporadic frontotemporal lobar degeneration (FTLD). The average life expectancy after symptom onset is 8.5 years, with a range from 1.5 to 26 years [15–17].
Disease phenotypes in patients with the same MAPT mutation may vary significantly within and between families, as well as between individuals with different mutations [16],[18],[19]. Thus, genetic modifiers and/or environmental factors may underlie the phenotypic variability in clinical presentation.
Genetics and molecular pathology
FTDP-17 MAPT is inherited in an autosomal-dominant manner. The MAPT gene, located on chromosome 17q21, encodes the tau protein, which was discovered in 1975 [20]. A decade later, the intraneuronal inclusions of AD and Pick disease were found to be immunoreactive for hyperphosphorylated tau [21–23]. The neurofibrillary tangles (NFTs) of AD are composed of paired helical and straight filaments. Their molecular characterization established that they are made of tau protein [24–26].
In the adult human brain, six tau isoforms are generated from MAPT, the tau gene, through alternative mRNA splicing (Figure 1) [27]. Alternative splicing of exon 10 gives rise to three isoforms with three microtubule-binding repeats (3R) each and three isoforms with four microtubule-binding repeats (4R) each. The repeats are 31 or 32 amino acids in length and are located towards the carboxy-terminus. In addition, the presence of inserts of 29 or 58 amino acids or no insert in the amino-terminus gives rise to 1 N, 2 N or 0 N forms of each 3R and 4R tau. Full-length tau assembles through the repeats that form the core of paired helical and straight filaments. In developing human brain, 3R tau predominates, while in adult brain, the concentrations of 3R and 4R tau are approximately equivalent. A normal ratio of wild-type 3R to 4R tau appears to be essential for preventing neurodegeneration and dementia in the human brain in mid-life.
Figure 1.

Schematic representation of the six tau isoforms generated by alternative mRNA splicing of exons 2, 3 and 10.
Between 1994 and 1997, familial forms of FTD were linked to chromosome 17q[21–22], a region that contains MAPT [28–30]. In parallel, neuropathological and biochemical studies showed abundant tau deposits in neurons and glia [31–34]. They highlighted the presence of a tauopathy affecting grey and white matter in the absence of amyloid beta deposition, and directed several laboratories towards the search for mutations in MAPT. In 1998, the first mutations were reported in exons 9, 10 and 13, as well as in the splice site of intron 10 [7–9]. The vast majority of known mutations occurring in the coding region are in the repeats, with the mutant tau proteins having a reduced ability to interact with microtubules [35–37].
Exonic mutations are missense, silent or deletion. All but two (R5H and R5L in exon 1) occur in exons [9–13]. Most intronic mutations are clustered in the 5′-splice site of the intron following exon 10. These intronic mutations and some exonic mutations located in exon 10 affect the alternative mRNA splicing of exon 10, causing a relative increase of 4R tau [8],[9],[38],[39]. They destabilize a stem-loop structure at the exon 10 5′-splice site intron junction or disrupt cis-acting elements in exon 10. Existence of a stem-loop structure was hypothesized [8],[9] at the time of the discovery of mutations in MAPT, in view of the self-complementarity of this region, with subsequent work supporting this hypothesis [40–42]. The determination of the solution structure of an oligonucleotide corresponding to the exon/intron junction refined the stem-loop model, with the identification of an adenosine bulge between the sixth and seventh base pairs [43]. Mutations S305I, S305N, S305S, +3, +4, +11, +12, +13, +14 and +16 destabilize the stem-loop, resulting in increased U1 snRNP binding, and enhanced exon 10 inclusion. Mutations in exon 10, located outside the stem-loop, can also increase exon 10 splicing, because of the strengthening of exon splicing enhancers or the weakening of exon splicing silencers [39],[44].
Thus, the primary effect of the coding region mutations may be equivalent to a partial loss of function. The net effect of mutations, whose primary effect is at the RNA level, is the overproduction of wild-type 4R tau, which interacts more strongly with microtubules than 3R tau [45]. Some mutations, such as P301L, P301S and P301T in exon 10, affect only [20–25]% of tau molecules, with [75–80]% being wild-type, arguing against a simple loss of function mechanism as an important disease determinant.
It is therefore possible that a partial loss of function of tau is necessary for setting in motion the gain of toxic function mechanism that will lead to neurodegeneration. For MAPT mutations with a primary effect at the RNA level, the overproduction of 4R tau may result in an excess of tau over available binding sites on microtubules, leading to the cytoplasmic accumulation of unbound 4R tau. This would probably require the existence of different binding sites on microtubules for 3R and 4R tau. Validation of this hypothesis will require structural information at the atomic level. An imbalance in isoform ratios could also affect tau aggregation directly. Studies in vitro have shown that filament assembly is decreased in reactions containing 3R and 4R tau when compared with those containing only 4R tau [46].
Figure 2 shows the 53 mutations that are currently known[6–9,14,16,33,34,38,14,39,47–132]. The most common are N279K, P301L and intron 10+16.
Figure 2.

Schematic representation of the exons and introns of the MAPT gene, where 53 mutations causing FTDP-17 have been found. Intronic mutations −15 and +4 occur together.
Soluble and insoluble Tau
A central question revolves around the process by which tau filaments form. In FTDP-17 MAPT, tau protein isoforms have biochemical characteristics that differ from those of the normal protein [133]. A mutation may result in a structurally abnormal protein, an abnormal ratio of 3R to 4R tau, or both. Normally, tau is a soluble protein; however, in FTDP-17 MAPT, it is found in both soluble and insoluble forms. Tau accumulates in the cytoplasm and becomes hyperphosphorylated, insoluble and assembles into filaments. However, the order of events in relation to hyperphosphorylation and filament formation is not clearly understood.
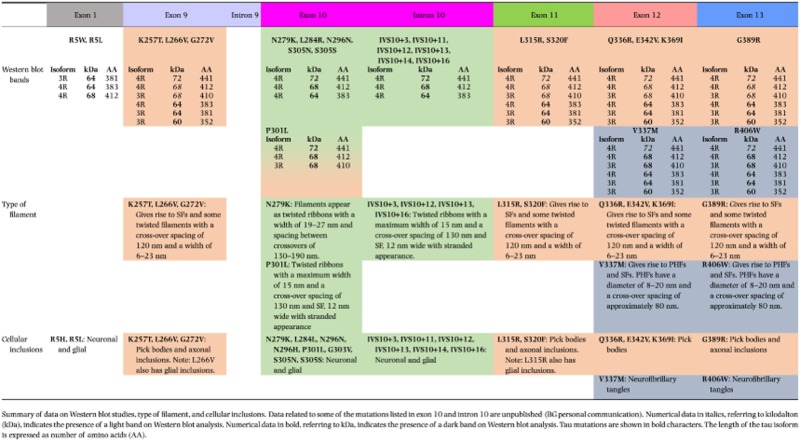
Missense mutations in exons 1, 9, 11, 12 and 13 affect all six tau isoforms. Missense and deletion mutations in exon 10 affect the alternative mRNA splicing of exon 10, altering isoform ratios in such a way that relatively more 4R than 3R tau is produced. A summary is given in Table 1, row 1.
Table 1.
Western blot analysis, filaments and cellular inclusions associated with of MAPT mutations
 |
Hyperphosphorylation of Tau and filament formation
Hyperphosphorylation of tau is believed to play a crucial part in the pathogenesis of human tauopathies [133]. In FTDP-17 MAPT, it is unlikely to be primary as none of the known mutations influence phosphorylation directly. Nevertheless, evidence has been adduced to suggest that some MAPT mutations can lead to enhanced phosphorylation [134], followed by filament formation. Morphological evidence for the presence of the insoluble form is provided by the finding that some tau deposits are fluorescent using Thioflavin S, tau filaments are found in neurons and glia and tau filaments can be visualized in sarkosyl-insoluble tissue preparations.
Filament morphologies have been studied using fixed tissues and preparations of dispersed filaments [135]. The latter are particularly informative as they allow one to correlate Western blot analysis with immunoelectron microscopy. Tau filaments can be straight, ribbon-like or paired helical. Table 1 summarizes the characteristics of abnormal tau as demonstrated by Western blot analysis, the type of tau filament and the nature of the intracytoplasmic inclusions.
Distribution of Tau inclusions
The neuropathological phenotypes associated with FTDP-17 MAPT vary substantially; however, the invariable hallmark is the presence of tau protein deposits in neurons or in both neurons and glia. No cases with only glial tau inclusions have been described. Tau deposits are abundant in cerebral cortex and white matter; subcortical and brain stem nuclei, as well as the spinal cord, are variably affected.
Inclusions are labelled by antibodies specific for the amino-terminus, the repeat region and the carboxy-terminus of tau. In addition, phosphorylation-dependent antibodies are used. According to the numbering of the longest human brain tau isoform, prominent phosphorylation sites are serines 202, 214, 235, 262, 356, 396, 404 and 422, and threonines 181, 205, 212 and 231. An antibody recognizing phosphorylated S262 and/or S356 labels NFTs, but not classical Pick bodies [136]. Antibody AT8, which recognizes tau phosphorylated at S202 and T205, labels tau deposits in neurons and glia. Some tau deposits are also immunoreactive for ubiquitin. RD3 and RD4 are anti-tau antibodies that recognize 3R and 4R tau, respectively.
Inclusions may resemble those of AD with filaments made of all six brain tau isoforms (see Table 1). This is the case of mutations V337M (Exon 12) and R406W (Exon 13), as illustrated in Figure 3. The images highlight neuronal involvement with tau immunopositivity revealed by an antibody specific for phosphorylated tau (AT8, Figure 3a,b), as well as by antibodies specific for 3R and 4R tau (Figure 3c–f). Inclusions similar to Pick bodies are often observed in association with mutations in exons 9, 11, 12 and 13. Straight filaments, with some twisted filaments, are characteristic of Pick body-like structures that are primarily composed of 3R tau, with a variable amount of 4R tau (Figure 4, Table 1). The images highlight Pick body-like inclusions immunopositive for phosphorylated tau (Figure 4a,b) and 3R tau (Figure 4c,d). There is occasional immunopositivity for 4R tau (Figure 4e,f). Mutations in exons 9, 11, 12 and 13 lead to deposits of tau filaments predominantly in neurons, while mutations in exons 1 and 10, as well as those in the introns following exons 9 and 10, are associated with neuronal and glial deposits. The glial pathology is in the form of coiled bodies in oligodendroglia, tufted astrocytes and astrocytic plaques, reminiscent of that of PSP and CBD. Cytoplasmic tau deposits affect the perikarya and dendrites of nerve cells. There is strong and diffuse cytoplasmic immunopositivity, but in Thioflavin S preparations, fluorescence is barely detectable, unlike what is seen in NFTs and Pick body-like inclusions. Twisted ribbon filaments characterize the neuronal and glial inclusions and are composed of 4R tau. Highlighted in Figure 5 are affected nerve cells and glial cells using antibodies specific for phosphorylated tau (Figure 5a,b) and 4R tau (Figure 5e,f). 3R tau staining was not observed (Figure 5c,d). Mutations in exon 10 only affect 4R tau; some of these mutations also affect exon 10 splicing, altering the ratio of 3R/4R tau. This is illustrated in Figure 6, where the immunohistochemical characteristics of neuronal and glial involvement in the hippocampus are revealed by antibodies specific for phosphorylated (a), 3R (b) and 4R (c) tau.
