

Summary
This review summarizes a presentation made at the retirement Symposium of Prof. dr. Cornelis Jakobs in November of 2011, highlighting the progress toward clinical trials in succinic semialdehyde dehydrogenase (SSADH) deficiency, a disorder first recognized in 1981. Active and potential clinical interventions, including vigabatrin, L-cycloserine, the GHB receptor antagonist NCS-382, and the ketogenic diet, are discussed. Several biomarkers to gauge clinical efficacy have been identified, including cerebrospinal fluid metabolites, neuropsychiatric testing, MRI, EEG, and measures of GABAergic function including (11C)flumazenil positron emission tomography (PET) and transcranial magnetic stimulation (TMS). Thirty years after its discovery, encompassing extensive studies in both patients and the corresponding murine model, we are now running an open-label trial of taurine intervention, and are poised to undertake a phase II trial of the GABAB receptor antagonist SGS742.
Introduction
The treatment of inborn errors of metabolism presents a host of challenges for the clinician, some of which include compartmentalization of the primary defect, alternative pathways for metabolism, interlinked metabolic pathways (e.g., oxidative phosphorylation with fat oxidation), the confounds of first-pass drug metabolism in the liver, the blood brain barrier, and many others. When considering neurometabolic disorders, these challenges magnify as we must entertain the largely anabolic and inaccessible brain. Does the primary metabolic lesion affect mainly the putamen, the amygdala, the striata or cortex, or cerebellum? Is the defect global or localized, and are there effective treatments that will cross the blood brain barrier? Are metabolites that form in the brain able to enter the peripheral metabolism, or are they intracellularly trapped? Adding to the complexity are neuronal-glial interactions, brain remodeling, and a number of other physiological parameters associated with brain metabolism such that it becomes a remarkable achievement that we can ever effectively treat a neurometabolic disorder.
SSADH deficiency mirrors all of these treatment challenges, and has posed additional considerations when entertaining effective treatment strategies. A defect of GABA metabolism, SSADH deficiency features accumulation of two neuromodulators, GABA and GHB, and paradoxically represents a hyperGABAergic seizure disorder (Fig. 1) (Wong et al 2004; Maitre 1997; Pearl et al 2011). One pathophysiological mechanism includes GABAA receptor downregulation, shown on positron emission tomography with 11C-flumazenil (Pearl et al 2009a). Clinical reports on dozens of patients reveal a neurological disorder that may often be misdiagnosed as autism or autism spectrum, oppositional defiance, ADHD or other behavioral disorders (Pearl et al 2003). Most therapies have been symptomatic, targeting occasional seizures, behavioral dysfunction, or other clinical manifestations, but few interventions target the primary lesion in this disease. Further complicating the situation is the fact that some GABA receptors in neural tissue are excitatory during development (as opposed to inhibitory, the common role for GABA in brain) (Rheims et al 2008; Herlenius and Lagercrantz 2001), the observation that GHB produced in the periphery (liver has about 70% of the SSADH activity of brain; Chambliss et al 1995) can freely traverse the blood brain barrier (BBB), and the probable main neurotoxin, GABA, can only be removed by enhanced metabolism or conjugation.
Fig. 1.

The GABA metabolic pathway. The block in SSADH deficiency is indicated by the crosshatched box. Upward arrows indicate metabolite elevations in patients and aldh5a1−/− mice. High levels of GHB will inhibit presynaptic DA (dopamine) release and enhance turnover. The site of VGB acting on GABA-transaminase (GABA-T) is indicated. Additional abbreviations: GAD, glutamic acid decarboxylase; TCA, tricarboxylic acid.
In this review, a synopsis of interventional strategies for SSADH deficiency is presented, highlighting what has been attempted, and what appears to be on the horizon (Knerr et al 2007). Most of the therapies that are planned, or studies in progress with patients, have springboarded from preclinical findings in the murine model of SSADH deficiency, aldh5a1−/− mice (Gibson et al 2002). Brief reference will be made to important findings in the mouse model that have revealed novel treatment concepts.
Treatment Strategies for SSADH Deficiency
Vigabatrin
Perhaps the most widely employed intervention for SSADH deficiency, at least in Europe, has been vigabatrin (VGB; gamma-vinylGABA; SabrilR; Lundbeck Corp.). VGB, an irreversible inhibitor of GABA-transaminase (Fig. 1), acts pharmacologically to increase brain GABA. VGB is employed for infantile spasms (IS) (especially in tuberous sclerosis patients), as well as refractory complex partial seizures. In the case of SSADH deficiency, the use of VGB is predicted to lead to decreased production of succinic semialdehyde (Fig. 1), the probable precursor of GHB (Gropman 2003). High-dose VGB in the aldh5a1−/− model led to significant extension of lifespan, although metabolic measurements (brain GHB/GABA) were not performed (Hogema et al 2001). Along these lines, the results of clinical intervention support decreased production of GHB, but a concern with VGB is the potential to further increase GABA in an already hyperGABAergic disorder (Pearl and Gropman 2004; Tables I & II).
Table 1.
Outcomes of VGB intervention in SSADH Deficiencya
| Age (yrs) | Sex | VGB (Months) | Clinical Improvement | Reference |
|---|---|---|---|---|
| 3 | F | 20–100 mg/kg (4 mo) | none | Gibson et al (1989) |
| 2 | F | 75 mg/kg/d (16 mo) | ataxia, EEG (swd)b | Jaeken et al(1989) |
| 12 | F | 500–3500 mg/d (9 mo) | ataxia, IQ | Jakobs et al (1992) |
| 1 | F | 1500 mg/d | hypotonia | Uziel et al (1993) |
| 12 | M | 25–75 mg/kg/d | cognition, behavior (low dose) | Matern et al (1996) |
| 5 | M | 1500 mg/d (6 mo) | hypotonia, myoclonus | Al-Essa et al (2000) |
| 10 | F | 22–34 mg/kg/d (57 mo) | hyperactivity, agitation behavior | Ergezinger et al (2003) |
| 2.5 | F | 20 mg/kg/d (12 mo) | hypotonia | Escalera et al (2010) |
| 8 | F | 25 mg/kg/d (9 mo) | communication | Casarano et al (2011) |
When reported, CSF GHB decreased 14–72% and CSF GABA increased 2.3 to 6-fold during VGB intervention (Ergezinger et al 2003);
Electroencephalogram, spike wave discharge. Two additional reports indicated clinical improvement in ~ 1/3 of patients treated with VGB, without clinical details (Rahbeeni et al 1994; Gibson et al 1997).
Table 2.
Cerebrospinal fluid (CSF) metabolites during VGB intervention in 4 SSADH-deficient patients
| Metabolites | Patient | Pre-treatment | Post-treatment | % Change |
|---|---|---|---|---|
| GHB: Total GABA | 1 | 116: 13.6 | 62: 31.2 | −47: +229 |
| 2 | 1110: 22.4 | 708: 32.1 | −36: +143 | |
| 3 | 615: 18.3 | 457: 38.8 | −26: +212 | |
| 4 | 525: 22.1 | 384: 33.6 | −27: +152 |
All metabolites in units of μmol/L. CSF control ranges: GHB, 0–2.6 μmol/L (n=10); total GABA, 4.7–11.8 μmol/L (n=10). Total GABA refers to free and “bound” GABA (e.g., homocarnosine, and other GABA compounds) (reprinted from Gibson et al 1995).
