

Abstract
To understand the molecular mechanism of oxidation-induced inhibition of muscle contractility, we have studied the effects of hydrogen peroxide on permeabilized rabbit psoas muscle fibers, focusing on changes in myosin purified from these fibers. Oxidation by 5 mM peroxide decreased fiber contractility (isometric force and shortening velocity) without significant changes in the enzymatic activity of myofibrils and isolated myosin. The inhibitory effects were reversed by treating fibers with dithiothreitol. Oxidation by 50 mM peroxide had a more pronounced and irreversible inhibitory effect on fiber contractility and also affected enzymatic activity of myofibrils, myosin, and actomyosin. Peroxide treatment also affected regulation of contractility, resulting in fiber activation in the absence of calcium. Electron paramagnetic resonance of spinlabeled myosin in muscle fibers showed that oxidation increased the fraction of myosin heads in the strong-binding structural state under relaxing conditions (low calcium) but had no effect under activating conditions (high calcium). This change in the distribution of structural states of myosin provides a plausible explanation for the observed changes in both contractile and regulatory functions. Mass spectroscopy analysis showed that 50 mM but not 5 mM peroxide induced oxidative modifications in both isoforms of the essential light chains and in the heavy chain of myosin subfragment 1 by targeting multiple methionine residues. We conclude that 1) inhibition of muscle fiber contractility via oxidation of myosin occurs at high but not low concentrations of peroxide and 2) the inhibitory effects of oxidation suggest a critical and previously unknown role of methionines in myosin function.
Keywords: oxidation, proteomics
reactive oxygen and nitrogen species are produced in living cells and are involved in the regulation of normal metabolic processes, but their excessive generation may lead to modifications of proteins and their physiological functions (34, 41, 74, 77, 92). For example, treatment of intact and permeabilized skeletal muscle fibers with such oxidants as nitric oxide, peroxynitrite, hydroxyl radicals, and hydrogen peroxide resulted in changes in force and Ca2+ sensitivity that were variable depending on muscle fiber type, conditions of modification, and the applied reagent (2, 3, 14, 30, 38, 60, 61). The results were discussed in terms of possible effects on myofibrillar Ca2+ sensitivity, state of contractile proteins, and oxidation of cysteines, but direct analysis of proteins purified from the affected muscle were not performed, so the molecular basis behind the observed changes remained unknown. Because individual oxidants can react with multiple amino acids and can form multiple products, understanding the molecular basis of oxidation-induced changes in muscle contractility in terms of chemical, functional, and structural changes at the level of individual proteins requires studies using well-defined in vitro model systems.
We have selected as our model oxidant hydrogen peroxide (H2O2). H2O2 is naturally produced in many tissues, including skeletal muscle. It is involved in multiple physiological processes necessary for normal functioning of cells (29, 69, 76), but it can also have deleterious effects and has been used to study the mechanism of cardiac and smooth muscle dysfunction (37, 57, 59, 98).
Previously reported physiological studies on the effects of H2O2 on skeletal muscle contractility have been performed with a variety of muscles, such as permeabilized extensor digitorum longus (EDL) and soleus muscle from rat, intact fibers from mouse foot, and permeabilized rat diaphragm fibers (2, 14, 38, 61) We have selected as our model muscle permeabilized rabbit psoas muscle fibers because these fibers have been routinely used to determine molecular changes in myosin during muscle contraction (20, 55), and because permeabilization diminishes the number of possible targets of peroxide by removing soluble proteins and the membrane-based excitation-contraction system. The majority of proteins remaining in the permeabilized psoas are actin (22%) and myosin (43%) (99). Also, the large size of the rabbit psoas muscle enables purification of myosin and actin in amounts required for biochemical and proteomic studies. In the present study we focused on oxidation-induced changes in myosin, and the changes in actin will be described in a separate paper.
Skeletal muscle myosin is a hexamer composed of two heavy chains, two regulatory light chains (RLC), and two essential light chains (ELC), of which there are two isoforms, ELC1 and ELC2. The specific regions essential to myosin's contractile and enzymatic activity are localized in the globular region of each heavy chain, named the myosin head. The light chains of skeletal muscle myosin have mainly regulatory functions involving calcium regulation and fiber shortening velocity (80), as well as regulating the extent of actin activation of myosin ATPase (75, 87, 93). Most current theories of muscle contraction postulate that hydrolysis of ATP by actomyosin is accompanied by structural changes in myosin and actin and force is generated during transition of the myosin head from the states of weak interaction to the states of strong interaction with actin (65, 83). We hypothesize that oxidation-induced decreases of contractile and enzymatic functions of muscle fibers will be linked to changes in structural transitions of myosin during the actomyosin ATPase cycle because of oxidation of specific regions in the myosin head.
The unique feature of the present study is that we tracked the functional effects of peroxide treatment from the level of the muscle fiber to individual amino acid residues of myosin purified from these fibers. The measurements on fibers included isometric force, resting tension, maximal shortening velocity, enzymatic activities, and the distribution of myosin structural states determined by electron paramagnetic resonance (EPR). Myosin purified from the fibers was used to determine Vmax and Km of actomyosin ATPase and the affinities (Kd) of weak and strong binding to actin. Chemical changes in functional domains of myosin and within the individual myosin residues were determined with mass spectroscopy (MS).
EXPERIMENTAL PROCEDURES
Preparation of Rabbit Psoas Muscle Fibers
All samples in this study were obtained from permeabilized rabbit psoas muscle. Animal care and use procedures were approved by the University of Minnesota institutional animal care and use committee and complied with guidelines set by the American Physiological Society. Strips of psoas muscle were freshly dissected from New Zealand White rabbits and permeabilized by incubation for 6 h at 4°C on a shaker in a glycerination buffer [in mM: 60 potassium propionate (KPr), 25 MOPS pH 7.0, 2 EGTA, and 1 NaN3, with 0.5% Triton X-100 and 25% glycerol]. Strips were then incubated for 20 h in a storage buffer (in mM: 60 KPr, 25 MOPS pH 7.0, 2 MgCl2, 1 EGTA, and 1 NaN3, with 50% glycerol) at 4°C and finally transferred to fresh storage buffer containing 0.1 mM DTT and stored at −20°C for up to 5 mo.