Figure 3.

Tau pathology in the hippocampus of a patient carrying the R406W mutation. Dentate gyrus (a, c, e) and pyramidal layers (b, d, f) of the hippocampus are immunolabeled with anti-tau antibodies, showing tau-immunoreactive neuropil threads and neurofibrillary tangles with antibodies AT8 (a, b), 3R tau (c, d) and 4R tau (e, f).
Figure 4.

Tau immunohistochemistry in the frontal cortex from a case with the G389R mutation. AT8 labelling demonstrates tau-immunoreactive deposits or Pick bodies in neurons of layers II-VI (a, b). The tau deposits are positive for 3R (c, d) and 4R (e, f) tau.
Figure 5.

Tau pathology in the hippocampus of a patient carrying the IVS10+16 mutation. Dentate gyrus (a, c, e) and pyramidal layers (b, d, f) of the hippocampus are immunolabeled with anti-tau antibodies, showing tau-immunoreactive inclusions with antibodies AT8 (a, b) and 4R tau (e, f), but not 3R tau (c, d).
Figure 6.
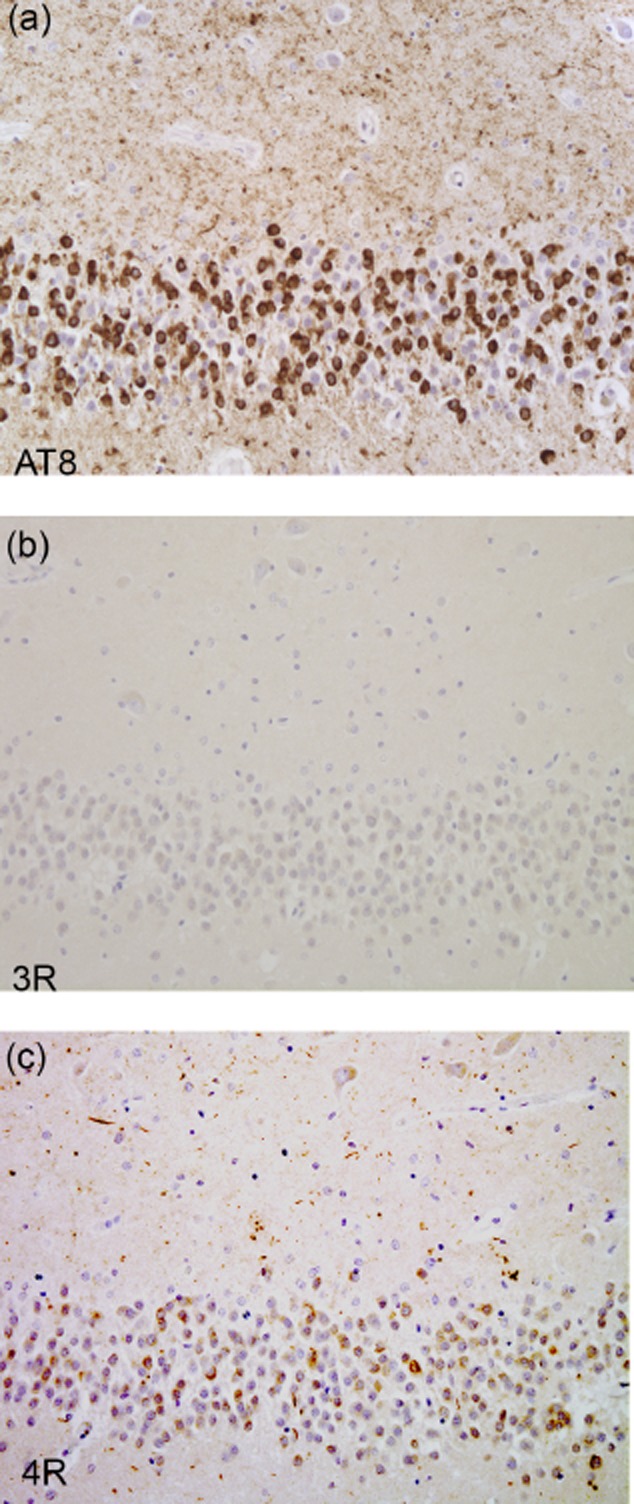
Tau pathology in the hippocampus of a patient carrying the P301L mutation. The dentate gyrus of the hippocampus is labeled with AT8 (a) and 4R tau (c), but not 3R tau (b).
The anatomical distribution of tau in the various regions of the central nervous system has been reported with different details in relation to individual mutations. In Table 2, the brain regions involved in FTDP-17 MAPT are presented according to the mutation and grey matter regions involved.
Table 2.
Brain areas affected in FTDP-17 MAPT according to mutation
 |
The data related to anatomical distribution are mostly obtained in intermediate and late stage of FTDP-17 MAPT. The degree of atrophy varies, with brain weights ranging from 654 to 1290 g. Little is known about the early neuropathologic stages. In the intermediate stages, atrophy of the cerebral hemispheres is mild, even though the characteristic histopathological changes in cerebral cortex, subcortical nuclei and white matter are already prominent. There may be mild atrophy of the caudate nucleus and a reduction in the pigmentation of the substantia nigra and the locus coeruleus. In advanced stages, the degree of atrophy varies and may be present throughout the frontal and temporal lobes, caudate nucleus, putamen, globus pallidus, amygdala, hippocampus and hypothalamus. Most often, the superior, middle and inferior frontal gyri, as well as the superior, middle and inferior temporal gyri, bear the brunt of the disease, with the anterior portion of the temporal lobe being particularly vulnerable. Brain atrophy may involve the frontal and temporal lobes asymmetrically and can be so severe that the gyri have a ‘knife edge’ appearance. The orbital, cingulate and parahippocampal gyri may also be involved. Parietal and occipital lobes are less frequently affected. The white matter of the centrum semiovale and the temporal lobes are often substantially reduced, as is the thickness of the corpus callosum. Midbrain and pons may also be reduced in bulk with particular involvement of the descending fibers of the fronto-pontine and temporo-pontine pathways. In addition, there is a reduction in the nigro-striatal projections. In some instances, mild atrophy of the cerebellar cortex and discoloration and atrophy of the dentate nucleus are present. The lateral ventricles and the third ventricle are enlarged.
Neuroimaging
Computerized tomography (CT) and magnetic resonance imaging (MRI) of patients with MAPT mutations reveal atrophy of the frontal and/or temporal lobes with occasional involvement of the parietal lobes, accompanied by enlargement of the lateral ventricles [16,74,82,96,121,137,138]. In some individuals, the cortical atrophy is asymmetrical, but the majority of cases have relatively symmetric patterns of atrophy. MRI T2*-weighted images may show accumulation of paramagnetic substances (iron) in mesencephalic nuclei [137]. Increased T2-weighted signal changes have been reported [139]; they are often seen in white matter, reflecting the prominent white matter pathology present in many cases. It is not yet clear if these changes are due to a loss of myelinated axons; additional radio-pathological studies are needed.
A few studies on familial FTD have begun to compare neuroimaging features resulting from mutations in different genes. MAPT mutations are associated with a relatively symmetric atrophy of the anterior temporal lobe, accompanied by lesser atrophy of orbitofrontal and lateral prefrontal cortices. Preliminary findings indicate that MAPT mutations affecting the splicing of exon 10 are predominantly associated with medial temporal lobe involvement, while mutations in the coding region are mainly associated with lateral temporal lobe involvement. This is important because it begins to differentiate patients with MAPT mutations from those with GRN or C9ORF72 mutations. GRN mutations tend to be associated with markedly asymmetric atrophy of the temporal, inferior frontal and inferior parietal lobes [138,140,141]. In contrast, C9ORF72 mutations tend to be associated with symmetric atrophy predominantly involving dorsolateral, medial and orbitofrontal lobes, with additional loss in anterior temporal, parietal and occipital lobes, as well as in the cerebellum [141].
An attempt to correlate structural brain imaging with the biological aspects of hereditary tauopathies may not be successful because of the different rates of atrophy and the sequences of anatomical involvement, which are highly variable even in cases with the same mutation. Figure 7 shows structural MRIs from patients who are carriers of MAPT mutations, V337M, G389R, IVS10+3 and P301L, which are respectively associated with inclusions containing 3R and 4R tau, predominantly 3R tau, predominantly 4R tau, and 4R with some 3R tau. An important confound in these comparisons is that these images are from individuals at different stages of disease, and specific details about the initial location of atrophy are no longer discernible.
Figure 7.

Coronal T1-weighted magnetic resonance imaging (MRI). Panel a is from a 65-year-old male with behavioural-variant frontotemporal dementia associated with the V337M MAPT mutation. Symptoms evolved over 20 years. Note the moderate to marked bilateral frontal and temporal cortical atrophy, with a severe anterior temporal lobe atrophy. Panel b is from a 25-year-old male with frontotemporal dementia and primary progressive aphasia associated with the G389R MAPT mutation. Symptoms rapidly developed over 1 year. Note the mild bilateral frontal and temporal cortical atrophy, with more pronounced medial and inferolateral anterior temporal atrophy. Panel c is from a 51-year-old male with behavioural-variant frontotemporal dementia associated with the IVS10+3 MAPT mutation. Symptoms evolved over 3 years. Note the mild bilateral frontal and temporal cortical atrophy with more pronounced mesial temporal atrophy. Panel d is from a 62-year-old female with severe behavioural-variant frontotemporal dementia associated with the P301L MAPT mutation. Symptoms evolved over 6 years. Note the striking bilateral prefrontal and anterior temporal atrophy with white matter changes.
Longitudinal MRI studies of brain atrophy suggest that MAPT mutations are associated with an atrophy rate intermediate between those of GRN and C9ORF72 [142,143].
Functional imaging studies, such as single photon emission CT (SPECT) and [F-18] fluorodeoxyglucose positron emissions tomography (FDG-PET), typically demonstrate substantial abnormalities. FDG-PET often shows reduced frontal and/or temporal uptake, similar to the patterns seen in sporadic FTD [144]. PET with dopaminergic (e.g. [F-18]-fluoro-L-dopa (6FD) and [C-11]-raclopride) tracers reveals uptake abnormalities different from those of Parkinson disease (PD) [145]. In the MSTD family, a study of multiple members carrying mutation IVS10+3 showed that structural changes, predominantly seen bilaterally in the medial temporal lobes, substantially overlapped with the hypometabolism observed with FDG-PET [146].
Investigations have begun to determine whether neuroimaging abnormalities are present in asymptomatic MAPT mutation carriers, with initial evidence suggesting that abnormalities of brain structure [147], connectivity [148],[149] and white matter tract integrity [148] may be detectable prior to the development of symptoms. Longitudinal changes in an asymptomatic MSTD mutation carrier showed that whole brain volume (WBV) changes were −0.47%/year in the first 2 years of assessment and −1.83%/year in the following 5 years, indicating an acceleration of the rate of brain atrophy and suggesting the approaching threshold of a clinically recognizable symptomatology [150]. In five symptomatic MSTD patients, the average WBV changes were −2.47%/year. Findings from the Genetic FTD Initiative suggest that structural changes can occur 25 years prior to symptom onset in the hippocampus, 15 years in the amygdala, 10 years in the temporal lobe and 5 years in insula and cingulate [151].