Despite a clear rationale for VGB in SSADH deficiency, surprisingly few literature reports have been presented, and no blinded, controlled trial has been published. The case reports of clinical improvements with VGB (Table 1) have been tempered by other reports of worsening of symptoms, including seizure control (Matern et al 1996). Although approved in Europe since the 1980s for intervention in IS, VGB was not approved until 2009 by the US FDA for treatment of IS in pediatric patients and uncontrolled complex partial seizures in adults. The delays in FDA approval related to early studies in animals, including rats and dogs, which suggested edema in myelin tracts and vacuolation of white matter (Butler et al 1987; Qiao et al 2000; Peyster et al 1995). VGB may have particular utility for IS in patients under 2 years of age, since these seizures are particularly challenging, and a common intervention for IS, ACTH (adrenocorticotropic hormone), may be difficult to obtain. Moreover, VGB showed adverse effects on visual fields in approximately 1/3 of patients receiving it for epilepsy (Krauss et al 1998; Spence and Sankar 2001; Vanhatalo et al 2002). The mechanism of retinal toxicity remains undefined, although reduced ornithine aminotransferase (OAT) activity observed with VGB consumption may elevate retinal ornithine and induce a form or gyrate atrophy (De Biase et al 1991; Sorri et al 2010). Moreover, GABA is a feedback regulator of OAT (Daune and Seiler 1988). These observations suggest that monitoring ornithine levels during VGB intervention would be prudent. In addition, 8 of 23 patients taking VGB had new-onset, reversible MRI T(2)-weighted hyperintensities and restricted diffusion in thalami, globus pallidus, dentate nuclei, brainstem, or corpus callosum (Pearl et al 2009b). Because of significant concerns, the FDA permits the use of VGB only for clinical instances in which the potential benefit against seizure outweighs the risk of visual loss. As well, VGB has a number of “black-box” warnings, and the FDA requires routine ocular exams/visual field testing.
Receptor studies in aldh5a1−/− mice have shown significant alterations of GABAB and GABAA receptors (GABABR and GABAAR), yet no alterations in GHB receptor (GHBR) binding or number (Cortez et al 2004; Buzzi et al 2006; Wu et al 2006; Mehta et al 2006). These data suggest that GHB is not necessarily the primary neurotoxin in SSADH deficiency, a role more likely fulfilled by GABA. The absence of GHBR alterations may associate with the capacity of GHB to freely traverse the blood-brain barrier, which GABA cannot. The cumulative data suggests that elevating brain GABA levels via VGB intervention in SSADH deficiency might not be prudent. Additionally, GABA-T exists in the periphery, including liver, kidney and even blood (Tillakaratne et al 1995), and the kinetic characteristics of the neural enzyme are different from those of GABA-T in the periphery. Thus, incomplete inactivation of peripheral GABA-T’s by VGB may still result in elevated intracerebral GHB. This hypothesis is supported by metabolic findings in patients receiving VGB (Table 2; Gibson et al 1995). Four patients treated with VGB (40–100 mg/kg/d) for 1–12 months underwent lumbar puncture for CSF collection. During treatment, there were variable improvements in cerebellar signs, concentration, hyperactivity, agility and cognitive function. As predicted, CSF total GABA increased and GHB decreased, although the absolute decrease in GHB was never as great as the GABA increase. Single body fluid measures such as these (Table 2) are limited by an absence of parallel measurements in other fluids, including plasma and urine. The fact that GHB levels did not decrease to the extent seen for total GABA increase (Table 2) is consistent with the hypothesis of peripheral resupply of GHB due to incomplete inactivation of peripheral GABA-T’s by VGB.
L-cycloserine
It is rational to attempt inhibition of GABA-T in patients with SSADH deficiency, as described above for VGB. However, if the primary source of pathology in this disease is elevated GABA, the expected decrease in GHB formation may be thwarted by further GABA elevations. L-Cycloserine (LCS) provides another potential inhibitor of GABA-T that warrants examination.
Cycloserine exists as two enantiomers: 1) the D-isomer (also known as Seromycin), used to treat tuberculosis and selected neurological disorders (associated with its potent NMDA receptor agonist activity (Lowther et al 2010)); and 2) the synthetic L-isomer. The latter, LCS, has been characterized in a number of model systems, yet the literature presents somewhat contradictory and conflicting findings. Early data highlighted the capacity of LCS to inactivate GABA-transaminase (see Fig. 1) as well as alanine aminotransferase (ALA-T; Beuster et al 2011). Linked to its capacity to raise intracerebral GABA levels (Fig. 1), LCS has been piloted in several seizure models, including petit-mal seizures in the rat (Marescaux et al 1985) and audiogenic seizures in mice (Polc et al 1986). Nonetheless, clinical development of LCS has been hampered because it also inhibits the first enzyme of sphingolipid formation, 3-ketodihydrosphingosine synthase (serine palmitoyltransferase; Cho 2007). The capacity of LCS to elevated intracerebral GABA is dose-dependent, with smaller doses (10–30 mg/kg p.o. or i.p. in rodents) reducing hyperexcitability, while higher doses (30–100 mg/kg) evoke central depressant actions (Polc et al 1986). As well, the literature indicates that inhibition of sphingomyelin formation may be dose-dependent, and may not be substantial at low pharmacologic doses of LCS.
Despite several rodent studies, there has been no report of LCS intervention in higher mammals or humans, which would make interventional studies in SSADH-deficient patients a very distant goal. Nonetheless, LCS may provide inhibition of GABA-T without the associated ocular side effects found with VGB, and preclinical studies in aldh5a1−/− mice would be of interest.
SGS742
SGS-742 (originally CGP-36742) is one of a long line of phosphinic-acid analogues of GABA that are antagonists of the GABABR (Farlow 2009). These antagonists were originally developed by Ciba-Geigy Corporation (Froestl et al 2004), and eventually the proprietary rights moved to other corporations. As mentioned above, studies in aldh5a1−/− mice have provided compelling evidence for use-dependent alterations of both GABAAR and GABABR (Gupta et al 2002; Cortez et al 2004; Wu et al 2006; Buzzi et al 2006). Accordingly, antagonists may prove beneficial as a treatment strategy in SSADH deficiency through blockade of supraphysiological agonist (GABA) levels. In preliminary studies in aldh5a1−/− mice, high-dose CGP 35348 (an early phosphinic acid progenitor of SGS742) provided significant extension of lifespan for mice that succumbed to early status epilepticus (Hogema et al 2001; Gupta et al 2002). The phosphinic acid analogue SGS742 was subsequently introduced by Saegis Pharmaceuticals and Novartis Corporation, and proposed for clinical trials in patients with mild cognitive impairment (MCI; Bowery 2006; Bullock 2005; Froestl et al 2004). The rationale for SGS742 in MCI patients centers on the known inhibition of learning and memory associated with functional activation of the GABABR by its ligand, GABA. SGS742 has undergone phase I/II safety and tolerability evaluation in humans, and has shown efficacy in MCI patients (Froestl 2004), yet extended trials have not been reported. Currently, the proprietary rights to SGS-742 have been obtained by the National Institutes of Health of the United States.