Oxidation of Muscle Fibers
Permeabilized strips were further dissected on ice into 0.5- to 1-mm-thick fiber bundles and collected in a 50-ml Falcon tube, and glycerol was washed out by three cycles of 2-min centrifugation at 1,800 g and resuspension of pelleted fiber bundles in rigor buffer (RB; in mM: 130 KPr, 25 MOPS pH 7.0, 6 MgCl2, and 1 EGTA) at 4°C. The final suspension of fiber bundles was divided, and one part was designated for a control unoxidized sample and the other part for oxidation. Oxidation was carried out by incubating fiber bundles for 30 min at 25°C in RB containing either 5 mM or 50 mM H2O2. To terminate oxidation, H2O2 was washed out by two cycles of 2-min centrifugation at 1,800 g and resuspension of pelleted fibers in RB at 4°C. Washed fiber bundles were then reduced by 15-min incubation at 4°C in RB supplemented with 10 mM DTT. Reduced fiber bundles were washed five times at 4°C in 20 mM Tris pH 7.5. Control unoxidized samples were always prepared and treated in parallel to the oxidized samples except for the omission of H2O2. Myofibrils from the permeabilized fiber bundles were prepared by homogenization (5 × 10 s) on ice in 20 mM Tris pH 7.5 with a PowerGen 700 homogenizer. Fiber bundles used for 4-(2-iodoacetamido)-2,2,6,6-tetramethyl-1-piperidinyloxy spin label (IASL) labeling and for isolation of single fibers were prepared following the same protocol as that for the fiber bundles described above for oxidation except that the fiber bundles were immediately bound to glass rods and washes were carried out by transferring the rods between tubes filled with 5 ml of RB. After oxidation and DTT reduction, rod-bound fiber bundles were washed three times in RB and then stored in the relaxing buffer (defined below) until contractile measurements were done on the day of oxidation. Control experiments showed that the effect of oxidation on myofibrillar ATPases was independent of whether oxidation was carried out on a large number of fiber bundles in suspension or on rod-bound fiber bundles.
Single-Fiber Contractile Measurements
Individual muscle fiber segments (~2 mm long) were isolated from unoxidized and oxidized fiber bundles and transferred to a relaxing buffer (in mM: 7.0 EGTA, 5.4 MgCl2, 20 imidazole pH 7.0, 14.5 creatine phosphate, and 4.7 ATP, with CaCl2 to achieve pCa 9 and KCl to achieve ionic strength of 180 mM). The sarcomere length of each fiber was set to 2.5 μm, and resting tension was recorded. Fibers were studied at 20°C for force-Ca2+ relationships (44, 48) or at 15°C for maximal isometric force at pCa 4.5 (Fmax), resting tension at pCa 9, and unloaded shortening velocity V0 (42–44, 64, 85).
EPR of IASL-Labeled Muscle Fiber Bundles
Unoxidized and oxidized fiber bundles were labeled with IASL, which specifically targets Cys707 in the catalytic domain of myosin subfragment 1 (S1), as described previously (43, 55).
EPR spectroscopy of spin-labeled muscle fiber bundles under conditions of rigor, relaxation, and contraction was performed on an E500 EleXsys spectrometer (Bruker Instruments) as described previously (43). The intensities of spectra were measured in isometric contraction (C) and relaxation (E) by averaging nine points of each spectrum at the position corresponding to the minimum of the rigor spectrum, which was defined to be zero (Fig. 1). The peak of the rigor spectrum (Rmax) was also measured. These intensities were used to determine the fraction of myosin heads in the strong-binding structural state during contraction (XC) and relaxation (XE). This calculation was simplified by observing that 1) the spectrum obtained in contraction is a linear combination of spectra obtained during relaxation and rigor, as shown previously (43), 2) E/Rmax is constant (0.63) for unoxidized fibers, 3) oxidation has no effect on the spectrum in rigor (R), and 4) in unoxidized fibers XE = 0. Thus, for unoxidized fibers, XC was calculated from
| (1) |
and for oxidized fibers, XE and XC were calculated from
| (2) |
which reduces to Eq. 1 for unoxidized fibers (E/Rmax = 0.63).
Fig. 1.

Low-field portion of electron paramagnetic resonance (EPR) spectra of iodoacetamide spin label-labeled unoxidized fiber bundle in rigor, relaxation (E), and contraction (C). Horizontal line is zero, defined as the minimum in rigor. Vertical line indicates the position where C and E are measured for use in Eqs. 1 and 2. Rmax, peak of rigor spectrum.
Preparation of Myosin, α-Chymotryptic Myosin S1, and Heavy Meromyosin from Muscle Fiber Bundles
All procedures were carried out on ice or at 4°C, unless stated otherwise. Oxidized and unoxidized bundles were pelleted by 5-min centrifugation at 1,800 g; the amount of pelleted fibers was in the range of 3–5 ml, depending on preparation. Myosin was extracted by incubating the pellets for 15 min on ice with three volumes of a high-salt buffer (0.6 M KCl, 4 mM MgCl2, 2 mM EGTA, 0.1 M K-phosphate pH 6.5, 2 mM ATP, and 5 mM DTT) followed by 10-min centrifugation at 3,200 g. The supernatants containing myosin were dialyzed overnight to 1 liter of 20 mM KCl-10 mM imidazole pH 7.0. Precipitated myosin was collected by 15-min centrifugation at 12,000 g, dissolved in 0.3 M KCl-10 mM imidazole pH 7.0, and centrifuged for 30 min at 300,000 g to remove actin and actomyosin. The purity of myosin was confirmed by SDS-PAGE. This myosin was used for ATPase measurements and preparation of S1.
S1 was prepared by α-chymotrypsin (Sigma) digestion of filamentous myosin (96) and stored in liquid nitrogen in the presence of 0.15 M sucrose. Control experiments performed on fresh and liquid nitrogen-stored S1 showed that the storage did not affect the high-salt ATPase activities or thiol content of S1. Typically, 1 g of pelleted fibers yielded ~2 mg of S1.