PET ligands to study tau pathology in vivo have been developed [152–157]. A series of compounds was tested for selectivity of binding to tau pathology in post-mortem brain tissue from patients with AD pathology [158]. Binding was compared against immunohistochemistry, and based on more than 25-fold greater binding to tissue sections with high tau burden relative to amyloid-β, [F-18]-T807 was selected; the first set of images and quantitative binding data of [F-18]-T807 to specific brain regions in a small group of patients with AD and normal controls was very encouraging [159].
A study at the Massachusetts General Hospital has begun to analyse MAPT mutation carriers with [F-18]-T807 PET. A 56-year-old man with the P301L mutation has been followed from prodromal FTD to bvFTD associated with an extrapyramidal syndrome. A [F-18]-T807 PET scan obtained 3.5 years from the onset of the behavioural symptoms (Figure 8) demonstrates robust signal in a classic frontotemporal distribution characteristic of inherited tauopathies and with remarkable similarity to the map of pathology described in the MSTD family by Spina and colleagues [108]. Sparing of the occipital cortex contrasts with severe involvement of the anterior and temporal regions of the telencephalon. Although involvement of the basal ganglia is variable in sporadic FTLD-tau, many MAPT cases have prominent pathology there. These studies are promising for the further characterization of patients with MAPT mutations.
Figure 8.

[F18]-T807 PET images from a 56-year-old individual with frontotemporal dementia and the P301L MAPT mutation. Coronal (top row), sagittal (middle row) and axial (bottom row) views of prefrontal and anterior temporal atrophy with white matter signal change on MRI (left column) and [F18]-T807 images (right column) showing elevated signal in frontal, anterior temporal and parietal cortex, as well as in basal ganglia, consistent with expected tau inclusions. The PET reference region was the cerebellar grey matter.
Comparative analysis of post-mortem tau immunohistochemistry with in vivo [F-18]-T807 PET is essential for understanding the sensitivity of the tracer and the evolution of hyperphosphorylated tau protein deposition. The post-mortem pattern of tau distribution, in the temporal cortex and hippocampus of a 62-year-old patient carrying the P301L MAPT mutation and symptomatic for 10 years, is shown in Figure 9. This image is compared with a PET scan obtained in vivo using the [F-18]-T807 tracer from the 56-year-old patient carrying the MAPT P301L mutation just described. Images obtained from immunohistochemistry and PET imaging reveal tau involvement in the middle temporal gyrus, parahippocampus, entorhinal cortex and hippocampus in both cases, as well as the sparing of the superior temporal gyrus.
Figure 9.

Post-mortem tau immunohistochemistry using AT8 (a) and in vivo [F-18]-T807 imaging (b) from two individuals with the P301L MAPT mutation. Tau-immunoreactivity is observed in the middle temporal gyrus, inferior temporal gyrus, fusiform gyrus, parahippocampus, entorhinal cortex and hippocampus. The superior temporal gyrus is spared (a). In vivo imaging using T807 demonstrates tau binding in the middle temporal gyrus, inferior temporal gyrus, fusiform gyrus, parahippocampus, entorhinal cortex and hippocampus, with no binding in the superior temporal gyrus (b).
In vivo tau imaging coupled with neuropathological investigation will improve our understanding of tau spreading in the brain and bring forward knowledge of the large number of disorders characterized by tau deposition [160],[161].
Clinical features
The onset of FTDP-17 MAPT is typically insidious. Individuals with fully developed clinical syndromes usually exhibit at least two of the three cardinal symptoms, which are behavioural and personality disturbances, cognitive impairment and/or motor dysfunction (most often in the form of an extrapyramidal/parkinsonism plus syndrome). Nevertheless, there is substantial heterogeneity. Moreover, clinical variability is seen in individuals with the same MAPT mutation, in different families or even within the same family (for details about clinical presentation, see Ghetti et al. [17]).
The behavioural and personality abnormalities include disinhibition, apathy, loss of empathy, emotional flatness, impulsive and/or compulsive behaviour, lack of regard for personal hygiene, hyperorality including excessive use of alcohol or other drugs, and in some cases verbal and/or physical aggressiveness. The cognitive symptoms commonly observed in early stages of disease include inattention and executive dysfunction (e.g. difficulty initiating or completing activities or tasks, disorganization, impaired judgment and decision making) with relative preservation of memory, orientation and visuospatial function, thus fulfilling criteria for behavioral variant FTD (bvFTD) [162]. Family members may report memory loss in daily life, but this is often a reflection of the effects of attentional or executive dysfunction on encoding or retrieval. However, some patients with FTDP-17 MAPT present with a profound amnestic syndrome [33]. Similarly, the literature contains statements about semantic dementia being a possible clinical phenotype of FTDP-17 MAPT, but all cases, except one, also had a behavioural phenotype [163]. A progressive loss of person-specific semantic memory with prominent anomia and right temporal polar atrophy, as well as other characteristics of semantic dementia, was described in an individual with the V363I MAPT mutation [119]. Thus, semantic memory in FTDP-17 deserves further investigation.
Progressive nonfluent aphasia may be seen initially [118], but more commonly, an adynamic aphasia syndrome occurs in which the patient speaks very little due to a loss of generative aspects of language. Later, progressive deterioration of memory, orientation and visuospatial function, as well as echolalia, palilalia, and verbal and vocal perseverations, are encountered. Finally, progressive dementia encompassing most cognitive domains develops, and patients often become mute. Motor signs are dominated by parkinsonism, which can be the presenting sign, with some patients being misdiagnosed as having PD or PSP. However, in some families, parkinsonism occurs late or not at all. Parkinsonism associated with FTDP-17 MAPT is characterized by symmetrical bradykinesia, postural instability and rigidity affecting axial and appendicular musculature, absence of resting tremor, and poor or no responsiveness to levodopa. Parkinsonism is an early feature of the N279K mutation, and asymmetric resting tremor and levodopa responsiveness have been observed [14]. Other motor disturbances may include dystonia, supranuclear gaze palsy, upper and lower motor neuron dysfunction, myoclonus, postural and action tremor, apraxia of eyelid opening and closing, dysphagia, and dysarthria.
Although essentially no systematic work has been published on genotype–phenotype correlations in FTDP-17 MAPT, anecdotal observations suggest that exonic mutations that do not affect the splicing of exon 10 are usually associated with a dementia-predominant phenotype. In contrast, intronic and exonic mutations that affect exon 10 splicing and lead to an overproduction of four-repeat tau tend to be associated with a parkinsonism plus-predominant phenotype.
Conclusion
This review emphasizes the protean nature of FTD associated with MAPT mutations, as well as the need for correlating longitudinal clinical and neuropsychological studies with neuroimaging. Ideally, this research should be carried out both before the onset of symptoms and during the disease in individuals with mutations that differentially affect tau isoforms. These studies, in conjunction with the neuropathological description of tau inclusions, will provide a precise characterization of phenotypic variants and may clarify the anatomical and cellular substrates of each phenotype, as well as the evolution of tau aggregate propagation.
Acknowledgments
We extend our appreciation to Clifford R. Jack, Jr. M.D. for providing some of the MR images and to Jill Murrell Ph.D., Matthew Baker B.S., Rosa Rademakers Ph.D. and Michael Hutton Ph.D. for performing some of the genetic analyses. We would like to thank Urs Kuederli, Rose Richardson, Brenda Dupree and Brad Glazier for their technical expertise and Diane Lucente for her genetic counselling contributions. We would like to thank the Department of Pathology and Laboratory Medicine at Indiana University School of Medicine, as well as the following grants, P30 AG010133 (BG), R21 NS084156 (BD), P50 AG016574 (BB) and R01 AG011378 (BB). The work of MG is supported by the UK Medical Research Council (U105184291).
Author contributions
BG, AO, BB, KJ, BD, MG: Contributed data and assisted in manuscript preparation.