Pilot trials of SGS742 in aldh5a1−/− mice have shown promising results. Moderate levels (30–100 mg/kg/d, i.p. administration) reduced spike-wave discharge, and led to near normalization of the electrocorticograph (Pearl et al 2009c). These data formed the foundation of a planned clinical intervention with SGS742 in SSADH-deficient patients. Two important hurdles must be cleared prior to undertaking this trial, however: 1) all safety and tolerability studies of SGS742 were performed in adults, and there are many adolescent patients with SSADH deficiency which could be enrolled in a blinded clinical trial; and 2) an application to the US Food and Drug Administration (FDA) for an IND (Investigational New Drug). Despite these challenges, we envision undertaking a blinded, placebo-controlled trial of SGS742 in SSADH-deficient patients shortly.
Ketogenic diet
Nylen and colleagues (2008; 2009; Stewart et al 2008) posited that early lethality in aldh5a1−/− mice occurred proximal to the weaning period. These investigators proposed that the high-fat content of dam’s milk was protective to the pups, and hypothesized that the use of the ketogenic diet (KD), a broadly employed dietary intervention for refractory epilepsy (Cusmai et al 2012; McNally and Hartman 2012), would lead to beneficial outcomes in the murine model (Nylen et al 2008, 2009; Stewart et al 2008). Implementation of a 4:1 (fat:carbohydrate) KD in aldh5a1−/− mice led to a significant improvement in seizure profiles, body weights and energy production, as well as extended lifespan. These findings have led a number of clinicians to consider using the KD in SSADH-deficient patients for whom control of convulsions has proven challenging. Nonetheless, no clinical report on its use has been presented. The other major drawback to use of the KD is the fact that most convulsions in SSADH-deficient patients can be controlled with standard antiepileptics (AEDs), such as lamotrigine, levetiracetam, or topiramate, and the KD is both challenging to implement and maintain. Thus, conventional seizure control has predominantly employed standard AEDs (Pearl et al 2011).
NCS-382
As noted above, one of the paradoxical findings in aldh5a1−/− mice is the absence of GHBR alterations despite high levels of GHB (Wu et al 2006; Mehta et al 2006). Nonetheless, in early studies on aldh5a1−/− mice, the drug most effective in rescuing these animals from early lethality was NCS-382 (6,7,8,9-tetrahydro-5-hydroxy-5H-benzocyclohept-6-ylidene acetic acid), a pure GHBR antagonist (Hogema et al 2001; Gupta et al 2002). These data suggest some role for GHB (and its receptor) in the process of early lethality, yet this remains paradoxical in the face of normal GHBR pharmacology observed in vivo (Mehta et al 2006). Despite positive outcomes with NCS-382 in aldh5a1−/− mice, no detailed studies on its use have been performed in these mice, and NCS-382 is not an FDA-approved drug. Considerable data on the safety and tolerability of NCS-382 would be required, not only in higher mammals (dog, primate, etc), but minimally phase I/II safety and tolerability data in humans, before any consideration of this compound could be undertaken in SSADH-deficient individuals.
Taurine
The high content of taurine in dam’s milk prompted Hogema and colleagues (2001) to examine this non-physiological amino acid as an intervention in aldh5a1−/− mice, and these investigators observed significant extension of truncated lifespan. Efficacy of taurine intervention in aldh5a1−/− mice prompted Saronwala and colleagues (2008) to perform a pilot investigation in an SSADH-deficient patient. The patient, a 2-year old male, was treated with 200 mg/kg/day taurine over a 12 month period, and tolerated it well. Plasma taurine levels ranged from 2–8 fold normal, and there was no correlation between taurine level and GHB excretion. By 9 months of treatment, marked behavioral improvements were noted, as well as improved peer interactions, level of activity and coordination. Repeat MRI obtained at 12 months of treatment was interpreted as resolution of prior restricted diffusion abnormality of the globus pallidus, with a very small residual T2 signal MRI abnormality.
Taurine is an abundant free amino acid in various tissues, and has important roles in neuromodulation and osmoregulation (Olive 2002). Taurine is known to interact with both GABAARs and GABABRs, and increases chloride conductance in excitable tissues. The neuroprotective action of taurine against beta-amyloid and glutamate receptor agonists in chick retinal neurons is blocked by the GABAAR antagonist picrotoxin (Louzada et al 2004). There is evidence that the taurine transporter is able to utilize GABA as a substrate, and that GABA competitively inhibits this transporter (Tomi et al 2008). There appears to be a reciprocal relationship between GABA and taurine, as taurine transporter null mice show increased GABAAR densities in the hippocampal dentate gyrus and in the cerebellum (Oermann et al 2005). Thus, since taurine appears to modulate GABA transmission, there may be beneficial competitive effects with respect to GABA in SSADH-deficient patients.
We extended the single-patient evaluation of taurine described above by enrolling patients into an open label trial in order to collect baseline data while preparing for a controlled trial with biomarkers used to evaluate this population without pharmacologic intervention. Subjects with confirmed SSADH deficiency were eligible for recruitment. Following informed consent, subjects were titrated weekly from 50mg/kg/day until reaching a target dose of 200 mg/kg/day to a maximum dosage of 16 grams/day. The Adaptive Behavior Assessment Scale-II (ABAS-II) was administered at baseline and requested at six and twelve months of therapy. The ABAS II measures adaptive skills from conceptual, social, and practical domains in study participants. This scale has normative data and is offered for the age ranges 0–5, 5–21, and 16–89 years. Patients initiated at age 16 years and older were administered the adult version. We used linear longitudinal modeling to compare ABAS scores during the period pre- to the time-averaged scores in the period during taurine use. The model adjusted variance estimates, analogous to a paired t-test, were used to account for the correlation of scores within the same participant.
Sixteen patients were recruited and provided baseline data: 6M/10F, age range at enrollment 2–28 yrs (mean 12 yrs). One patient had a serious adverse event on 16 grams/day (200 mg/kg): hospitalization for hypersomnia. This led to a dose lowering paradigm with a new maximum daily dose of 10 grams. The taurine has been otherwise well-tolerated. Six subjects have completed the trial; 3 withdrew early due to perceived lack of efficacy. For patients who provided follow-up data during active therapy, there were no significant changes between pre- and Rx-composite scores in general adaptive, conceptual, practical, or social skills. However, a borderline lower score was observed on the conceptual (p=0.1) index during taurine use. Our open-label pilot trial of taurine in patients with SSADH deficiency did not reveal demonstrable improvement in adaptive neurobehavioral functioning (Pearl et al 2012). Nonetheless, additional biomarker studies during taurine intervention are in progress.