Heavy meromyosin (HMM) was prepared by α-chymotrypsin digestion of solubilized myosin as described previously (64) and used exclusively for detecting RLCs in MS experiments.
Protein Concentration
Concentration of myofibrils was measured by the Bradford (12) protein assay (Bio-Rad) with bovine serum albumin (Sigma) as a standard. The concentration of purified proteins was measured by ultraviolet absorption, assuming molar extinction coefficients of 0.63 mg·ml−1·cm−1 for actin at 290 nm, 0.53 mg·ml−1·cm−1 for myosin at 280 nm, and 0.75 mg·ml−1·cm−1 for S1 at 280 nm. Protein concentrations of S1 determined by ultraviolet absorption were the same as those determined by Bradford protein assay, showing that the molar extinction coefficients of purified proteins were not affected by oxidation.
Interaction of S1 With Actin: Strong and Weak Binding Affinity and Actin-Activated ATPase
Actin used in these experiments was extracted from rabbit skeletal muscle acetone powder, purified as described previously (65), and dissolved in F-actin (FA) buffer (mM): 3 MgCl2-10 Tris pH 7.5 supplemented with 0.2 ATP.
Binding of S1 to actin was measured at 25°C in FA buffer containing 1) 1 mM ADP + 20 μM adenylate kinase inhibitor P1,P5-di(adenosine-5′)pentaphosphate (Ap5A) in samples for strong binding (in the absence of ATP) or 2) 2.7 mM ATP in samples for weak binding (in the presence of ATP), as described previously (63, 65). The actin-activated S1 ATPase was measured at 25°C in FA buffer and 2.2 mM ATP, as described previously (64).
The high-salt ATPase activities of myofibrils, myosin, and S1 were measured at 25°C in 4 mM ATP and 0.6 M KCl, 50 mM Tris pH 7.5, and 5 mM EDTA (K-ATPase), or 5 mM CaCl2 (Ca-ATPase). The physiological ATPase activities of myofibrils were measured at 25°C in (mM) 3 ATP, 121 KPr, 25 Tris, 3 MgCl2, and 1.5 CaCl2, pH 7.0 (activating conditions, pCa 4.5) and (mM) 3 ATP, 124 KPr, 25 Tris, 3 MgCl2, and 2 EGTA, pH 7.0 (relaxing conditions, pCa 9). The concentration of the liberated phosphate was determined by the malachite green method (39).
Determination of Reactive Cysteine
Reactive cysteines were quantitated in 2.7 μM S1 in the presence of 0.1 M NaHPO4 pH 8.0, 1 mM EDTA, 1% SDS, and 10 mM Tris (pH 7.5) by the 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) method. The cysteine content (mol cysteine/mol protein) was calculated by using the absorption coefficient of the TNB anion at 412 nm as 14,150 M −1cm−1 (68).
Fluorescein Iodoacetamide and Isotope-Coded Affinity Tag Labeling of S1
In experiments using native S1, 100 μM fluorescein iodoacetamide (IAF; Invitrogen) dissolved in dimethylformamide was added to 10 μM S1 in 60 mM KCl-50 mM Tris pH 7.5 and incubated for 3 h at 0°C (81). In experiments using denatured S1, 350 μM IAF dissolved in dimethylformamide was added to 3.2 μM S1 in 25 mM Tris pH 8.5–0.1% SDS-10% acetonitrile and incubated for 2 h at 37°C (denatured S1). Each reaction was terminated by loading samples on Sephadex G-25 columns and eluting with 25 mM NH4HCO3.
Isotope-coded affinity tag (ICAT) labeling of S1 with light 12C reagent (unoxidized S1) and heavy 13C reagent (oxidized S1) was carried out according to the manufacturer's instructions and using the manufacturer's buffers and columns [Applied Biosystems (ABI) Cleavable ICAT Reagent 10-Assay Kit, part no. 4339036].
Mass Spectroscopy
Electrospray analysis of heavy and light chains of S1
S1 (1–2 mg/ml) was dialyzed for 12–24 h at 4°C to 10 mM ammonium bicarbonate (pH 7.9). One aliquot of S1 was used directly for MS analysis of the intact protein, and another aliquot was subjected to limited trypsin (Worthington) digest into 20-kDa, 23-kDa, and 50-kDa domains by 30-min incubation at 23°C with a substrate-to-enzyme ratio of 100:1 (52). The digestion was terminated by the addition of a twofold excess of soybean trypsin inhibitor relative to trypsin, and samples were immediately analyzed by MS. S1 from three independently prepared pairs of unoxidized and 50 mM H2O2-oxidized fibers was used for analysis, and analysis of one S1 pair was performed in duplicate.
The masses of the S1 heavy chain, the heavy chain tryptic domains, and light chains were determined with a QSTAR quadrupole-TOF mass spectrometer (ABI) with an electrospray ionization (ESI) source. Before infusion of the intact S1 solution, a solution of 50:50 acetonitrile-water and 0.1% formic acid (load solution) was infused at 50 μl/min with an integrated syringe pump. After a stable, baseline total ion current (TIC) was established for the load solution, the protein solution (20 μM protein, 10 mM NH4HCO3 pH 7.9) was introduced into the solvent stream with a 10-μl injection loop installed in the integrated loop injector. After the TIC returned to baseline intensity, four more consecutive loop injections of the protein solution were made, for a total of five injections per sample. Data were acquired continuously during load buffer infusion and protein infusions over the range 500–2,000 mass-to-charge ratio (m/z).
Matrix-assisted laser desorption ionization MS and ESI analysis of S1 peptides
Exhaustive digestion of S1 into peptides was performed in 25 mM ammonium bicarbonate (pH 7.9) and 5% acetonitrile, using an protein-to-enzyme ratio of 20:1 (wt/wt) for trypsin (Promega), Glu-C (Roche), or an equal mixture of Glu C and Lys-C (Roche). After 16-h incubation at 37°C (trypsin) or at 25°C (Glu-C, Glu-C/Lys-C), the digestion was terminated by lowering the pH to 3 with 10% trifluoroacetic acid (TFA) for matrix-assisted laser desorption ionization (MALDI) MS analysis or 10% formic acid for the ESI analysis. S1 from two to four independently prepared pairs of unoxidized and 50 mM H2O2-oxidized fibers were used for analysis.