References
- 1.Vermaart W. Over de ziekte van pick. Nederl Tijdschr Geneesk. 1930;74:13. [Google Scholar]
- 2.Schenk VW. Re-examination of a family with Pick's disease. Ann Hum Genet. 1959;23:325–333. doi: 10.1111/j.1469-1809.1959.tb01476.x. [DOI] [PubMed] [Google Scholar]
- 3.Groen JJ, Endtz LJ. Hereditary Pick's disease: second re-examination of the large family and discussion of other hereditary cases, with particular reference to electroencephalography, a computerized tomography. Brain. 1982;105(Pt 3):443–459. doi: 10.1093/brain/105.3.443. [DOI] [PubMed] [Google Scholar]
- 4.Heston LL. The clinical genetics of Pick's disease. Acta Psychiatr Scand. 1978;57:202–206. doi: 10.1111/j.1600-0447.1978.tb06886.x. [DOI] [PubMed] [Google Scholar]
- 5.Foster NL, Wilhelmsen K, Sima AA, Jones MZ, D'Amato CJ, Gilman S. Frontotemporal dementia and parkinsonism linked to chromosome 17: a consensus conference. Conference participants. Ann Neurol. 1997;41:706–715. doi: 10.1002/ana.410410606. [DOI] [PubMed] [Google Scholar]
- 6.Spillantini MG, Goedert M, Crowther RA, Murrell JR, Farlow MR, Ghetti B. Familial multiple system tauopathy with presenile dementia: a disease with abundant neuronal and glial tau filaments. Proc Natl Acad Sci U S A. 1997;94:4113–4118. doi: 10.1073/pnas.94.8.4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- 8.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Che LK, Norton J, Morris JC, Reed LA, Trojanowski J, Basun H, Lannfelt L, Neystat M, Fahn S, Dark F, Tannenberg T, Dodd PR, Hayward N, Kwok JB, Schofield PR, Andreadis A, Snowden J, Craufurd D, Neary D, Owen F, Oostra BA, Hardy J, Goate A, van Swieten J, Mann D, Lynch T, Heutink P. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 9.Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci U S A. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- 11.Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, Dickey CA, Crook R, McGowan E, Mann D, Boeve B, Feldman H, Hutton M. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 12.Boeve BF, Hutton M. Refining frontotemporal dementia with parkinsonism linked to chromosome 17: introducing FTDP-17 (MAPT) and FTDP-17 (PGRN) Arch Neurol. 2008;65:460–464. doi: 10.1001/archneur.65.4.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van der Zee J, Van Broeckhoven C. Dementia in 2013: frontotemporal lobar degeneration-building on breakthroughs. Nat Rev Neurol. 2014;10:70–72. doi: 10.1038/nrneurol.2013.270. [DOI] [PubMed] [Google Scholar]
- 14.Tsuboi Y, Baker M, Hutton ML, Uitti RJ, Rascol O, Delisle MB, Soulages X, Murrell JR, Ghetti B, Yasuda M, Komure O, Kuno S, Arima K, Sunohara N, Kobayashi T, Mizuno Y, Wszolek ZK. Clinical and genetic studies of families with the tau N279K mutation (FTDP-17) Neurology. 2002;59:1791–1793. doi: 10.1212/01.wnl.0000038909.49164.4b. [DOI] [PubMed] [Google Scholar]
- 15.Reed LA, Wszolek ZK, Hutton M. Phenotypic correlations in FTDP-17. Neurobiol Aging. 2001;22:89–107. doi: 10.1016/s0197-4580(00)00202-5. [DOI] [PubMed] [Google Scholar]
- 16.Wszolek ZK, Tsuboi Y, Farrer M, Uitti RJ, Hutton ML. Hereditary tauopathies and parkinsonism. Adv Neurol. 2003;91:153–163. [PubMed] [Google Scholar]
- 17.Ghetti BG, Wszolek ZK, Boeve BF, Spina S, Goedert M. Frontotemporal Dementia and Parkinsonism Linked to Chromosome 17. In: Dickson D, Weller R, editors. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. Second edn. Oxford, UK: Wiley-Blackwell; 2011. pp. 110–134. [Google Scholar]
- 18.Bugiani O, Murrell JR, Giaccone G, Hasegawa M, Ghigo G, Tabaton M, Morbin M, Primavera A, Carella F, Solaro C, Grisoli M, Savoiardo M, Spillantini MG, Tagliavini F, Goedert M, Ghetti B. Frontotemporal dementia and corticobasal degeneration in a family with a P301S mutation in tau. J Neuropathol Exp Neurol. 1999;58:667–677. doi: 10.1097/00005072-199906000-00011. [DOI] [PubMed] [Google Scholar]
- 19.Forman MS. Genotype-phenotype correlations in FTDP-17: does form follow function? Exp Neurol. 2004;187:229–234. doi: 10.1016/j.expneurol.2004.01.031. [DOI] [PubMed] [Google Scholar]
- 20.Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A. 1975;72:1858–1862. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brion JP, Passareiro H, Nunez J, Flament-Durand J. Mise en évidence immunologique de la protéine tau au niveau des lésions de dégénérescence neurofibrillaires de la maladie d'Alzheimer. Arch Bio. 1985;95:7. [Google Scholar]
- 22.Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pollock NJ, Mirra SS, Binder LI, Hansen LA, Wood JG. Filamentous aggregates in Pick's disease, progressive supranuclear palsy, and Alzheimer's disease share antigenic determinants with microtubule-associated protein, tau. Lancet. 1986;2:1211. doi: 10.1016/s0140-6736(86)92212-9. [DOI] [PubMed] [Google Scholar]
- 24.Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci U S A. 1988;85:4051–4055. doi: 10.1073/pnas.85.11.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wischik CM, Novak M, Edwards PC, Klug A, Tichelaar W, Crowther RA. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A. 1988;85:4884–4888. doi: 10.1073/pnas.85.13.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wischik CM, Novak M, Thogersen HC, Edwards PC, Runswick MJ, Jakes R, Walker JE, Milstein C, Roth M, Klug A. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A. 1988;85:4506–4510. doi: 10.1073/pnas.85.12.4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 28.Wilhelmsen KC, Lynch T, Pavlou E, Higgins M, Nygaard TG. Localization of disinhibition-dementia-parkinsonism-amyotrophy complex to 17q21-22. Am J Hum Genet. 1994;55:1159–1165. [PMC free article] [PubMed] [Google Scholar]
- 29.Heutink P, Stevens M, Rizzu P, Bakker E, Kros JM, Tibben A, Niermeijer MF, van Duijn CM, Oostra BA, van Swieten JC. Hereditary frontotemporal dementia is linked to chromosome 17q21-q22: a genetic and clinicopathological study of three Dutch families. Ann Neurol. 1997;41:150–159. doi: 10.1002/ana.410410205. [DOI] [PubMed] [Google Scholar]
- 30.Murrell JR, Koller D, Foroud T, Goedert M, Spillantini MG, Edenberg HJ, Farlow MR, Ghetti B. Familial multiple-system tauopathy with presenile dementia is localized to chromosome 17. Am J Hum Genet. 1997;61:1131–1138. doi: 10.1086/301594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghetti B, Farlow M. Familial dementia and multiple system degeneration with neurofibrillary tangles in hypothalamus, brainstem and spinal cord. Clinical Neuropathology. 1994;13:152. [Google Scholar]
- 32.Ghetti B, Goedert M, Spillantini MG, Farlow M. Hereditary multiple system degeneration with presenile dementia: abnormally phosphorylated τ in nerve cells and glia. 1995. p. 32. Fourth Joint American Society for Cell Biology/European Molecular Biology Organization Conference, Cambridge, England.
- 33.Reed LA, Grabowski TJ, Schmidt ML, Morris JC, Goate A, Solodkin A, Van Hoesen GW, Schelper RL, Talbot CJ, Wragg MA, Trojanowski JQ. Autosomal dominant dementia with widespread neurofibrillary tangles. Ann Neurol. 1997;42:564–572. doi: 10.1002/ana.410420406. [DOI] [PubMed] [Google Scholar]
- 34.Spillantini MG, Bird TD, Ghetti B. Frontotemporal dementia and Parkinsonism linked to chromosome 17: a new group of tauopathies. Brain Pathol. 1998;8:387–402. doi: 10.1111/j.1750-3639.1998.tb00162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dayanandan R, Van Slegtenhorst M, Mack TG, Ko L, Yen SH, Leroy K, Brion JP, Anderton BH, Hutton M, Lovestone S. Mutations in tau reduce its microtubule binding properties in intact cells and affect its phosphorylation. FEBS Lett. 1999;446:228–232. doi: 10.1016/s0014-5793(99)00222-7. [DOI] [PubMed] [Google Scholar]
- 36.Hasegawa M, Smith MJ, Goedert M. Tau proteins with FTDP-17 mutations have a reduced ability to promote microtubule assembly. FEBS Lett. 1998;437:207–210. doi: 10.1016/s0014-5793(98)01217-4. [DOI] [PubMed] [Google Scholar]
- 37.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VM. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–1917. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- 38.Clark LN, Poorkaj P, Wszolek Z, Geschwind DH, Nasreddine ZS, Miller B, Li D, Payami H, Awert F, Markopoulou K, Andreadis A, D'Souza I, Lee VM, Reed L, Trojanowski JQ, Zhukareva V, Bird T, Schellenberg G, Wilhelmsen KC. Pathogenic implications of mutations in the tau gene in pallido-ponto-nigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc Natl Acad Sci U S A. 1998;95:13103–13107. doi: 10.1073/pnas.95.22.13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.D'Souza I, Poorkaj P, Hong M, Nochlin D, Lee VM, Bird TD, Schellenberg GD. Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci U S A. 1999;96:5598–5603. doi: 10.1073/pnas.96.10.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grover A, Houlden H, Baker M, Adamson J, Lewis J, Prihar G, Pickering-Brown S, Duff K, Hutton M. 5′ splice site mutations in tau associated with the inherited dementia FTDP-17 affect a stem-loop structure that regulates alternative splicing of exon 10. J Biol Chem. 1999;274:15134–15143. doi: 10.1074/jbc.274.21.15134. [DOI] [PubMed] [Google Scholar]