Other potential interventions in SSADH deficiency
As mentioned above, interventions targeting reduced GHB production through inhibition of GABA-T may be complicated by concomitant GABA elevations. One approach to mitigating this side-effect may be co-application of 3-mercaptopropionate, an inhibitor of glutamic acid decarboxylase (Netopilova et al 1997; Roberts et al 1978). Such studies could be piloted in aldh5a1−/− mice, but the intracerebral levels of both glutamate and GABA would require cautious evaluation. A further mechanism to mitigate GABA elevation in SSADH deficiency, in the presence or absence of drug intervention, might be supplementation with L-histidine with the objective of enhancing homocarnosine (the GABA:L-histidine dipeptide). Homocarnosine is a major component of excitable tissues, with roles in neuromodulation, osmoregulation, and neuroprotection (Bauer et al 2005). Increasing intracerebral homocarnosine in SSADH deficiency may have additional therapeutic benefit, in relation to its capacity to quench 4-hydroxy-2-nonenal (4-HNE), a major lipid peroxidation product and cytotoxic carbonyl agent (Aldini et al 2005). Since SSADH is the major aldehyde dehydrogenase responsible for degradation of 4-HNE in mammals (Murphy et al 2003), the quenching role of homocarnosine might have therapeutic relevance. Quantitation of 4-HNE levels in neural tissue of aldh5a1−/− mice, or fluids/tissues derived from SSADH-deficient patients, however, has not been reported.
Outcome Measures for Clinical Trials
A challenge for any effective clinical trial is the identification of surrogate biomarker outcomes that can be quantified to gauge efficacy. Along these lines, numerous studies in aldh5a1−/− mice, and SSADH-deficient patients, have highlighted potential biomarkers, some of which have already been clinically validated. Along these lines, GABAergic anomalies (both GABABR and GABAAR) in aldh5a1−/− mice have led to corollary studies of GABA receptor function in patients (Pearl et al 2009a; Reis et al, in press). For example, (11C)flumazenil (FMZ) binding (a marker for GABAAR function) is decreased throughout all brain regions in patients with SSADH deficiency. Similar anomalies have been documented for GABABR function in patients employing transcranial magnetic stimulation (TMS), a technique in which depolarization or hyperpolarization of the neurons is induced using weak electric currents from a magnetic field (de Vries et al 2012). Both (11C)FMZ binding and TMS will be considered as outcome measures in clinical trials of SSADH-deficient patients.
Additional standard clinical measures that can be employed for clinical trials include neurobehavioral evaluation (as described for taurine intervention), as well as EEG and MRI. Several reports of EEG in SSADH-deficient patients have revealed slowing in both hemispheres and spike-wave discharges (Vanadia et al, in press), an observation consistent with the effect of GHB on EEG (Snead 1978; 2000; Snead and Gibson 2005). As well, a consistent finding in patients has been hyperintensity on the T1-weighted MRI images in the globus pallidi bilaterally (Pearl et al 2009a; Yalcinkaya et al 2000; Ziyeh et al 2002). This finding is non-specific, and most likely represents localized cytotoxicity associated with metabolite accumulation and/or downstream metabolic disruptions. Nonetheless, brain MRI remains a valid clinical biomarker to test therapeutic efficacy, and was employed as an outcome marker in the index patient treated with taurine (Saronwala et al 2008). Optimally, a phase II trial (targeting tolerability, dosing, safety, and other features without assessing efficacy) could be employed to identify which of the biomarkers outlined above is the most responsive and reproducible for interventional evaluation in SSADH-deficient patients.
Pathophysiological considerations in SSADH deficiency
Most data from the aldh5a1−/− mouse model points to GABA as the primary mediator of neuropathology in SSADH deficiency (Pearl et al 2009c). Insights into discrete metabolic effects (GABA, GHB) might be gleaned, therefore, by contrasting the phenotypes of SSADH and GABA-T deficiencies. The index siblings with GABA-T deficiency presented with severe psychomotor retardation, hypotonia, hyperreflexia and growth acceleration (Jaeken et al 1984), while a recently described third patient (Tsuji et al 2010) presented with intractable seizures. SSADH deficiency features intellectual disability with disproportionate deficit in expressive language, hypotonia, ataxia, infrequent seizures, and a component of neuropsychiatric morbidity in adolescence and adulthood. The severity of GABA-T deficiency supports a prominent neuropathological role for increased GABA in both diseases, although both disorders feature other metabolic disturbances, such as elevated β-alanine and homocarnosine in GABA-T deficiency, and increased 4,5-dihydroxyhexanoic acid, D-2-hydroxyglutaric acid, homocarnosine and succinic semialdehyde in SSADH deficiency.
Conclusions
One of many challenges with SSADH deficiency that will remain to be negotiated includes the fact that both GHB and GABA accumulate, and likely each plays a role in the pathophysiology of the disorder. Along these lines, combinatorial therapy (e.g., VGB/SGS742, or NCS-382/SGS742, etc) may be optimal, if regulatory hurdles can be cleared. Peripheral therapy (whether cell-based or gene therapeutic, targeting the liver) could have value, and preclinical studies in aldh5a1−/− mice using adenoviral hepatic gene therapy have shown efficacy (Gupta et al 2004a; 2004b). Intracerebral cell or gene therapy also represents an attractive objective in SSADH deficiency, especially since the phenotype is primarily neurological. Finally, some consideration must be given to in utero therapy, since studies in the murine model have revealed that GABA and GHB accumulate in early aldh5a1−/− embryos, and the fact that the GABAAR is excitatory during early development (Jansen et al 2008; Waagepetersen et al 1999; Herlenius and Lagercrantz 2001). Thus, consideration may need to be given to the earliest possible intervention that can mitigate long-term neurological complications.
Looking back on the progress made in SSADH deficiency, it has clearly not been fast enough for the benefit of the patients. Nonetheless, once we traverse into the realms of both animal and human research, multiple regulatory considerations come into play. Today, however, we have a validated animal model, an IRB-approved patient registry, and multiple websites that have enabled patients and parents to interact and exchange information (www.pndassoc.org; www.waveofhope-ssadh.org; www.liamslinks.org). The Pediatric Neurotransmitter Disease Association, a parent support group focused solely on disorders of neurotransmitter metabolism, is supporting research through a competitive grant program. Our goal remains to continue working as a team, focused on identifying better, long-term therapies for patients and families with SSADH deficiency, so that all can enjoy an enhanced quality of life.
Fig. 2.
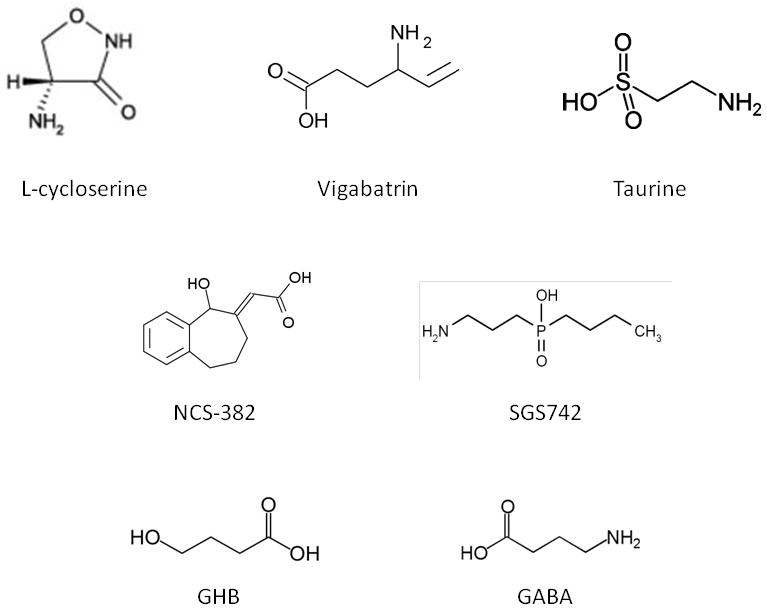
Chemical structures of the compounds discussed in the text, including the species accumulating to pathological levels in SSADH deficiency, GHB and GABA (bottom).