MALDI MS analysis
Peptides from IAF- and ICAT-labeled S1 were desalted with C-18 ZipTips (Millipore) according to the manufacturer's protocol. Eluted peptides were mixed 1:1 with a saturated solution of α-cyano-4-hydroxycinnamic acid in 50% acetonitrile and 0.1% TFA. One microliter of the peptide-matrix solution was spotted on a MALDI target and allowed to dry on the benchtop. Analysis of peptides was performed on a QSTAR-XL Hybrid Quadrupole TOF mass spectrometer (ABI) over an m/z range of 500–3,500, using a pulser frequency of 4.993 kHz. A 337-nm nitrogen laser was used, with 8 μJ of energy and a repetition rate of 20 Hz. The instrument was calibrated with commercially available peptides (angiotensin II, 1046.5417, monoisotopic m/z, and ACTH clip [18–39], 2465.1989 monoisotopic m/z) (Sigma). The relative amount of cysteine-containing peptides in IAF-labeled S1 was quantitated by normalizing the total peak area of the cysteine-containing peptide {using all measurable [signal-to-noise ratio (S/N) >3] peaks in the isotope envelope} to the total peak area of a reference peptide. The reference peptide was chosen as one that did not contain oxidizable residues (C, N, W, or Y) and should therefore be unaffected by the H2O2 treatment. One peptide met this criterion: GSSFQRVSALFR (mass 1298.66 Da) in the trypsin digestion and GSSFQRVSALFRE (mass 1427.71 Da) in the Glu-C/Lys-C digestion. This quantitation procedure was validated by analyzing a single sample multiple times with multiple acquisition times, and the ratio of the labeled peptide area to the reference peptide area remained constant (within ±5% variability) for each replicate. The normalized areas for all labeled peptides from the oxidized samples were expressed as a fraction of the normalized areas for the same IAF-labeled peptide from the unoxidized samples, giving a relative content of reactive cysteines.
ESI analysis
Peptides from unlabeled S1 were desalted with a 3-ml C18 Sep-Pak cartridge (Waters, Milford, MA) according to the manufacturer's protocol and separated on a strong-cation exchange column (PolyLC) into 28 fractions collected in 3-min time intervals. The peptides were eluted during a 60-min linear gradient of 0–500 mM KCl in aqueous K2HPO4 (pH 3), 20% acetonitrile. Fractions with absorbance at 280 nm lower than 2 milliabsorbance units were discarded. Fractions with a UV intensity above the baseline level (except for the flow-through fractions between 0 and 10 min) were separated by C18 reverse-phase capillary liquid chromatography (LC) online with a QSTAR Pulsar i quadrupole-TOF MS instrument (ABI) with a Proxeon nano-ESI source. Details of the LC-MS/MS method were described previously (35).
Analysis of mass spectroscopy data
The ESI spectra of S1 heavy and light chains and the MALDI spectra of S1 peptides were analyzed with BioAnalyst QS (ABI) software (version 1.1.5). The Bayesian Protein or Peptide Reconstruction tool was used to generate peak lists from the spectra, using a S/N threshold of 20 for intact S1 and its tryptic domains and 8 for S1 peptides. Error of protein reconstruction was calculated with a propagated error formula described previously (58). Individual reconstruction error ranges were for intact S1 0.1–0.5 Da (heavy chain) and 0.01–0.1 Da (ELC) and for tryptic domains 0.07–0.2 Da (23 kDa), 0.08–0.2 Da (20 kDa), and 0.01–1 Da (50 kDa).
Theoretical protein masses for known sequences of myosin heavy and light chains (sequence identifiers in the NCBI protein database: gi 13431707, gi 127130, gi 127135, and gi 127176) and of S1 domains were compared with experimental peak masses. The experimental masses of heavy chain, its tryptic domains, and ELCs given in the text represent averages of two or three independent measurements and standard deviations. All results of MS measurements (protein and peptides) are reported as a mass M.
The ESI spectra of S1 peptides were analyzed with Protein Pilot software (version 1, ABI) with the following parameters: ID type: peptide; enzyme: trypsin or Glu-C; biological modifications: invoked; thorough search: invoked; cysteine modification: none; minimum protein confidence: 90%; database: rabbit proteins extracted from NCBI nonredundant database on November 29, 2006, merged with common contaminants to create a database containing 4,926 proteins. Peptides with a confidence score <95% were excluded from the analysis. The MS/MS spectra of the peptides highlighted by the software as modified were inspected manually for accuracy. The “no cysteine alkylation” modification set automatically included searches for cysteic acid and dehydroalanine (from cysteine), and the biological modifications set included searches for oxidations of methionine, tryptophan, and histidine.
Statistical Analysis of Data
Results are presented as means ± SE, unless otherwise indicated. The effects of oxidation were evaluated by one-way ANOVA, with Tukey post hoc tests performed for multiple comparisons. Differences were considered significant when P < 0.05.
RESULTS
Selecting Conditions of Muscle Fiber Oxidation by H2O2
The conditions of oxidation were selected in a series of preliminary experiments in which fibers were oxidized with increasing concentrations of H2O2 (0–200 mM) in RB. Previous works from our laboratories (6, 43, 55) documented that this buffer supports optimal contractile function of fibers associated with structural transitions in myosin, detectable by EPR spectroscopy; buffers of similar composition were also used in other studies on oxidation of permeabilized muscle fibers (14, 38, 61).
The effects of the increasing concentrations of H2O2 were assessed by measuring changes in all four myofibrillar ATPase activities. On the basis of these results two concentrations of H2O2, 5 and 50 mM, were selected for detailed functional, structural, biochemical, and proteomic studies. Oxidation by 5 mM H2O2 initiated measurable changes in ATPases, and 50 mM H2O2 induced ~50% change in at least one of the ATPase activities (data not shown), indicating that at this concentration we would be able to detect significant biochemical and structural effects. Since molecular changes induced by H2O2 could also depend on the physiological state of muscle during oxidation (30, 38), we compared the enzymatic effects of muscle fiber oxidation by 50 mM H2O2 in the absence (rigor) and the presence of 10 mM K-pyrophosphate (stable condition like relaxation). Oxidation-induced changes in myofibrillar ATPase activities were independent of the presence of pyrophosphate; therefore in the present study H2O2 treatment was performed on fibers in the rigor state.