- 41.Jiang Z, Cote J, Kwon JM, Goate AM, Wu JY. Aberrant splicing of tau pre-mRNA caused by intronic mutations associated with the inherited dementia frontotemporal dementia with parkinsonism linked to chromosome 17. Mol Cell Biol. 2000;20:4036–4048. doi: 10.1128/mcb.20.11.4036-4048.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Donahue CP, Muratore C, Wu JY, Kosik KS, Wolfe MS. Stabilization of the tau exon 10 stem loop alters pre-mRNA splicing. J Biol Chem. 2006;281:23302–23306. doi: 10.1074/jbc.C600143200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Varani L, Hasegawa M, Spillantini MG, Smith MJ, Murrell JR, Ghetti B, Klug A, Goedert M, Varani G. Structure of tau exon 10 splicing regulatory element RNA and destabilization by mutations of frontotemporal dementia and parkinsonism linked to chromosome 17. Proc Natl Acad Sci U S A. 1999;96:8229–8234. doi: 10.1073/pnas.96.14.8229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.D'Souza I, Schellenberg GD. tau Exon 10 expression involves a bipartite intron 10 regulatory sequence and weak 5′ and 3′ splice sites. J Biol Chem. 2002;277:26587–26599. doi: 10.1074/jbc.M203794200. [DOI] [PubMed] [Google Scholar]
- 45.Goedert M, Jakes R. Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990;9:4225–4230. doi: 10.1002/j.1460-2075.1990.tb07870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Adams SJ, DeTure MA, McBride M, Dickson DW, Petrucelli L. Three repeat isoforms of tau inhibit assembly of four repeat tau filaments. PLoS ONE. 2010;5:e10810. doi: 10.1371/journal.pone.0010810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rosso SM, van Herpen E, Deelen W, Kamphorst W, Severijnen LA, Willemsen R, Ravid R, Niermeijer MF, Dooijes D, Smith MJ, Goedert M, Heutink P, van Swieten JC. A novel tau mutation, S320F, causes a tauopathy with inclusions similar to those in Pick's disease. Ann Neurol. 2002;51:373–376. doi: 10.1002/ana.10140. [DOI] [PubMed] [Google Scholar]
- 48.Poorkaj P, Muma NA, Zhukareva V, Cochran EJ, Shannon KM, Hurtig H, Koller WC, Bird TD, Trojanowski JQ, Lee VM, Schellenberg GD. An R5L tau mutation in a subject with a progressive supranuclear palsy phenotype. Ann Neurol. 2002;52:511–516. doi: 10.1002/ana.10340. [DOI] [PubMed] [Google Scholar]
- 49.Spillantini MG, Crowther RA, Kamphorst W, Heutink P, van Swieten JC. Tau pathology in two Dutch families with mutations in the microtubule-binding region of tau. Am J Pathol. 1998;153:1359–1363. doi: 10.1016/S0002-9440(10)65721-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pickering-Brown S, Baker M, Yen SH, Liu WK, Hasegawa M, Cairns N, Lantos PL, Rossor M, Iwatsubo T, Davies Y, Allsop D, Furlong R, Owen F, Hardy J, Mann D, Hutton M. Pick's disease is associated with mutations in the tau gene. Ann Neurol. 2000;48:859–867. [PubMed] [Google Scholar]
- 51.Rizzini C, Goedert M, Hodges JR, Smith MJ, Jakes R, Hills R, Xuereb JH, Crowther RA, Spillantini MG. Tau gene mutation K257T causes a tauopathy similar to Pick's disease. J Neuropathol Exp Neurol. 2000;59:990–1001. doi: 10.1093/jnen/59.11.990. [DOI] [PubMed] [Google Scholar]
- 52.Kobayashi T, Ota S, Tanaka K, Ito Y, Hasegawa M, Umeda Y, Motoi Y, Takanashi M, Yasuhara M, Anno M, Mizuno Y, Mori H. A novel L266V mutation of the tau gene causes frontotemporal dementia with a unique tau pathology. Ann Neurol. 2003;53:133–137. doi: 10.1002/ana.10447. [DOI] [PubMed] [Google Scholar]
- 53.Hogg M, Grujic ZM, Baker M, Demirci S, Guillozet AL, Sweet AP, Herzog LL, Weintraub S, Mesulam MM, LaPointe NE, Gamblin TC, Berry RW, Binder LI, de Silva R, Lees A, Espinoza M, Davies P, Grover A, Sahara N, Ishizawa T, Dickson D, Yen SH, Hutton M, Bigio EH. The L266V tau mutation is associated with frontotemporal dementia and Pick-like 3R and 4R tauopathy. Acta Neuropathol. 2003;106:323–336. doi: 10.1007/s00401-003-0734-x. [DOI] [PubMed] [Google Scholar]
- 54.Bronner IF, ter Meulen BC, Azmani A, Severijnen LA, Willemsen R, Kamphorst W, Ravid R, Heutink P, van Swieten JC. Hereditary Pick's disease with the G272V tau mutation shows predominant three-repeat tau pathology. Brain. 2005;128:2645–2653. doi: 10.1093/brain/awh591. [DOI] [PubMed] [Google Scholar]
- 55.Van Deerlin VM, Forman MS, Farmer JM, Grossman M, Joyce S, Crowe A, Trojanowski JQ, Lee VM, Chatterjee A. Biochemical and pathological characterization of frontotemporal dementia due to a Leu266Val mutation in microtubule-associated protein tau in an African American individual. Acta Neuropathol. 2007;113:471–479. doi: 10.1007/s00401-006-0155-8. [DOI] [PubMed] [Google Scholar]
- 56.de Silva R, Lashley T, Strand C, Shiarli AM, Shi J, Tian J, Bailey KL, Davies P, Bigio EH, Arima K, Iseki E, Murayama S, Kretzschmar H, Neumann M, Lippa C, Halliday G, MacKenzie J, Ravid R, Dickson D, Wszolek Z, Iwatsubo T, Pickering-Brown SM, Holton J, Lees A, Revesz T, Mann DM. An immunohistochemical study of cases of sporadic and inherited frontotemporal lobar degeneration using 3R- and 4R-specific tau monoclonal antibodies. Acta Neuropathol. 2006;111:329–340. doi: 10.1007/s00401-006-0048-x. [DOI] [PubMed] [Google Scholar]
- 57.Grover A, England E, Baker M, Sahara N, Adamson J, Granger B, Houlden H, Passant U, Yen SH, DeTure M, Hutton M. A novel tau mutation in exon 9 (1260V) causes a four-repeat tauopathy. Exp Neurol. 2003;184:131–140. doi: 10.1016/s0014-4886(03)00393-5. [DOI] [PubMed] [Google Scholar]
- 58.van der Zee J, Rademakers R, Engelborghs S, Gijselinck I, Bogaerts V, Vandenberghe R, Santens P, Caekebeke J, De Pooter T, Peeters K, Lubke U, Van den Broeck M, Martin JJ, Cruts M, De Deyn PP, Van Broeckhoven C, Dermaut B. A Belgian ancestral haplotype harbours a highly prevalent mutation for 17q21-linked tau-negative FTLD. Brain. 2006;129:841–852. doi: 10.1093/brain/awl029. [DOI] [PubMed] [Google Scholar]
- 59.Ghetti B, Murrell JR, Hagen MC, Geldmacher D, Foroud T, Goedert M, Spina S. Clinicopathologic characterization of frontotemporal dementia associated with the IVS10-10G > T MAPT gene mutation. J Neuropathol Exp Neurol. 2010;69:551. [Google Scholar]
- 60.Wszolek ZK, Pfeiffer RF, Bhatt MH, Schelper RL, Cordes M, Snow BJ, Rodnitzky RL, Wolters EC, Arwert F, Calne DB. Rapidly progressive autosomal dominant parkinsonism and dementia with pallido-ponto-nigral degeneration. Ann Neurol. 1992;32:312–320. doi: 10.1002/ana.410320303. [DOI] [PubMed] [Google Scholar]
- 61.Reed LA, Schmidt ML, Wszolek ZK, Balin BJ, Soontornniyomkij V, Lee VM, Trojanowski JQ, Schelper RL. The neuropathology of a chromosome 17-linked autosomal dominant parkinsonism and dementia (‘pallido-ponto-nigral degeneration’) J Neuropathol Exp Neurol. 1998;57:588–601. doi: 10.1097/00005072-199806000-00006. [DOI] [PubMed] [Google Scholar]
- 62.Delisle MB, Murrell JR, Richardson R, Trofatter JA, Rascol O, Soulages X, Mohr M, Calvas P, Ghetti B. A mutation at codon 279 (N279K) in exon 10 of the Tau gene causes a tauopathy with dementia and supranuclear palsy. Acta Neuropathol. 1999;98:62–77. doi: 10.1007/s004010051052. [DOI] [PubMed] [Google Scholar]
- 63.Yasuda M, Kawamata T, Komure O, Kuno S, D'Souza I, Poorkaj P, Kawai J, Tanimukai S, Yamamoto Y, Hasegawa H, Sasahara M, Hazama F, Schellenberg GD, Tanaka C. A mutation in the microtubule-associated protein tau in pallido-nigro-luysian degeneration. Neurology. 1999;53:864–868. doi: 10.1212/wnl.53.4.864. [DOI] [PubMed] [Google Scholar]
- 64.Arima K, Kowalska A, Hasegawa M, Mukoyama M, Watanabe R, Kawai M, Takahashi K, Iwatsubo T, Tabira T, Sunohara N. Two brothers with frontotemporal dementia and parkinsonism with an N279K mutation of the tau gene. Neurology. 2000;54:1787–1795. doi: 10.1212/wnl.54.9.1787. [DOI] [PubMed] [Google Scholar]
- 65.Rizzu P, Van Swieten JC, Joosse M, Hasegawa M, Stevens M, Tibben A, Niermeijer MF, Hillebrand M, Ravid R, Oostra BA, Goedert M, van Duijn CM, Heutink P. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am J Hum Genet. 1999;64:414–421. doi: 10.1086/302256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Momeni P, Pittman A, Lashley T, Vandrovcova J, Malzer E, Luk C, Hulette C, Lees A, Revesz T, Hardy J, de Silva R. Clinical and pathological features of an Alzheimer's disease patient with the MAPT Delta K280 mutation. Neurobiol Aging. 2009;30:388–393. doi: 10.1016/j.neurobiolaging.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van Swieten JC, Bronner IF, Azmani A, Severijnen LA, Kamphorst W, Ravid R, Rizzu P, Willemsen R, Heutink P. The DeltaK280 mutation in MAP tau favors exon 10 skipping in vivo. J Neuropathol Exp Neurol. 2007;66:17–25. doi: 10.1097/nen.0b013e31802c39a4. [DOI] [PubMed] [Google Scholar]
- 68.Spillantini MG, Yoshida H, Rizzini C, Lantos PL, Khan N, Rossor MN, Goedert M, Brown J. A novel tau mutation (N296N) in familial dementia with swollen achromatic neurons and corticobasal inclusion bodies. Ann Neurol. 2000;48:939–943. doi: 10.1002/1531-8249(200012)48:6<939::aid-ana17>3.3.co;2-t. [DOI] [PubMed] [Google Scholar]
- 69.Iseki E, Matsumura T, Marui W, Hino H, Odawara T, Sugiyama N, Suzuki K, Sawada H, Arai T, Kosaka K. Familial frontotemporal dementia and parkinsonism with a novel N296H mutation in exon 10 of the tau gene and a widespread tau accumulation in the glial cells. Acta Neuropathol. 2001;102:285–292. doi: 10.1007/s004010000333. [DOI] [PubMed] [Google Scholar]
- 70.Pastor P, Pastor E, Carnero C, Vela R, Garcia T, Amer G, Tolosa E, Oliva R. Familial atypical progressive supranuclear palsy associated with homozigosity for the delN296 mutation in the tau gene. Ann Neurol. 2001;49:263–267. doi: 10.1002/1531-8249(20010201)49:2<263::aid-ana50>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 71.Ferrer I, Pastor P, Rey MJ, Munoz E, Puig B, Pastor E, Oliva R, Tolosa E. Tau phosphorylation and kinase activation in familial tauopathy linked to deln296 mutation. Neuropathol Appl Neurobiol. 2003;29:23–34. doi: 10.1046/j.1365-2990.2003.00435.x. [DOI] [PubMed] [Google Scholar]