Concise 1 sentence take-home message.
An overview of current and potential treatment interventions in heritable succinic semialdehyde dehydrogenase deficiency is presented, with a highlight on preclinical studies obtained from the corresponding murine model.
Prelude.
It is doubtful that any reader of the Journal would question the concept that the care and treatment of patients with inborn errors of metabolism functions optimally through a “team” approach. The team is generally defined as the clinician, supported by clinical chemists, dieticians, physical and/or occupational therapists, clinical psychologists, nurse practitioners, and other colleagues. This “team” approach represents the foundation of the current article, when a young analytical chemist named Cornelis Jakobs came to the University of California, San Diego, to pursue his Doctoral studies in stable-isotope dilution mass spectrometry with Dr. Lawrence Sweetman and Dr. William Nyhan. Jakobs had identified three patients who excreted gamma-hydroxybutyric acid in urine, and he hypothesized that the defect in these patients was in succinic semialdehyde dehydrogenase (SSADH, or ALDH5A1 (aldehyde dehydrogenase 5a1) (Jakobs et al 1981). What he needed was an enzymologist to test this hypothesis, and the senior author was fortunate enough to have been that individual (Gibson et al 1983). The path to the current paper highlights the team approach, with clinicians, molecular biologists, mammalian geneticists, neurophysiologists, and so many other scientists that it is challenging to identify (much less remember) them all. The good news is that we now possess sufficient knowledge of the pathophysiology of this disorder to institute clinical trials! Where these trials take us remains to be seen, but we can say with certainty that without Cornelis, we would never have started. The senior author will miss the daily interactions by e mail, but a 30-year friendship is not easily lost or forgotten.
Acknowledgments
The authors acknowledge Dr. Boris M. Hogema, who developed the murine model. The funding and support of the NIH (HD 58553) and the Pediatric Neurotransmitter Disease Association are gratefully acknowledged. This research was also supported by awards UL1RR031988 and P30HD40677 from the NIH National Center for Research Resources and NIH Intellectual and Developmental Disabilities Research Center, respectively.
Abbreviations employed
- ABAS-II
adaptive behavior assessment scale II
- ACTH
adrenocorticotropic hormone
- ADHD
attention deficit hyperactivity disorder
- AED
antiepileptic drug
- BBB
blood brain barrier
- CSF
cerebrospinal fluid
- DA
dopamine
- EEG
electroencephalography
- FDA
Food and Drug Administration (of the USA)
- FMZ
flumazenil (GABAAR ligand, benzodiazepine binding site)
- GABA
4-aminobutyric acid
- GABAAR
GABAA receptor
- GABABR
GABAB receptor
- GABA-T
GABA-transaminase
- GAD
glutamic acid decarboxylase
- GHB
4-hydroxybutyric acid
- GHBR
GHB receptor
- IND
investigational new drug
- IQ
intelligence quotient
- IS
infantile spasms
- KD
ketogenic diet
- LCS
L-cycloserine
- MCI
mild cognitive impairment
- MRI
magnetic resonance imaging
- NCS-382
(6,7,8,9-tetrahydro-5-hydroxy-5H-benzocyclohept-6-ylidene)acetic acid
- SGS742
phosphinic acid, (3-aminopropyl)butyl-
- SSADH
succinic semialdehyde dehydrogenase (or ALDH5A1, aldehyde dehydrogenase 5a1)
- TMS
transcranial magnetic stimulation
- TCA
tricarboxylic acid
- VGB
vigabatrin (gamma-vinyl GABA; SabrilR (Lundbeck Corporation))
References
- Aldini G, Facino RM, Beretta G, Carini M. Carnosine and related dipeptides as quenchers of reactive carbonyl species: from structural studies to therapeutic perspectives. Biofactors. 2005;24:77–87. doi: 10.1002/biof.5520240109. [DOI] [PubMed] [Google Scholar]
- Al-Essa MA, Bakheet SM, Patay ZJ, Powe JE, Ozand PT. Clinical, fluorine-18 labeled 2-fluoro-2-deoxyglucose positron emission tomography (FDG PET), MRI of the brain and biochemical observations in a patient with 4-hydroxybutyric aciduria; a progressive neurometabolic disease. Brain Dev. 2000;22:127–31. doi: 10.1016/s0387-7604(99)00121-7. [DOI] [PubMed] [Google Scholar]
- Bauer K. Carnosine and homocarnosine, the forgotten, enigmatic peptides of the brain. Neurochem Res. 2005;30:1339–45. doi: 10.1007/s11064-005-8806-z. [DOI] [PubMed] [Google Scholar]
- Beuster G, Zarse K, Kaleta C, et al. Inhibition of alanine aminotransferase in silico and in vivo promotes mitochondrial metabolism to impair malignant growth. J Biol Chem. 2011;286:22323–30. doi: 10.1074/jbc.M110.205229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowery NG. GABAB receptor: a site of therapeutic benefit. Curr Opin Pharmacol. 2006;6:37–43. doi: 10.1016/j.coph.2005.10.002. [DOI] [PubMed] [Google Scholar]
- Bullock R. SGS-742 Novartis. Curr Opin Investig Drugs. 2005;6:108–13. [PubMed] [Google Scholar]
- Butler WH, Ford GP, Newberne JW. A study of the effects of vigabatrin on the central nervous system and retina of Sprague Dawley and Lister-Hooded rats. Toxicol Pathol. 1987;15:143–8. doi: 10.1177/019262338701500203. [DOI] [PubMed] [Google Scholar]
- Buzzi A, Wu Y, Frantseva MV, et al. Succinic semialdehyde dehydrogenase deficiency: GABAB receptor-mediated function. Brain Res. 2006;1090:15–22. doi: 10.1016/j.brainres.2006.02.131. [DOI] [PubMed] [Google Scholar]
- Casarano M, Alessandri MG, Salomons GS, et al. Efficacy of vigabatrin intervention in a mild phenotypic expression of succinic semialdehyde dehydrogenase deficiency. J Inherit Metab Dis (JIMD Reports) 2011;2:119–123. doi: 10.1007/8904_2011_60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambliss KL, Zhang YA, Rossier E, Vollmer B, Gibson KM. Enzymatic and immunologic identification of succinic semialdehyde dehydrogenase in rat and human neural and nonneural tissues. J Neurochem. 1995;65:851–5. doi: 10.1046/j.1471-4159.1995.65020851.x. [DOI] [PubMed] [Google Scholar]