Effects of H2O2 Oxidation on Contractile Function of Muscle Fibers
The contractile parameters (Fig. 2) of control unoxidized fibers, Fmax = 115.7 ± 4.4 kN/m2 and V0 = 3.82 ± 0.15 fl/s (n = 24), were similar to those reported from other laboratories (17, 46). Oxidation of fibers by 5 and 50 mM H2O2 had significant concentration-dependent effects on these parameters (Fig. 2). Oxidation by 5 mM H2O2 decreased Fmax by ~31% (down to 79.8 ± 4.9 kN/m2, n = 19; Fig. 2A) and V0 by ~54% (down to 1.69 ± 0.13 fl/s, n = 19; Fig. 2B). Increasing the concentration of H2O2 to 50 mM had much more pronounced inhibitory effects: Fmax decreased by ~85% (down to 19.5 ± 1.2 kN/m2, n = 10), and V0 approached 0.
Fig. 2.

Effects of 5 and 50 mM H2O2 on muscle fiber contractility. A: maximal isometric force (Fmax). B: maximal unloaded shortening velocity (V0). Data are means ± SE. *P < 0.001, significantly different from unoxidized.
Measurements at submaximal Ca2+ showed that oxidation significantly affected Ca2+ sensitivity. The Ca2+ threshold for force development (assessed from Hill plots; Ref. 48) increased from pCa 6.89 ± 0.05 in unoxidized fibers to 7.51 ± 0.15 in fibers oxidized by 5 mM H2O2 and to 10.13 ± 0.19 (n = 12, 6, and 10, respectively) in fibers oxidized by 50 mM H2O2, indicating oxidation-induced loss of Ca2+ regulation (Fig. 3A). In contrast, a decrease was observed in pCa50 (where 50% of the Fmax is developed), with no significant difference between the effects of 5 and 50 mM H2O2 (Fig. 3B). Thus peroxide treatment decreased the Ca2+ concentration ([Ca2+]) required for the threshold of force activation while increasing the [Ca2+] required for 50% of the maximum force.
Fig. 3.

Effects of 5 and 50 mM H2O2 on Ca2+ regulation of muscle fiber contractility. A: Ca2+ threshold. B: pCa50. C: resting tension. Data are means ± SE. *P < 0.001, significantly different from unoxidized.
These oxidation-induced changes in Ca2+ regulation of contractility were also reflected in the changes of force under relaxing conditions (pCa 9), i.e., resting tension. Resting tension in unoxidized fibers (0.97 ± 0.16 kN/m2, n = 6) and fibers oxidized by 5 mM H2O2 (1.57 ± 0.09 kN/m2, n = 3) was only a small fraction of the Fmax, but in fibers oxidized by 50 mM H2O2, the resting tension significantly increased (to 14.04 ± 1.34 kN/m2, n = 8), reaching ~70% of Fmax, consistent with the loss of Ca2+ regulation in these fibers (Fig. 3C).
The Fmax and V0 of 5 mM and 50 mM H2O2-oxidized fibers was not affected by 10 mM DTT reduction applied during our standard preparation procedure. However, when fibers oxidized by 5 mM H2O2 were further reduced by 100 mM DTT (15-min incubation at 0°C), Fmax, resting tension, and V0 returned to the levels observed in unoxidized fibers. In contrast, after treatment with 50 mM H2O2, these contractile parameters were not fully recovered by 100 mM DTT reduction: Fmax (59.7 ± 5.1 kN/m2, n = 11) remained ~50% lower than that of unoxidized fibers (110.5 ± 3.2 kN/m2, n = 12), resting tension (6.7 ± 1.3 kN/m2, n = 8) remained about sixfold higher than that of the unoxidized fibers (0.92 ± 0.02 kN/m2, n = 8), and V0 remained negligible.
Effect of H2O2 on Structural States of Myosin in Muscle Fibers
We determined directly the effect of oxidation on the structural state of myosin by performing EPR measurements on oxidized and unoxidized muscle fibers. EPR spectra of unoxidized fibers acquired under conditions of rigor, relaxation, and contraction were essentially the same as reported in the previous works from our and other laboratories, showing that myosin undergoes nucleotide-dependent structural transitions associated with the contractile cycle of the fibers (20, 43, 55). Monitoring of force during EPR measurements confirmed that the inhibitory effects of oxidation on fiber contractility were not affected by the subsequent IASL labeling. The reported results were obtained on fibers that were first oxidized and then spin labeled. Control experiments showed that reversal of the modification order—first spin labeling and then oxidation–made no difference in the observed spectral changes.
EPR spectra acquired under rigor and contraction conditions were not significantly affected by peroxide treatment at either 5 or 50 mM, i.e., there was no significant effect of oxidation on XC (P = 0.094, Fig. 4), so the observed decreases in Fmax and V0 (Fig. 2) cannot be attributed to decreases in XC. However, oxidation had a significant effect on the spectra in relaxation conditions, increasing XE from 0 (0 mM H2O2) to 27% (5 mM H2O2) and 34% (50 mM H2O2) (Fig. 4), correlating with the observed decrease in Ca2+ regulation (Fig. 3).
Fig. 4.

Effects of 5 and 50 mM H2O2 on the structural states of myosin in muscle fibers. Top: EPR spectra under rigor (R), relaxation (E), and contraction (C). Bottom: fraction of myosin heads in strongly bound structural state in relaxation (XE) and contraction (XC) as calculated with Eqs. 1 and 2. Data are means ± SE. *P < 0.05, significantly different from unoxidized.