- 72.Rossi G, Gasparoli E, Pasquali C, Di Fede G, Testa D, Albanese A, Bracco F, Tagliavini F. Progressive supranuclear palsy and Parkinson's disease in a family with a new mutation in the tau gene. Ann Neurol. 2004;55:448. doi: 10.1002/ana.20006. [DOI] [PubMed] [Google Scholar]
- 73.Houlden H, Baker M, Adamson J, Grover A, Waring S, Dickson D, Lynch T, Boeve B, Petersen RC, Pickering-Brown S, Owen F, Neary D, Craufurd D, Snowden J, Mann D, Hutton M. Frequency of tau mutations in three series of non-Alzheimer's degenerative dementia. Ann Neurol. 1999;46:243–248. doi: 10.1002/1531-8249(199908)46:2<243::aid-ana14>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 74.Dumanchin C, Camuzat A, Campion D, Verpillat P, Hannequin D, Dubois B, Saugier-Veber P, Martin C, Penet C, Charbonnier F, Agid Y, Frebourg T, Brice A. Segregation of a missense mutation in the microtubule-associated protein tau gene with familial frontotemporal dementia and parkinsonism. Hum Mol Genet. 1998;7:1825–1829. doi: 10.1093/hmg/7.11.1825. [DOI] [PubMed] [Google Scholar]
- 75.Bird TD, Nochlin D, Poorkaj P, Cherrier M, Kaye J, Payami H, Peskind E, Lampe TH, Nemens E, Boyer PJ, Schellenberg GD. A clinical pathological comparison of three families with frontotemporal dementia and identical mutations in the tau gene (P301L) Brain. 1999;122(Pt 4):741–756. doi: 10.1093/brain/122.4.741. [DOI] [PubMed] [Google Scholar]
- 76.Mirra SS, Murrell JR, Gearing M, Spillantini MG, Goedert M, Crowther RA, Levey AI, Jones R, Green J, Shoffner JM, Wainer BH, Schmidt ML, Trojanowski JQ, Ghetti B. Tau pathology in a family with dementia and a P301L mutation in tau. J Neuropathol Exp Neurol. 1999;58:335–345. doi: 10.1097/00005072-199904000-00004. [DOI] [PubMed] [Google Scholar]
- 77.Nasreddine ZS, Loginov M, Clark LN, Lamarche J, Miller BL, Lamontagne A, Zhukareva V, Lee VM, Wilhelmsen KC, Geschwind DH. From genotype to phenotype: a clinical pathological, and biochemical investigation of frontotemporal dementia and parkinsonism (FTDP-17) caused by the P301L tau mutation. Ann Neurol. 1999;45:704–715. doi: 10.1002/1531-8249(199906)45:6<704::aid-ana4>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 78.Sperfeld AD, Collatz MB, Baier H, Palmbach M, Storch A, Schwarz J, Tatsch K, Reske S, Joosse M, Heutink P, Ludolph AC. FTDP-17: an early-onset phenotype with parkinsonism and epileptic seizures caused by a novel mutation. Ann Neurol. 1999;46:708–715. doi: 10.1002/1531-8249(199911)46:5<708::aid-ana5>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 79.van Swieten JC, Stevens M, Rosso SM, Rizzu P, Joosse M, de Koning I, Kamphorst W, Ravid R, Spillantini MG, Niermeijer MF, Heutink P. Phenotypic variation in hereditary frontotemporal dementia with tau mutations. Ann Neurol. 1999;46:617–626. doi: 10.1002/1531-8249(199910)46:4<617::aid-ana10>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 80.Kodama K, Okada S, Iseki E, Kowalska A, Tabira T, Hosoi N, Yamanouchi N, Noda S, Komatsu N, Nakazato M, Kumakiri C, Yazaki M, Sato T. Familial frontotemporal dementia with a P301L tau mutation in Japan. J Neurol Sci. 2000;176:57–64. doi: 10.1016/s0022-510x(00)00288-4. [DOI] [PubMed] [Google Scholar]
- 81.Tanaka R, Kobayashi T, Motoi Y, Anno M, Mizuno Y, Mori H. A case of frontotemporal dementia with tau P301L mutation in the Far East. J Neurol. 2000;247:705–707. doi: 10.1007/s004150070115. [DOI] [PubMed] [Google Scholar]
- 82.Yasuda M, Takamatsu J, D'Souza I, Crowther RA, Kawamata T, Hasegawa M, Hasegawa H, Spillantini MG, Tanimukai S, Poorkaj P, Varani L, Varani G, Iwatsubo T, Goedert M, Schellenberg DG, Tanaka C. A novel mutation at position +12 in the intron following exon 10 of the tau gene in familial frontotemporal dementia (FTD-Kumamoto) Ann Neurol. 2000;47:422–429. [PubMed] [Google Scholar]
- 83.Walker RH, Friedman J, Wiener J, Hobler R, Gwinn-Hardy K, Adam A, DeWolfe J, Gibbs R, Baker M, Farrer M, Hutton M, Hardy J. A family with a tau P301L mutation presenting with parkinsonism. Parkinsonism Relat Disord. 2002;9:121–123. doi: 10.1016/s1353-8020(02)00003-2. [DOI] [PubMed] [Google Scholar]
- 84.Werber E, Klein C, Grunfeld J, Rabey JM. Phenotypic presentation of frontotemporal dementia with Parkinsonism-chromosome 17 type P301S in a patient of Jewish-Algerian origin. Mov Disord. 2003;18:595–598. doi: 10.1002/mds.10401. [DOI] [PubMed] [Google Scholar]
- 85.Lossos A, Reches A, Gal A, Newman JP, Soffer D, Gomori JM, Boher M, Ekstein D, Biran I, Meiner Z, Abramsky O, Rosenmann H. Frontotemporal dementia and parkinsonism with the P301S tau gene mutation in a Jewish family. J Neurol. 2003;250:733–740. doi: 10.1007/s00415-003-1074-4. [DOI] [PubMed] [Google Scholar]
- 86.Alberici A, Gobbo C, Panzacchi A, Nicosia F, Ghidoni R, Benussi L, Hock C, Papassotiropoulos A, Liberini P, Growdon JH, Frisoni GB, Villa A, Zanetti O, Cappa S, Fazio F, Binetti G. Frontotemporal dementia: impact of P301L tau mutation on a healthy carrier. J Neurol Neurosurg Psychiatry. 2004;75:1607–1610. doi: 10.1136/jnnp.2003.021295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Casseron W, Azulay JP, Guedj E, Gastaut JL, Pouget J. Familial autosomal dominant cortico-basal degeneration with the P301S mutation in the tau gene: an example of phenotype variability. J Neurol. 2005;252:1546–1548. doi: 10.1007/s00415-005-0880-2. [DOI] [PubMed] [Google Scholar]
- 88.Yasuda M, Nakamura Y, Kawamata T, Kaneyuki H, Maeda K, Komure O. Phenotypic heterogeneity within a new family with the MAPT p301s mutation. Ann Neurol. 2005;58:920–928. doi: 10.1002/ana.20668. [DOI] [PubMed] [Google Scholar]
- 89.Llado A, Ezquerra M, Sanchez-Valle R, Rami L, Tolosa E, Molinuevo JL. A novel MAPT mutation (P301T) associated with familial frontotemporal dementia. Eur J Neurol. 2007;14:e9–10. doi: 10.1111/j.1468-1331.2007.01763.x. [DOI] [PubMed] [Google Scholar]
- 90.Miyasaka T, Morishima-Kawashima M, Ravid R, Kamphorst W, Nagashima K, Ihara Y. Selective deposition of mutant tau in the FTDP-17 brain affected by the P301L mutation. J Neuropathol Exp Neurol. 2001;60:872–884. doi: 10.1093/jnen/60.9.872. [DOI] [PubMed] [Google Scholar]
- 91.Rizzu P, Joosse M, Ravid R, Hoogeveen A, Kamphorst W, van Swieten JC, Willemsen R, Heutink P. Mutation-dependent aggregation of tau protein and its selective depletion from the soluble fraction in brain of P301L FTDP-17 patients. Hum Mol Genet. 2000;9:3075–3082. doi: 10.1093/hmg/9.20.3075. [DOI] [PubMed] [Google Scholar]
- 92.Ros R, Thobois S, Streichenberger N, Kopp N, Sanchez MP, Perez M, Hoenicka J, Avila J, Honnorat J, de Yebenes JG. A new mutation of the tau gene, G303V, in early-onset familial progressive supranuclear palsy. Arch Neurol. 2005;62:1444–1450. doi: 10.1001/archneur.62.9.1444. [DOI] [PubMed] [Google Scholar]
- 93.Kovacs GG, Pittman A, Revesz T, Luk C, Lees A, Kiss E, Tariska P, Laszlo L, Molnar K, Molnar MJ, Tolnay M, de Silva R. MAPT S305I mutation: implications for argyrophilic grain disease. Acta Neuropathol. 2008;116:103–118. doi: 10.1007/s00401-007-0322-6. [DOI] [PubMed] [Google Scholar]
- 94.Iijima M, Tabira T, Poorkaj P, Schellenberg GD, Trojanowski JQ, Lee VM, Schmidt ML, Takahashi K, Nabika T, Matsumoto T, Yamashita Y, Yoshioka S, Ishino H. A distinct familial presenile dementia with a novel missense mutation in the tau gene. Neuroreport. 1999;10:497–501. doi: 10.1097/00001756-199902250-00010. [DOI] [PubMed] [Google Scholar]
- 95.Kobayashi K, Kidani T, Ujike H, Hayashi M, Ishihara T, Miyazu K, Kuroda S, Koshino Y. Another phenotype of frontotemporal dementia and parkinsonism linked to chromosome-17 (FTDP-17) with a missense mutation of S305N closely resembling Pick's disease. J Neurol. 2003;250:990–992. doi: 10.1007/s00415-003-1137-6. [DOI] [PubMed] [Google Scholar]
- 96.Boeve BF, Tremont-Lukats IW, Waclawik AJ, Murrell JR, Hermann B, Jack CR, Jr, Shiung MM, Smith GE, Nair AR, Lindor N, Koppikar V, Ghetti B. Longitudinal characterization of two siblings with frontotemporal dementia and parkinsonism linked to chromosome 17 associated with the S305N tau mutation. Brain. 2005;128:752–772. doi: 10.1093/brain/awh356. [DOI] [PubMed] [Google Scholar]
- 97.Stanford PM, Halliday GM, Brooks WS, Kwok JB, Storey CE, Creasey H, Morris JG, Fulham MJ, Schofield PR. Progressive supranuclear palsy pathology caused by a novel silent mutation in exon 10 of the tau gene: expansion of the disease phenotype caused by tau gene mutations. Brain. 2000;123(Pt 5):880–893. doi: 10.1093/brain/123.5.880. [DOI] [PubMed] [Google Scholar]
- 98.Halliday GM, Song YJ, Creasey H, Morris JG, Brooks WS, Kril JJ. Neuropathology in the S305S tau gene mutation. Brain. 2006;129:E40. doi: 10.1093/brain/awh720. [DOI] [PubMed] [Google Scholar]
- 99.Janssen JC, Warrington EK, Morris HR, Lantos P, Brown J, Revesz T, Wood N, Khan MN, Cipolotti L, Fox NC, Rossor MN. Clinical features of frontotemporal dementia due to the intronic tau 10(+16) mutation. Neurology. 2002;58:1161–1168. doi: 10.1212/wnl.58.8.1161. [DOI] [PubMed] [Google Scholar]
- 100.Goedert M, Spillantini MG, Crowther RA, Chen SG, Parchi P, Tabaton M, Lanska DJ, Markesbery WR, Wilhelmsen KC, Dickson DW, Petersen RB, Gambetti P. Tau gene mutation in familial progressive subcortical gliosis. Nat Med. 1999;5:454–457. doi: 10.1038/7454. [DOI] [PubMed] [Google Scholar]