- Cho JY. Effect of L-cycloserine on cellular responses mediated by macrophages and T cells. Biol Pharm Bull. 2007;30:2105–12. doi: 10.1248/bpb.30.2105. [DOI] [PubMed] [Google Scholar]
- Cortez MA, Wu Y, Gibson KM, Snead OC. Absence seizures in succinic semialdehyde dehydrogenase deficient mice: a model of juvenile absence epilepsy. Pharmacol Biochem Behav. 2004;79:547–53. doi: 10.1016/j.pbb.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Cusmai R, Martinelli D, Moavero R, et al. Ketogenic diet in early myoclonic encephalopathy due to non ketotic hyperglycinemia. Eur J Paediatr Neurol. 2012 Jan 17; doi: 10.1016/j.ejpn.2011.12.015. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Daune G, Seiler N. Interrelationships between ornithine, glutamate, and GABA. II. Consequences of inhibition of GABA-T and ornithine aminotransferase in brain. Neurochem Res. 1988;13:69–75. doi: 10.1007/BF00971857. [DOI] [PubMed] [Google Scholar]
- De Biase D, Simmaco M, Barra D, Bossa F, Hewlins M, John RA. Mechanism of inactivation and identification of sites of modification of ornithine aminotransferase by 4-aminohex-5-ynoate. Biochemistry. 1991;30:2239–46. doi: 10.1021/bi00222a029. [DOI] [PubMed] [Google Scholar]
- de Vries PM, de Jong BM, Bohning DE, Hinson VK, George MS, Leenders KL. Reduced parietal activation in cervical dystonia after parietal TMS interleaved with fMRI. Clin Neurol Neurosurg. 2012 Mar 1; doi: 10.1016/j.clineuro.2012.02.006. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Ergezinger K, Jeschke R, Frauendienst-Egger G, Korall H, Gibson KM, Schuster VH. Monitoring of 4-hydroxybutyric acid levels in body fluids during vigabatrin treatment in succinic semialdehyde dehydrogenase deficiency. Ann Neurol. 2003;54:686–689. doi: 10.1002/ana.10752. [DOI] [PubMed] [Google Scholar]
- Escalera GI, Ferrer I, Marina LC, et al. Succinic semialdehyde dehydrogenase deficiency: decrease in 4-OH-butyric acid levels with low doses of vigabatrin. An Pediatr (Barc) 2010;72:128–32. doi: 10.1016/j.anpedi.2009.09.018. [DOI] [PubMed] [Google Scholar]
- Farlow MR. Treatment of mild cognitive impairment (MCI) Curr Alzheimer Res. 2009;6:362–7. doi: 10.2174/156720509788929282. [DOI] [PubMed] [Google Scholar]
- Froestl W, Gallagher M, Jenkins H, et al. SGS742: the first GABA(B) receptor antagonist in clinical trials. Biochem Pharmacol. 2004;68:1479–87. doi: 10.1016/j.bcp.2004.07.030. [DOI] [PubMed] [Google Scholar]
- Gibson KM, Sweetman L, Nyhan WL, et al. Succinic semialdehyde dehydrogenase deficiency: an inborn error of gamma-aminobutyric acid metabolism. Clin Chim Acta. 1983;133:33–42. doi: 10.1016/0009-8981(83)90018-9. [DOI] [PubMed] [Google Scholar]
- Gibson KM, DeVivo DC, Jakobs C. Vigabatrin therapy in patient with succinic semialdehyde dehydrogenase deficiency. Lancet. 1989;2:1105–6. doi: 10.1016/s0140-6736(89)91126-4. [DOI] [PubMed] [Google Scholar]
- Gibson KM, Jakobs C, Ogier H, et al. Vigabatrin therapy in six patients with succinic semialdehyde dehydrogenase deficiency. J Inherit Metab Dis. 1995;18:143–6. doi: 10.1007/BF00711750. [DOI] [PubMed] [Google Scholar]
- Gibson KM, Christensen E, Jakobs C, et al. The clinical phenotype of succinic semialdehyde dehydrogenase deficiency (4-hydroxybutyric aciduria): case reports of 23 new patients. Pediatrics. 1997;99:567–74. doi: 10.1542/peds.99.4.567. [DOI] [PubMed] [Google Scholar]
- Gibson KM, Schor DS, Gupta M, et al. Focal neurometabolic alterations in mice deficient for succinate semialdehyde dehydrogenase. J Neurochem. 2002;81:71–9. doi: 10.1046/j.1471-4159.2002.00784.x. [DOI] [PubMed] [Google Scholar]
- Gropman A. Vigabatrin and newer interventions in succinic semialdehyde dehydrogenase deficiency. Ann Neurol. 2003;54:S66–S72. doi: 10.1002/ana.10626. [DOI] [PubMed] [Google Scholar]
- Gupta M, Greven R, Jansen EE, et al. Therapeutic intervention in mice deficient for succinate semialdehyde dehydrogenase (gamma-hydroxybutyric aciduria) J Pharmacol Exp Ther. 2002;302:180–7. doi: 10.1124/jpet.302.1.180. [DOI] [PubMed] [Google Scholar]
- Gupta M, Polinsky M, Senephansiri H, et al. Seizure evolution and amino acid imbalances in murine succinate semialdehyde dehydrogenase (SSADH) deficiency. Neurobiol Dis. 2004a;16:556–62. doi: 10.1016/j.nbd.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Gupta M, Jansen EE, Senephansiri H, et al. Liver-directed adenoviral gene transfer in murine succinate semialdehyde dehydrogenase deficiency. Mol Ther. 2004b;9:527–39. doi: 10.1016/j.ymthe.2004.01.013. [DOI] [PubMed] [Google Scholar]
- Herlenius E, Lagercrantz H. Neurotransmitters and neuromodulators during early human development. Early Hum Dev. 2001;65:21–37. doi: 10.1016/s0378-3782(01)00189-x. [DOI] [PubMed] [Google Scholar]
- Hogema BM, Gupta M, Senephansiri H, et al. Pharmacologic rescue of lethal seizures in mice deficient in succinate semialdehyde dehydrogenase. Nat Genet. 2001;29:212–6. doi: 10.1038/ng727. [DOI] [PubMed] [Google Scholar]
- Jaeken J, Casaer P, de Cock P, et al. Gamma-aminobutyric acid-transaminase deficiency: a newly recognized inborn error of neurotransmitter metabolism. Neuropediatrics. 1984;15:165–9. doi: 10.1055/s-2008-1052362. [DOI] [PubMed] [Google Scholar]
- Jaeken J, Casaer P, de Cock P, Francois B. Vigabatrin in GABA metabolism disorders. Lancet. 1989;1:1074. doi: 10.1016/s0140-6736(89)92466-5. [DOI] [PubMed] [Google Scholar]
- Jakobs C, Bojasch M, Mönch E, Rating D, Siemes H, Hanefeld F. Urinary excretion of gamma-hydroxybutyric acid in a patient with neurological abnormalities. The probability of a new inborn error of metabolism. Clin Chim Acta. 1981;111:169–78. doi: 10.1016/0009-8981(81)90184-4. [DOI] [PubMed] [Google Scholar]
- Jakobs C, Michael T, Jaeger E, Jaeken J, Gibson KM. Further evaluation of vigabatrin therapy in 4-hydroxybutyric aciduria. Eur J Pediatr. 1992;151:466. doi: 10.1007/BF01959366. [DOI] [PubMed] [Google Scholar]