Effect of H2O2 on Functional State of Myosin in Muscle Fibers: Myofibrillar ATPase Activities Measured Under Activating and Relaxing Conditions
ATPase activities of myofibrils under activating conditions (pCa 4.5) were much less affected by oxidation than changes in the contractile parameters of fibers. Oxidation of fibers by 5 mM H2O2 had no inhibitory effect on myofibrillar ATPase activity (P = 0.207), and oxidation by 50 mM H2O2 decreased ATPase activity by 30% (from 2.21 ± 0.15 to 1.48 ± 0.06 s−1, n = 8) (Fig. 5), but this decrease can provide only a partial explanation for the 85% decrease in Fmax and 99% decrease in V0 (Fig. 2). These ATPase effects were not reversed by 100 mM DTT.
Fig. 5.

Effects of 5 and 50 mM H2O2 on ATPase activities of myofibrils under activating conditions at pCa = 4.5 (top) and under relaxing conditions at pCa = 9 (bottom). Data are means ± SE. *P < 0.001, significantly different from unoxidized.
ATPase activity measured under relaxing conditions (pCa 9) was enhanced from 0.11 ± 0.01 to 0.29 ± 0.03 s−1 after oxidation by 5 mM H2O2 and to 0.78 ± 0.05 s−1 (n = 8) after oxidation by 50 mM H2O2 (Fig. 5). This increased ATPase activity in the absence of Ca2+ (Fig. 5) is consistent with the loss of Ca2+ regulation of fiber contractility (Fig. 3). These changes in ATPase activity at pCa 9 were decreased, but not fully reversed, by 100 mM DTT reduction of fibers.
Effect of H2O2 on Functional Interaction of Myosin Purified from Muscle Fibers With Actin
The effect of oxidation on functional interaction of myosin with actin was studied by using α-chymotryptic S1 of myosin from fibers and actin purified from rabbit skeletal muscle. The high-salt ATPase activities of S1 from unoxidized fibers were essentially the same as those of our standard preparations of skeletal muscle S1: Ca-ATPase 0.80 ± 0.01 s−1 and K-ATPase 8.37 ± 0.27 s−1 (n = 11; Fig. 6); this 10-fold difference in these activities is typical for unmodified myosin heads (72, 82). Oxidation-induced changes in these high-salt ATPase activities, which report directly the activity of myosin, were similar for myofibrils, isolated myosin, and S1 (Fig. 6). The values observed in Fig. 6 were not affected by treatment of fibers with 10 or 100 mM DTT. Thus oxidation-induced changes in the enzymatic properties of muscle fibers are due to changes in the myosin head S1, and this conclusion justifies the use of myosin S1 in further studies on the biochemical and chemical effects of oxidation.
Fig. 6.

Effects of 5 and 50 mM H2O2 on high-salt ATPase activities of myofibrils, purified myosin, and subfragment 1 (S1). Top: K-ATPase. Bottom: Ca-ATPase. Data are means ± SE. *P < 0.05, significantly different from unoxidized. MF, myofibrils; M, myosin; S1, myosin head.
Sedimentation assays showed that the Kd values of weak binding (+ATP) of actin for S1 from unoxidized and 5 mM H2O2 and 50 mM H2O2-oxidized fibers were essentially the same (Table 1) and comparable to values reported in the previous studies from our (65) and other (16, 91, 94) laboratories. Strong binding (no ATP), measured in the presence of ADP, was in all cases near stoichiometric, with Kd in the nanomolar range. Thus peroxide treatment did not inhibit binding of myosin heads to actin. Peroxide treatment did, however, have a concentration-dependent effect on the actin-activated S1 ATPase. Actin-activated ATPase activity of S1 from unoxidized fibers was similar to the previously reported actin-activated activity of skeletal muscle S1, where Vmax ranged from 10 to 23 s−1, depending on reaction conditions (15, 21, 62, 71, 94). Oxidation by 5 mM H2O2 had no effect on the extent of actin activation of fiber S1 ATPase; however, oxidation by 50 mM H2O2 had a large inhibitory effect on S1, decreasing Vmax by ~60% and Km by ~27% (Table 1). These results show directly that 50 mM but not 5 mM peroxide induced changes in the enzymatic function of fibers by affecting enzymatic function of myosin head S1.
Table 1.
Effect of H2O2 oxidation of muscle fibers on functional interactions of actin and S1
| H2O2, mM | Kd(+ATP) μM | Vmax, S−1 | Km, μM |
|---|---|---|---|
| 0 | 30.97±1.98 | 16.93±0.35 | 15.48±0.97 |
| 5 | 26.91±2.32 | 16.18±0.44 | 16.75±1.34 |
| 50 | 27.22±2.56 | 6.10±0.21 | 10.40±1.32 |
Kd of binding in the presence of ATP [Kd(+ATP)] was calculated by fitting the data to the equation Slb/S1t = [A]/([A] + Kd), where S1b/S1t is the fraction of subfragment 1 (S1) bound to actin and [A] is the concentration of unoccupied actin protomers. Vmax and Km of acto-S1 ATPase were determined by fitting the data to the Michaelis-Menten equation with the software package Origin 7.0. Uncertainties were determined from statistical analysis of the fits. Data were obtained from 4 independent preparations of S1.
Mass Spectroscopy Analysis of Global Chemical Changes in Heavy and Light Chain of S1 Isolated from H2O2-Oxidized Muscle Fibers
We analyzed peroxide's global chemical effect by measuring oxidation-induced changes in the mass of fiber myosin head (S1) with ESI-MS. MS data for purified S1 showed mass peaks that corresponded to its heavy and essential light chains (ELC1 and ELC2) (Figs. 7 and 8). The mass of intact S1 (~110 kDa) was not detected, which suggests complete dissociation of S1 into heavy and light chains during MS analysis.
Fig. 7.

Mass profiles of heavy chain (HC) and heavy chain tryptic domains of myosin S1 purified from unoxidized (top) and 50 mM H2O2-oxidized (bottom) muscle fibers.
Fig. 8.

Mass profiles of essential light chains (ELC1, ELC2) of S1 and regulatory light chain (RLC) of heavy meromyosin from unoxidized (top) and 50 mM H2O2-oxidized (bottom) muscle fibers.