- 101.Hulette CM, Pericak-Vance MA, Roses AD, Schmechel DE, Yamaoka LH, Gaskell PC, Welsh-Bohmer KA, Crowther RA, Spillantini MG. Neuropathological features of frontotemporal dementia and parkinsonism linked to chromosome 17q21–22 (FTDP-17): Duke Family 1684. J Neuropathol Exp Neurol. 1999;58:859–866. doi: 10.1097/00005072-199908000-00008. [DOI] [PubMed] [Google Scholar]
- 102.Tolnay M, Spillantini MG, Rizzini C, Eccles D, Lowe J, Ellison D. A new case of frontotemporal dementia and parkinsonism resulting from an intron 10 +3-splice site mutation in the tau gene: clinical and pathological features. Neuropathol Appl Neurobiol. 2000;26:368–378. doi: 10.1046/j.1365-2990.2000.00109.x. [DOI] [PubMed] [Google Scholar]
- 103.Miyamoto K, Kowalska A, Hasegawa M, Tabira T, Takahashi K, Araki W, Akiguchi I, Ikemoto A. Familial frontotemporal dementia and parkinsonism with a novel mutation at an intron 10+11-splice site in the tau gene. Ann Neurol. 2001;50:117–120. doi: 10.1002/ana.1083. [DOI] [PubMed] [Google Scholar]
- 104.Lantos PL, Cairns NJ, Khan MN, King A, Revesz T, Janssen JC, Morris H, Rossor MN. Neuropathologic variation in frontotemporal dementia due to the intronic tau 10(+16) mutation. Neurology. 2002;58:1169–1175. doi: 10.1212/wnl.58.8.1169. [DOI] [PubMed] [Google Scholar]
- 105.Pickering-Brown SM, Richardson AM, Snowden JS, McDonagh AM, Burns A, Braude W, Baker M, Liu WK, Yen SH, Hardy J, Hutton M, Davies Y, Allsop D, Craufurd D, Neary D, Mann DM. Inherited frontotemporal dementia in nine British families associated with intronic mutations in the tau gene. Brain. 2002;125:732–751. doi: 10.1093/brain/awf069. [DOI] [PubMed] [Google Scholar]
- 106.Morris HR, Osaki Y, Holton J, Lees AJ, Wood NW, Revesz T, Quinn N. Tau exon 10+16 mutation FTDP-17 presenting clinically as sporadic young onset PSP. Neurology. 2003;61:102–104. doi: 10.1212/01.wnl.0000072325.27824.a5. [DOI] [PubMed] [Google Scholar]
- 107.Neumann M, Mittelbronn M, Simon P, Vanmassenhove B, de Silva R, Lees A, Klapp J, Meyermann R, Kretzschmar HA. A new family with frontotemporal dementia with intronic 10+3 splice site mutation in the tau gene: neuropathology and molecular effects. Neuropathol Appl Neurobiol. 2005;31:362–373. doi: 10.1111/j.1365-2990.2005.00629.x. [DOI] [PubMed] [Google Scholar]
- 108.Spina S, Farlow MR, Unverzagt FW, Kareken DA, Murrell JR, Fraser G, Epperson F, Crowther RA, Spillantini MG, Goedert M, Ghetti B. The tauopathy associated with mutation +3 in intron 10 of Tau: characterization of the MSTD family. Brain. 2008;131:72–89. doi: 10.1093/brain/awm280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.van Herpen E, Rosso SM, Serverijnen LA, Yoshida H, Breedveld G, van de Graaf R, Kamphorst W, Ravid R, Willemsen R, Dooijes D, Majoor-Krakauer D, Kros JM, Crowther RA, Goedert M, Heutink P, van Swieten JC. Variable phenotypic expression and extensive tau pathology in two families with the novel tau mutation L315R. Ann Neurol. 2003;54:573–581. doi: 10.1002/ana.10721. [DOI] [PubMed] [Google Scholar]
- 110.Zarranz JJ, Ferrer I, Lezcano E, Forcadas MI, Eizaguirre B, Atares B, Puig B, Gomez-Esteban JC, Fernandez-Maiztegui C, Rouco I, Perez-Concha T, Fernandez M, Rodriguez O, Rodriguez-Martinez AB, de Pancorbo MM, Pastor P, Perez-Tur J. A novel mutation (K317M) in the MAPT gene causes FTDP and motor neuron disease. Neurology. 2005;64:1578–1585. doi: 10.1212/01.WNL.0000160116.65034.12. [DOI] [PubMed] [Google Scholar]
- 111.Spina S, Murrell JR, Yoshida H, Ghetti B, Bermingham N, Sweeney B, Dlouhy SR, Crowther RA, Goedert M, Keohane C. The novel Tau mutation G335S: clinical, neuropathological and molecular characterization. Acta Neuropathol. 2007;113:461–470. doi: 10.1007/s00401-006-0182-5. [DOI] [PubMed] [Google Scholar]
- 112.Neumann M, Diekmann S, Bertsch U, Vanmassenhove B, Bogerts B, Kretzschmar HA. Novel G335V mutation in the tau gene associated with early onset familial frontotemporal dementia. Neurogenetics. 2005;6:91–95. doi: 10.1007/s10048-005-0210-y. [DOI] [PubMed] [Google Scholar]
- 113.Pickering-Brown SM, Baker M, Nonaka T, Ikeda K, Sharma S, Mackenzie J, Simpson SA, Moore JW, Snowden JS, de Silva R, Revesz T, Hasegawa M, Hutton M, Mann DM. Frontotemporal dementia with Pick-type histology associated with Q336R mutation in the tau gene. Brain. 2004;127:1415–1426. doi: 10.1093/brain/awh147. [DOI] [PubMed] [Google Scholar]
- 114.Spillantini MG, Crowther RA, Goedert M. Comparison of the neurofibrillary pathology in Alzheimer's disease and familial presenile dementia with tangles. Acta Neuropathol. 1996;92:42–48. doi: 10.1007/s004010050487. [DOI] [PubMed] [Google Scholar]
- 115.Sumi SM, Bird TD, Nochlin D, Raskind MA. Familial presenile dementia with psychosis associated with cortical neurofibrillary tangles and degeneration of the amygdala. Neurology. 1992;42:120–127. doi: 10.1212/wnl.42.1.120. [DOI] [PubMed] [Google Scholar]
- 116.Lippa CF, Zhukareva V, Kawarai T, Uryu K, Shafiq M, Nee LE, Grafman J, Liang Y, St George-Hyslop PH, Trojanowski JQ, Lee VM. Frontotemporal dementia with novel tau pathology and a Glu342Val tau mutation. Ann Neurol. 2000;48:850–858. [PubMed] [Google Scholar]
- 117.Nicholl DJ, Greenstone MA, Clarke CE, Rizzu P, Crooks D, Crowe A, Trojanowski JQ, Lee VM, Heutink P. An English kindred with a novel recessive tauopathy and respiratory failure. Ann Neurol. 2003;54:682–686. doi: 10.1002/ana.10747. [DOI] [PubMed] [Google Scholar]
- 118.Munoz DG, Ros R, Fatas M, Bermejo F, de Yebenes JG. Progressive nonfluent aphasia associated with a new mutation V363I in tau gene. Am J Alzheimers Dis Other Demen. 2007;22:294–299. doi: 10.1177/1533317507302320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bessi V, Bagnoli S, Nacmias B, Tedde A, Sorbi S, Bracco L. Semantic dementia associated with mutation V363I in the tau gene. J Neurol Sci. 2010;296:112–114. doi: 10.1016/j.jns.2010.06.007. [DOI] [PubMed] [Google Scholar]
- 120.Neumann M, Schulz-Schaeffer W, Crowther RA, Smith MJ, Spillantini MG, Goedert M, Kretzschmar HA. Pick's disease associated with the novel Tau gene mutation K369I. Ann Neurol. 2001;50:503–513. doi: 10.1002/ana.1223. [DOI] [PubMed] [Google Scholar]
- 121.Murrell JR, Spillantini MG, Zolo P, Guazzelli M, Smith MJ, Hasegawa M, Redi F, Crowther RA, Pietrini P, Ghetti B, Goedert M. Tau gene mutation G389R causes a tauopathy with abundant pick body-like inclusions and axonal deposits. J Neuropathol Exp Neurol. 1999;58:1207–1226. doi: 10.1097/00005072-199912000-00002. [DOI] [PubMed] [Google Scholar]
- 122.Bermingham N, Cowie TF, Paine M, Storey E, McLean C. Frontotemporal dementia and Parkinsonism linked to chromosome 17 in a young Australian patient with the G389R Tau mutation. Neuropathol Appl Neurobiol. 2008;34:366–370. doi: 10.1111/j.1365-2990.2007.00918.x. [DOI] [PubMed] [Google Scholar]
- 123.Rossi G, Marelli C, Farina L, Laura M, Maria Basile A, Ciano C, Tagliavini F, Pareyson D. The G389R mutation in the MAPT gene presenting as sporadic corticobasal syndrome. Mov Disord. 2008;23:892–895. doi: 10.1002/mds.21970. [DOI] [PubMed] [Google Scholar]
- 124.Saito Y, Geyer A, Sasaki R, Kuzuhara S, Nanba E, Miyasaka T, Suzuki K, Murayama S. Early-onset, rapidly progressive familial tauopathy with R406W mutation. Neurology. 2002;58:811–813. doi: 10.1212/wnl.58.5.811. [DOI] [PubMed] [Google Scholar]
- 125.Lindquist SG, Holm IE, Schwartz M, Law I, Stokholm J, Batbayli M, Waldemar G, Nielsen JE. Alzheimer disease-like clinical phenotype in a family with FTDP-17 caused by a MAPT R406W mutation. Eur J Neurol. 2008;15:377–385. doi: 10.1111/j.1468-1331.2008.02069.x. [DOI] [PubMed] [Google Scholar]
- 126.Miyasaka T, Morishima-Kawashima M, Ravid R, Heutink P, van Swieten JC, Nagashima K, Ihara Y. Molecular analysis of mutant and wild-type tau deposited in the brain affected by the FTDP-17 R406W mutation. Am J Pathol. 2001;158:373–379. doi: 10.1016/S0002-9440(10)63979-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Giaccone G, Rossi G, Farina L, Marcon G, Di Fede G, Catania M, Morbin M, Sacco L, Bugiani O, Tagliavini F. Familial frontotemporal dementia associated with the novel MAPT mutation T427M. J Neurol. 2005;252:1543–1545. doi: 10.1007/s00415-005-0879-8. [DOI] [PubMed] [Google Scholar]
- 128.Kouri N, Parisi JE, Petersen RB, Baker M, Rademakers R, Dickson D. FTDP-17 with MAPT Exon 13 mutations: comparison of neuropathologic features of Gly389Arg to a novel mutation, Glu372Gly. J Neuropathol Exp Neurol. 2012;71:1. [Google Scholar]
- 129.Di Fonzo A, Ronchi D, Gallia F, Cribiu FM, Trezzi I, Vetro A, Della Mina E, Limongelli I, Bellazzi R, Ricca I, Micieli G, Fassone E, Rizzuti M, Bordoni A, Fortunato F, Salani S, Mora G, Corti S, Ceroni M, Bosari S, Zuffardi O, Bresolin N, Nobile-Orazio E, Comi GP. Lower motor neuron disease with respiratory failure caused by a novel MAPT mutation. Neurology. 2014;82:1990–1998. doi: 10.1212/WNL.0000000000000476. [DOI] [PubMed] [Google Scholar]
- 130.Goedert M, Ghetti B, Spillantini MG. Frontotemporal dementia: implications for understanding Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006254. doi: 10.1101/cshperspect.a006254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kouri N, Carlomagno Y, Baker M, Liesinger AM, Caselli RJ, Wszolek ZK, Petrucelli L, Boeve BF, Parisi JE, Josephs KA, Uitti RJ, Ross OA, Graff-Radford NR, DeTure MA, Dickson DW, Rademakers R. Novel mutation in MAPT exon 13 (p.N410H) causes corticobasal degeneration. Acta Neuropathol. 2014;127:271–282. doi: 10.1007/s00401-013-1193-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rossi G, Bastone A, Piccoli E, Mazzoleni G, Morbin M, Uggetti A, Giaccone G, Sperber S, Beeg M, Salmona M, Tagliavini F. New mutations in MAPT gene causing frontotemporal lobar degeneration: biochemical and structural characterization. Neurobiol Aging. 2012;33:834. doi: 10.1016/j.neurobiolaging.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 133.Goedert M, Spillantini MG. A century of Alzheimer's disease. Science. 2006;314:777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- 134.Alonso Adel C, Mederlyova A, Novak M, Grundke-Iqbal I, Iqbal K. Promotion of hyperphosphorylation by frontotemporal dementia tau mutations. J Biol Chem. 2004;279:34873–34881. doi: 10.1074/jbc.M405131200. [DOI] [PubMed] [Google Scholar]