- Jansen EE, Struys E, Jakobs C, Hager E, Snead OC, Gibson KM. Neurotransmitter alterations in embryonic succinate semialdehyde dehydrogenase (SSADH) deficiency suggest a heightened excitatory state during development. BMC Dev Biol. 2008;8:112. doi: 10.1186/1471-213X-8-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knerr I, Pearl PL, Bottiglieri T, Snead OC, Jakobs C, Gibson KM. Therapeutic concepts in succinate semialdehyde dehydrogenase (SSADH; ALDH5a1) deficiency (gamma-hydroxybutyric aciduria). Hypotheses evolved from 25 years of patient evaluation, studies in Aldh5a1−/− mice and characterization of gamma-hydroxybutyric acid pharmacology. J Inherit Metab Dis. 2007;30:279–94. doi: 10.1007/s10545-007-0574-2. [DOI] [PubMed] [Google Scholar]
- Krauss GL, Johnson MA, Miller NR. Vigabatrin-associated retinal cone system dysfunction: electroretinogram and ophthalmologic findings. Neurology. 1998;50:614–8. doi: 10.1212/wnl.50.3.614. [DOI] [PubMed] [Google Scholar]
- Louzada PR, Paula Lima AC, Mendonca-Silva DL, Noël F, De Mello FG, Ferreira ST. Taurine prevents the neurotoxicity of beta-amyloid and glutamate receptor agonists: activation of GABA receptors and possible implications for Alzheimer’s disease and other neurological disorders. FASEB J. 2004;18:511–8. doi: 10.1096/fj.03-0739com. [DOI] [PubMed] [Google Scholar]
- Lowther J, Yard BA, Johnson KA, et al. Inhibition of the PLP-dependent enzyme serine palmitoyltransferase by cycloserine: evidence for a novel decarboxylative mechanism of inactivation. Mol Biosyst. 2010;6:1682–93. doi: 10.1039/c003743e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitre M. The gamma-hydroxybutyrate signalling system in brain: organization and functional implications. Prog Neurobiol. 1997;51:337–61. doi: 10.1016/s0301-0082(96)00064-0. [DOI] [PubMed] [Google Scholar]
- Marescaux C, Micheletti G, Vergnes M, Rumbach L, Warter JM. Diazepam antagonizes GABAmimetics in rats with spontaneous petit mal-like epilepsy. Eur J Pharmacol. 1985;113:19–24. doi: 10.1016/0014-2999(85)90338-3. [DOI] [PubMed] [Google Scholar]
- Matern D, Lehnert W, Gibson KM, Korinthenberg R. Seizures in a boy with succinic semialdehyde dehydrogenase deficiency treated with vigabatrin (gamma-vinyl-GABA) J Inherit Metab Dis. 1996;19:313–8. doi: 10.1007/BF01799261. [DOI] [PubMed] [Google Scholar]
- McNally MA, Hartman AL. Ketone bodies in epilepsy. J Neurochem. 2012 Jan 23; doi: 10.1111/j.1471-4159.2012.07670.x. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta AK, Gould GG, Gupta M, Carter LP, Gibson KM, Ticku MK. Succinate semialdehyde dehydrogenase deficiency does not down-regulate gamma-hydroxybutyric acid binding sites in the mouse brain. Mol Genet Metab. 2006;88:86–9. doi: 10.1016/j.ymgme.2005.11.012. [DOI] [PubMed] [Google Scholar]
- Murphy TC, Amarnath V, Gibson KM, Picklo MJ., Sr Oxidation of 4-hydroxy-2-nonenal by succinic semialdehyde dehydrogenase (ALDH5A1) J Neurochem. 2003;86:298–305. doi: 10.1046/j.1471-4159.2003.01839.x. [DOI] [PubMed] [Google Scholar]
- Netopilová M, Drsata J, Haugvicová R, Kubová H, Mares P. Inhibition of glutamate decarboxylase activity by 3-mercaptopropionic acid has different time course in the immature and adult rat brains. Neurosci Lett. 1997;226:68–70. doi: 10.1016/s0304-3940(97)00241-3. [DOI] [PubMed] [Google Scholar]
- Nylen K, Velazquez JL, Likhodii SS, et al. A ketogenic diet rescues the murine succinic semialdehyde dehydrogenase deficient phenotype. Exp Neurol. 2008;210:449–57. doi: 10.1016/j.expneurol.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nylen K, Velazquez JL, Sayed V, Gibson KM, Burnham WM, Snead OC. The effects of a ketogenic diet on ATP concentrations and the number of hippocampal mitochondria in Aldh5a1(−/−) mice. Biochim Biophys Acta. 2009;1790:208–12. doi: 10.1016/j.bbagen.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oermann E, Warskulat U, Heller-Stilb B, Häussinger D, Zilles K. Taurine-transporter gene knockout-induced changes in GABA(A), kainate and AMPA but not NMDA receptor binding in mouse brain. Anat Embryol (Berl) 2005;210:363–72. doi: 10.1007/s00429-005-0024-6. [DOI] [PubMed] [Google Scholar]
- Olive MF. Interactions between taurine and ethanol in the central nervous system. Amino Acids. 2002;23:345–57. doi: 10.1007/s00726-002-0203-1. [DOI] [PubMed] [Google Scholar]
- Pearl PL, Gropman A. Monitoring gamma-hydroxybutyric acid levels in succinate-semialdehyde dehydrogenase deficiency. Ann Neurol. 2004;55:599–599. doi: 10.1002/ana.20084. [DOI] [PubMed] [Google Scholar]
- Pearl PL, Gibson KM, Quezado Z, et al. Decreased GABA-A binding on FMZ-PET in succinic semialdehyde dehydrogenase deficiency. Neurology. 2009a;73:423–9. doi: 10.1212/WNL.0b013e3181b163a5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl PL, Vezina LG, Saneto RP, et al. Cerebral MRI abnormalities associated with vigabatrin therapy. Epilepsia. 2009b;50:184–94. doi: 10.1111/j.1528-1167.2008.01728.x. [DOI] [PubMed] [Google Scholar]
- Pearl PL, Gibson KM, Cortez MA, et al. Succinic semialdehyde dehydrogenase deficiency: lessons from mice and men. J Inherit Metab Dis. 2009c;32:343–52. doi: 10.1007/s10545-009-1034-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl PL, Shukla L, Theodore WH, Jakobs C, Michael Gibson K. Epilepsy in succinic semialdehyde dehydrogenase deficiency, a disorder of GABA metabolism. Brain Dev. 2011;33:796–805. doi: 10.1016/j.braindev.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl PL, Theodore WH, McCarter R, McGavin C, Sweetman L, Gibson KM. Taurine intervention in succinic semialdehyde dehydrogenase (SSADH) deficiency: an open label trial. Molec Genet Metab. 2012;105:346. (abstract) [Google Scholar]