Heavy chain
Deconvoluted data for S1 heavy chain from unoxidized fibers produced a broad mass profile (Fig. 7) with a major peak at 92,350.9 ± 3.4 Da (mean ± SD, n = 3), comparable to the mass of our (data not shown) and others' (97) preparations of α-chymotryptic S1. The broadening of the mass profile is likely due to the sequence microheterogeneity (88, 89) and noise inherent in the raw mass spectral data for such a large protein.
The mass profile of the S1 heavy chain was not affected by oxidation of fibers by 5 mM H2O2 (data not shown). In contrast, oxidation of fibers by 50 mM H2O2 caused a general shift of the heavy chain profile to higher mass values (Fig. 7). Its most intense peak, labeled as 92,441.9 ± 1.1 (mean ± SD, n = 2) was shifted by 91 Da relative to the main peak of S1 from unoxidized fibers and may represent the addition of 6 oxygens on the heavy chain (theoretical mass of 1 oxygen is 16 Da).
Oxidation-affected regions of the heavy chain were further determined by analysis of the mass shifts of the NH2-terminal 23-kDa, central 50-kDa, and COOH-terminal 20-kDa domains, obtained by limited tryptic proteolysis of S1 (51, 52). Deconvoluted ESI-MS data for the three tryptic domains of S1 from unoxidized fibers detected a mass peak at 19,556.7 ± 0.5 Da (mean ± SD, n = 3), corresponding to the 20-kDa domain, and at 22,982.0 ± 0.8 Da (mean ± SD, n = 3), corresponding to the 23-kDa domain (Fig. 7). The most intense peak in a broad mass profile at 48,048.9 ± 1.3 Da was assigned to the 50-kDa domain, and the two additional peaks with masses of 48,069.4 ± 0.7 and 48,091.6 ± 1.2 Da most likely represent sodium adducts on the protein (Fig. 7).
Deconvoluted ESI-MS data for the three tryptic domains of S1 from 50 mM H2O2-oxidized fibers show peaks corresponding to the domains from unoxidized fibers, as well as additional mass peaks (Fig. 7). The data for the 20-kDa domain showed a peak at 19,556.9 ± 0.3 Da and a peak at 19,572.9 ± 0.3 Da (mean ± SD, n = 3), which is a 16.0 ± 0 Da mass shift, suggesting addition of a single oxygen. The data for the 23-kDa domain showed a peak at 22,982.3 ± 0.8 Da and a second peak at 22,998.4 ± 0.3 Da, which is also a 16.1 ± 0.1 Da mass shift, suggesting the addition of a single oxygen. This second peak domain was followed by unresolved shoulder peaks extending into higher masses, which could not be clearly resolved. Shoulder peaks were not present in the sample from unoxidized fibers; therefore we concluded that the sample from oxidized fibers may contain heterogeneous products of oxidation.
The extent of oxidation of each of the 20-kDa and 23-kDa domains was estimated by comparing the areas of peaks corresponding to the oxidized and unoxidized masses. The peak area of the oxidized form of the 20-kDa domain was 1.2 ± 0.2 (mean ± SD, n = 3) times greater than the peak area of the unoxidized, and the peak area of the oxidized form of the 23-kDa domain was 1.4 ± 0.1 (mean ± SD, n = 3) times greater than the peak area of the unoxidized, indicating that in both cases the oxidized form was predominant. Since the peak for the oxidized 23-kDa domain showed unresolved shoulder peaks extending into higher masses, the calculated extent of oxidation of this domain may be underestimated.
The deconvoluted data for the 50-kDa domain of S1 from oxidized fibers showed a general shift in mass profile to higher values compared with the unoxidized profile (Fig. 7). The peak at 48,114.6 ± 7.3 Da shows a mass shift of 64.0 ± 6.1 Da (mean ± SD, n = 3) relative to the protein from unoxidized fibers and may represent the addition of 4 oxygens. The addition of 6 oxygens to all three tryptic domains (total added mass of 96 Da) is consistent with the detected mass shift of the intact heavy chain by 91 Da (Fig. 7).
Light chains
Besides heavy chain, data deconvolution for the S1 from unoxidized fibers produced peaks at 20,860.5 ± 0.2 Da and 16,569.2 ± 0.1 Da, assigned to ELC1 and ELC2, respectively (Fig. 8). These masses are identical to the theoretical masses for rabbit fast skeletal muscle ELCs calculated from mRNA sequences: 20,859 Da (gi 127130) for ELC1 and 16,568 (gi 127135) for ELC2 (in the theoretical calculations the initial methionine was removed and the acetyl group at the NH2 terminus was added as the blocking group; Ref. 27). The RLC, which is absent in α-chymotryptic S1 (96), was detected by ESI-MS of the two-headed proteolytic fragment of myosin, HMM. The deconvoluted peak mass of 16,967.8 Da (Fig. 8) corresponds to the theoretical mass of rabbit fast skeletal muscle RLC (gi 127176) spanning residues 20 to 169, which represents an α-chymotryptic fragment from cleavage at F19.
The analysis of S1 from unoxidized and 5 mM H2O2-oxidized fibers showed that oxidation by this concentration of peroxide had no effect on the mass profiles of the ELCs (data not shown). We concluded that the extent of oxidative modification of S1 by 5 mM H2O2 was minimal and any potentially oxidized forms were below the detection limit of the applied method.
The mass spectrum of the ELCs of S1 from the 50 mM H2O2-oxidized fibers contained peaks corresponding to the light chains of S1 from unoxidized fibers, as well as additional mass peaks. In the spectrum of ELC1, the additional masses were 20,876.9 ± 0.2, 20,891.6 ± 1.8, and 20,907.7 ± 1.4 Da (means ± SD, n = 2), which is 16.4 ± 0.1, 31.1 ± 1.5, and 47.1 ± 1.1 Da (means ± SD, n = 2) higher than the mass of the unoxidized form, suggesting the additions of 1, 2, and 3 oxygens. The sum of peak areas of these three oxidized forms was 2.04 ± 0.10 times greater than the peak area of the unoxidized form, indicating that the oxidized form of ELC1 was the predominant form.