- 135.Crowther RA, Goedert M. Abnormal tau-containing filaments in neurodegenerative diseases. J Struct Biol. 2000;130:271–279. doi: 10.1006/jsbi.2000.4270. [DOI] [PubMed] [Google Scholar]
- 136.Probst A, Tolnay M, Langui D, Goedert M, Spillantini MG. Pick's disease: hyperphosphorylated tau protein segregates to the somatoaxonal compartment. Acta Neuropathol. 1996;92:588–596. doi: 10.1007/s004010050565. [DOI] [PubMed] [Google Scholar]
- 137.Whitwell JL, Jack CR, Jr, Boeve BF, Senjem ML, Baker M, Ivnik RJ, Knopman DS, Wszolek ZK, Petersen RC, Rademakers R, Josephs KA. Atrophy patterns in IVS10+16, IVS10+3, N279K, S305N, P301L, and V337M MAPT mutations. Neurology. 2009;73:1058–1065. doi: 10.1212/WNL.0b013e3181b9c8b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Whitwell JL, Jack CR, Jr, Boeve BF, Senjem ML, Baker M, Rademakers R, Ivnik RJ, Knopman DS, Wszolek ZK, Petersen RC, Josephs KA. Voxel-based morphometry patterns of atrophy in FTLD with mutations in MAPT or PGRN. Neurology. 2009;72:813–820. doi: 10.1212/01.wnl.0000343851.46573.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Cordes M, Wszolek ZK, Calne DB, Rodnitzky RL, Pfeiffer RF. Magnetic resonance imaging studies in rapidly progressive autosomal dominant parkinsonism and dementia with pallido-ponto-nigral degeneration. Neurodegeneration. 1992;1:8. doi: 10.1002/ana.410320303. [DOI] [PubMed] [Google Scholar]
- 140.Rohrer JD, Ridgway GR, Modat M, Ourselin S, Mead S, Fox NC, Rossor MN, Warren JD. Distinct profiles of brain atrophy in frontotemporal lobar degeneration caused by progranulin and tau mutations. Neuroimage. 2010;53:1070–1076. doi: 10.1016/j.neuroimage.2009.12.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Whitwell JL, Weigand SD, Boeve BF, Senjem ML, Gunter JL, DeJesus-Hernandez M, Rutherford NJ, Baker M, Knopman DS, Wszolek ZK, Parisi JE, Dickson DW, Petersen RC, Rademakers R, Jack CR, Jr, Josephs KA. Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain. 2012;135:794–806. doi: 10.1093/brain/aws001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Mahoney CJ, Downey LE, Ridgway GR, Beck J, Clegg S, Blair M, Finnegan S, Leung KK, Yeatman T, Golden H, Mead S, Rohrer JD, Fox NC, Warren JD. Longitudinal neuroimaging and neuropsychological profiles of frontotemporal dementia with C9ORF72 expansions. Alzheimers Res Ther. 2012;4:41. doi: 10.1186/alzrt144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Whitwell JL, Weigand SD, Gunter JL, Boeve BF, Rademakers R, Baker M, Knopman DS, Wszolek ZK, Petersen RC, Jack CR, Jr, Josephs KA. Trajectories of brain and hippocampal atrophy in FTD with mutations in MAPT or GRN. Neurology. 2011;77:393–398. doi: 10.1212/WNL.0b013e318227047f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Frank A, Wszolek Z, Jack CR, Boeve BF. Distinctive MRI findings in pallido-ponto-nigral degeneration (PPND) Neurology. 2007;68:2. doi: 10.1212/01.wnl.0000254614.39759.3d. [DOI] [PubMed] [Google Scholar]
- 145.Foster NL. Validating FDG-PET as a biomarker for frontotemporal dementia. Exp Neurol. 2003;184(Suppl. 1):S2–8. doi: 10.1016/s0014-4886(03)00360-1. [DOI] [PubMed] [Google Scholar]
- 146.Deters K, Risacher S, Farlow MR, Unverzagt FW, Kareken DA, Hutchins G, Yoder K, Murrell JR, Spina S, Epperson F, Gao S, Saykin A, Ghetti B. Cerebral hypometabolism and grey matter density in MAPT intron 10 +3 mutation carriers. Am J Neurodegener Dis. 2014;3:103–114. [PMC free article] [PubMed] [Google Scholar]
- 147.Miyoshi M, Shinotoh H, Wszolek ZK, Strongosky AJ, Shimada H, Arakawa R, Higuchi M, Ikoma Y, Yasuno F, Fukushi K, Irie T, Ito H, Suhara T. In vivo detection of neuropathologic changes in presymptomatic MAPT mutation carriers: a PET and MRI study. Parkinsonism Relat Disord. 2010;16:404–408. doi: 10.1016/j.parkreldis.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 148.Dopper EG, Rombouts SA, Jiskoot LC, den Heijer T, de Graaf JR, de Koning I, Hammerschlag AR, Seelaar H, Seeley WW, Veer IM, van Buchem MA, Rizzu P, van Swieten JC. Structural and functional brain connectivity in presymptomatic familial frontotemporal dementia. Neurology. 2014;83:e19–26. doi: 10.1212/WNL.0000000000000583. [DOI] [PubMed] [Google Scholar]
- 149.Whitwell JL, Josephs KA, Avula R, Tosakulwong N, Weigand SD, Senjem ML, Vemuri P, Jones DT, Gunter JL, Baker M, Wszolek ZK, Knopman DS, Rademakers R, Petersen RC, Boeve BF, Jack CR., Jr Altered functional connectivity in asymptomatic MAPT subjects: a comparison to bvFTD. Neurology. 2011;77:866–874. doi: 10.1212/WNL.0b013e31822c61f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ghetti B, Farlow MR, Murrell JR, Oblak AL, Johnson CB, Risacher SL, Richardson RM, Epperson F, Unverzagt FW, Saykin AJ, Spina S. The tauopathy associated with mutation +3 in intron 10 of Tau gene: characterization of the MSTD family. Am J Neurodegener Dis. 2014;3:126. [Google Scholar]
- 151.Rohrer JD. Imaging in familial FTD: results of the Genetic FTD Initiative (GENFI) Am J Neurodegener Dis. 2014;3:13. [Google Scholar]
- 152.Fodero-Tavoletti MT, Okamura N, Furumoto S, Mulligan RS, Connor AR, McLean CA, Cao D, Rigopoulos A, Cartwright GA, O'Keefe G, Gong S, Adlard PA, Barnham KJ, Rowe CC, Masters CL, Kudo Y, Cappai R, Yanai K, Villemagne VL. 18F-THK523: a novel in vivo tau imaging ligand for Alzheimer's disease. Brain. 2011;134:1089–1100. doi: 10.1093/brain/awr038. [DOI] [PubMed] [Google Scholar]
- 153.Maruyama M, Shimada H, Suhara T, Shinotoh H, Ji B, Maeda J, Zhang MR, Trojanowski JQ, Lee VM, Ono M, Masamoto K, Takano H, Sahara N, Iwata N, Okamura N, Furumoto S, Kudo Y, Chang Q, Saido TC, Takashima A, Lewis J, Jang MK, Aoki I, Ito H, Higuchi M. Imaging of tau pathology in a tauopathy mouse model and in Alzheimer patients compared to normal controls. Neuron. 2013;79:1094–1108. doi: 10.1016/j.neuron.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Okamura N, Fodero-Tavoletti MT, Kudo Y, Rowe CC, Furumoto S, Arai H, Masters CL, Yanai K, Villemagne VL. Advances in molecular imaging for the diagnosis of dementia. Expert Opin Med Diagn. 2009;3:705–716. doi: 10.1517/17530050903133790. [DOI] [PubMed] [Google Scholar]
- 155.Okamura N, Furumoto S, Harada R, Tago T, Yoshikawa T, Fodero-Tavoletti M, Mulligan RS, Villemagne VL, Akatsu H, Yamamoto T, Arai H, Iwata R, Yanai K, Kudo Y. Novel 18F-labeled arylquinoline derivatives for noninvasive imaging of tau pathology in Alzheimer disease. J Nucl Med. 2013;54:1420–1427. doi: 10.2967/jnumed.112.117341. [DOI] [PubMed] [Google Scholar]
- 156.Rojo LE, Alzate-Morales J, Saavedra IN, Davies P, Maccioni RB. Selective interaction of lansoprazole and astemizole with tau polymers: potential new clinical use in diagnosis of Alzheimer's disease. J Alzheimers Dis. 2010;19:573–589. doi: 10.3233/JAD-2010-1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Small GW, Kepe V, Ercoli LM, Siddarth P, Bookheimer SY, Miller KJ, Lavretsky H, Burggren AC, Cole GM, Vinters HV, Thompson PM, Huang SC, Satyamurthy N, Phelps ME, Barrio JR. PET of brain amyloid and tau in mild cognitive impairment. N Engl J Med. 2006;355:2652–2663. doi: 10.1056/NEJMoa054625. [DOI] [PubMed] [Google Scholar]
- 158.Xia CF, Arteaga J, Chen G, Gangadharmath U, Gomez LF, Kasi D, Lam C, Liang Q, Liu C, Mocharla VP, Mu F, Sinha A, Su H, Szardenings AK, Walsh JC, Wang E, Yu C, Zhang W, Zhao T, Kolb HC. [(18)F]T807, a novel tau positron emission tomography imaging agent for Alzheimer's disease. Alzheimers Dement. 2013;9:666–676. doi: 10.1016/j.jalz.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 159.Chien DT, Bahri S, Szardenings AK, Walsh JC, Mu F, Su MY, Shankle WR, Elizarov A, Kolb HC. Early clinical PET imaging results with the novel PHF-tau radioligand [F-18]-T807. J Alzheimers Dis. 2013;34:457–468. doi: 10.3233/JAD-122059. [DOI] [PubMed] [Google Scholar]
- 160.Villemagne VL, Fodero-Tavoletti MT, Masters CL, Rowe CC. Tau imaging: early progress and future directions. Lancet Neurol. 2015;14:114–124. doi: 10.1016/S1474-4422(14)70252-2. [DOI] [PubMed] [Google Scholar]
- 161.Ossenkoppele R, Schonhaut DR, Baker SL, O'Neil JP, Janabi M, Ghosh PM, Santos M, Miller ZA, Bettcher BM, Gorno-Tempini ML, Miller BL, Jagust WJ, Rabinovici GD. Tau, amyloid, and hypometabolism in a patient with posterior cortical atrophy. Ann Neurol. 2014 doi: 10.1002/ana.24321. [e-pub] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, Boone K, Miller BL, Cummings J, Benson DF. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 163.Pickering-Brown SM, Rollinson S, Du Plessis D, Morrison KE, Varma A, Richardson AM, Neary D, Snowden JS, Mann DM. Frequency and clinical characteristics of progranulin mutation carriers in the Manchester frontotemporal lobar degeneration cohort: comparison with patients with MAPT and no known mutations. Brain. 2008;131:721–731. doi: 10.1093/brain/awm331. [DOI] [PubMed] [Google Scholar]