- Peyster RG, Sussman NM, Hershey BL, et al. Use of ex vivo magnetic resonance imaging to detect onset of vigabatrin-induced intramyelinic edema in canine brain. Epilepsia. 1995;36:93–100. doi: 10.1111/j.1528-1157.1995.tb01672.x. [DOI] [PubMed] [Google Scholar]
- Polc P, Pieri L, Bonetti EP, et al. L-cycloserine: behavioural and biochemical effects after single and repeated administration to mice, rats and cats. Neuropharmacology. 1986;25:411–8. doi: 10.1016/0028-3908(86)90236-4. [DOI] [PubMed] [Google Scholar]
- Qiao M, Malisza KL, Del Bigio MR, Kozlowski P, Seshia SS, Tuor UI. Effect of long-term vigabatrin administration on the immature rat brain. Epilepsia. 2000;41:655–65. doi: 10.1111/j.1528-1157.2000.tb00225.x. [DOI] [PubMed] [Google Scholar]
- Rahbeeni Z, Ozand PT, Rashed M, et al. 4-Hydroxybutyric aciduria. Brain Dev. 1994;16(Suppl):64–71. doi: 10.1016/0387-7604(94)90098-1. [DOI] [PubMed] [Google Scholar]
- Reis J, Cohen LG, Pearl PL, et al. GABA(B)ergic motor dysfunction in SSADH deficiency. Neurology. doi: 10.1212/WNL.0b013e31825dcf71. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rheims S, Minlebaev M, Ivanov A, et al. Excitatory GABA in rodent developing neocortex in vitro. J Neurophysiol. 2008;100:609–19. doi: 10.1152/jn.90402.2008. [DOI] [PubMed] [Google Scholar]
- Roberts F, Taberner PV, Hill RG. The effect of 3-mercaptopropionate, an inhibitor of glutamate decarboxylase, on the levels of GABA and other amino acids, and on presynaptic inhibition in the rat cuneate nucleus. Neuropharmacology. 1978;17:715–20. doi: 10.1016/0028-3908(78)90085-0. [DOI] [PubMed] [Google Scholar]
- Saronwala A, Tournay A, Gargus JJ. Taurine treatment of succinate semialdehyde dehydrogenase (SSADH) deficiency reverses MRI-documented globus lesion and clinical syndrome. Am Coll Med Genet; 15th Ann Clinical Genet Meeting; March 12–16; Phoenix AZ USA. 2008. p. 103. [Google Scholar]
- Snead OC. Gamma hydroxybutyrate in the monkey. I. Electroencephalographic, behavioral, and pharmacokinetic studies. Neurology. 1978;28:636–42. doi: 10.1212/wnl.28.7.636. [DOI] [PubMed] [Google Scholar]
- Snead OC. Evidence for a G protein-coupled gamma-hydroxybutyric acid receptor. J Neurochem. 2000;75:1986–96. doi: 10.1046/j.1471-4159.2000.0751986.x. [DOI] [PubMed] [Google Scholar]
- Snead OC, Gibson KM. Gamma-hydroxybutyric acid. N Engl J Med. 2005;352:2721–32. doi: 10.1056/NEJMra044047. [DOI] [PubMed] [Google Scholar]
- Sorri I, Brigell MG, Mályusz M, Mahlamäki E, de Meynard C, Kälviäinen R. Is reduced ornithine-δ-aminotransferase activity the cause of vigabatrin-associated visual field defects? Epilepsy Res. 2010;92:48–53. doi: 10.1016/j.eplepsyres.2010.08.006. [DOI] [PubMed] [Google Scholar]
- Spence SJ, Sankar R. Visual field defects and other ophthalmological disturbances associated with vigabatrin. Drug Saf. 2001;24:385–404. doi: 10.2165/00002018-200124050-00005. [DOI] [PubMed] [Google Scholar]
- Stewart LS, Nylen KJ, Persinger MA, Cortez MA, Gibson KM, Snead OC. Circadian distribution of generalized tonic-clonic seizures associated with murine succinic semialdehyde dehydrogenase deficiency, a disorder of GABA metabolism. Epilepsy Behav. 2008;13:290–4. doi: 10.1016/j.yebeh.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillakaratne NJ, Medina-Kauwe L, Gibson KM. γ-Aminobutyric acid (GABA) metabolism in mammalian neural and nonneural tissues. Comp Biochem Physiol A Physiol. 1995;112:247–63. doi: 10.1016/0300-9629(95)00099-2. [DOI] [PubMed] [Google Scholar]
- Tomi M, Tajima A, Tachikawa M, Hosoya K. Function of taurine transporter (Slc6a6/TauT) as a GABA transporting protein and its relevance to GABA transport in rat retinal capillary endothelial cells. Biochim Biophys Acta. 2008;1778:2138–42. doi: 10.1016/j.bbamem.2008.04.012. [DOI] [PubMed] [Google Scholar]
- Tsuji M, Aida N, Obata T, et al. A new case of GABA transaminase deficiency facilitated by proton MR spectroscopy. J Inherit Metab Dis. 2010;33:85–90. doi: 10.1007/s10545-009-9022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uziel G, Bardelli P, Pantaleoni C, Rimoldi M, Savoiardo M. 4-Hydroxybutyric aciduria: clinical findings and vigabatrin therapy. J Inherit Metab Dis. 1993;16:520–2. doi: 10.1007/BF00711670. [DOI] [PubMed] [Google Scholar]
- Vanadia E, Gibson KM, Pearl PL, Trapolino E, Mangano S, Vanadia F. Therapeutic efficacy of magnesium valproate in succinic semialdehyde dehydrogenase deficiency. J Inherit Metab Dis. doi: 10.1007/8904_2012_170. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhatalo S, Nousiainen I, Eriksson K, et al. Visual field constriction in 91 Finnish children treated with vigabatrin. Epilepsia. 2002;43:748–756. doi: 10.1046/j.1528-1157.2002.17801.x. [DOI] [PubMed] [Google Scholar]
- Waagepetersen HS, Sonnewald U, Schousboe A. The GABA paradox: multiple roles as metabolite, neurotransmitter, and neurodifferentiative agent. J Neurochem. 1999;73:1335–42. doi: 10.1046/j.1471-4159.1999.0731335.x. [DOI] [PubMed] [Google Scholar]
- Wong CG, Gibson KM, Snead OC. From the street to the brain: neurobiology of the recreational drug gamma-hydroxybutyric acid. Trends Pharmacol Sci. 2004;25:29–34. doi: 10.1016/j.tips.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Wu Y, Buzzi A, Frantseva M, et al. Status epilepticus in mice deficient for succinate semialdehyde dehydrogenase: GABAA receptor-mediated mechanisms. Ann Neurol. 2006;59:42–52. doi: 10.1002/ana.20686. [DOI] [PubMed] [Google Scholar]
- Yalçinkaya C, Gibson KM, Gündüz E, Koçer N, Fiçicio lu C, Küçükercan I. MRI findings in succinic semialdehyde dehydrogenase deficiency. Neuropediatrics. 2000;31:45–6. doi: 10.1055/s-2000-15298. [DOI] [PubMed] [Google Scholar]
- Ziyeh S, Berlis A, Korinthenberg R, Spreer J, Schumacher M. Selective involvement of the globus pallidus and dentate nucleus in succinic semialdehyde dehydrogenase deficiency. Pediatr Radiol. 2002;32:598–600. doi: 10.1007/s00247-002-0717-4. [DOI] [PubMed] [Google Scholar]