In the deconvoluted data for ELC2 from the oxidized fibers, the peak at 16,585.9 ± 0.2 Da was shifted in mass by 16.5 ± 0.1 Da relative to ELC2 from unoxidized fibers, which suggested the addition of 1 oxygen. The peak area of this unoxidized form was 1.2 times greater than the peak area of the oxidized form; however, the mass peak of the oxidized ELC2 was followed by unresolved shoulder peaks extending into the higher mass region, indicating that this calculated extent of oxidation is underestimated.
In contrast to the two ELCs, H2O2 treatment of fibers did not affect the mass of the RLC, suggesting that structural environment of this light chain in muscle fiber protected it against action of H2O2.
Mass Spectroscopy Analysis of Local Chemical Changes in S1 Isolated from H2O2-Oxidized Muscle Fibers
We analyzed peroxide's local chemical effects by ESI-MS of S1 peptides obtained by exhaustive trypsin and Glu-C digestions. Two independently prepared pairs of S1 from unoxidized and 50 mM H2O2-oxidized fibers were used, and each trypsin and Glu-C digest was performed in duplicate. Thus the final analysis was performed on four sets of peptides of S1 from unoxidized fibers and four sets of peptides of S1 from oxidized fibers. At the peptide confidence level ≥95%, peptides detected by MS covered 78.3% of the sequence of S1 heavy chain (gi 13431707) and 100% of the sequence of the essential light chain ELC1. Peptides unique to ELC1 or ELC2 could not be distinguished, since they were located within the stretch of 141 COOH-terminal residues that are identical in ELC1 and ELC2 (27).
ESI analysis of S1 peptides detected oxidation exclusively on methionines, and only those that reside in peptides consistently found in all four sets of peptides from given S1 samples are reported. Oxidations not consistent within the four sets of peptides were considered a result of accidental oxidation and are not reported. Thirteen methionine residues were found consistently oxidized to methionine sulfoxide, and one methionine residue was oxidized to methionine sulfone (Table 2). These sites of oxidative modifications represent only a fraction of the total S1 methionines, since the known sequences of S1 heavy chain contain 22 (88) or 25 (gi 13431707) methionines and each of the ELCs contains 6 methionines (27). However, 4 methionines of the heavy chain are located in peptides that were undetected because of one or more of the following reasons: poor ionization, signal suppression in ESI, or loss during purification process. All methionines of ELC were detected. The identity of each oxidation-containing peptide was confirmed by manual inspection of all relevant MS/MS spectra; an example MS/MS spectrum that confirms the oxidation of Met189 in ELC of S1 from the oxidized fibers is shown in Fig. 9.
Table 2.
Peptides containing oxidized residues in S1 from unoxidized and 50 mM H2O2-oxidized muscle fibers as determined by LC-MS/MS
| Subunit | Residue | Exp. Mass, Da | Δ | Charge | Sequence | 50 mM H2O2 | 0 mM H2O2 |
|---|---|---|---|---|---|---|---|
| ELC | 100 | 2,143.08 | −0.05 | 4 | VKKVLGNPSNEE(M-ox)NAKKIE | Yes | No |
| 113 | 2,037.05 | 0.03 | 3 | IEFEQFLP(M-ox)LQAISNNK | Yes | No | |
| 189 | 1,060.57 | −0.01 | 2 | AFVKHI(M-ox)SI | Yes | No | |
| 146 | 1,737.78 | 0.01 | 3 | VFDKEGNGTV(M-ox)GAELR | Yes | Yes | |
| 160 | 1,907.95 | −0.05 | 4 | LRHVLATLGEK(M-ox)KEEE | Yes | Yes | |
| HC 23 kDa | 94 | 2,422.10 | −0.01 | 3 | YDKIEDMAM(M-ox)THLHEPAVLY | Yes | No |
| 166 | 1,804.77 | −0.03 | 2 | SISDNAYQF(M-ox)LTDRE | Yes | No | |
| 80 | 1,835.84 | −0.03 | 3 | DQVFP(M-ox)NPPKYDKIE | Yes | Yes | |
| HC 50 kDa | 496 | 2,510.18 | −0.02 | 4 | LQQFFNHH(M-ox)FVLEQEEYKK | Yes | No |
| 542 | 2,079.94 | −0.05 | 4 | C(M-ox)FPKATDTSFKNKLYE | Yes | No | |
| 531 | 1,504.74 | −0.09 | 2 | LIEKP(M-ox)GIFSILE | Yes | Yes | |
| HC 20 kDa | 779 | 1,590.79 | 0.00 | 3 | AGLLGLLEE(M-2ox)RDDK | Yes | No |
| 687 | 1,745.89 | 0.01 | 4 | TPGA(M-ox)EHELVLHQLR | Yes | Yes | |
| 779 | 1,574.79 | −0.01 | 3 | AGLLGLLEE(M-ox)RDEK | Yes | Yes |
Methionine residues found oxidized exclusively in S1 from 50 mM H2O2-oxidized fibers are shown in bold. LC, liquid chromatography; MS, mass spectroscopy; Δ, difference between experimental (Exp.) and theoretical mass of a peptide; ELC, essential light chain; HC, heavy chain. “Yes” and “no” indicate whether oxidized methionine was found in given sample.
Fig. 9.
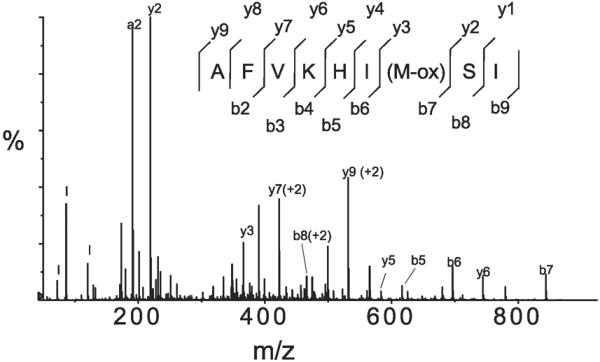
Mass spectroscopy (MS)/MS spectrum of the COOH-terminal peptide of ELC of S1 isolated from 50 mM H2O2-oxidized muscle fibers, confirming oxidation of Met189. m/z, Mass-to-charge ratio.
Eight of 13 oxidized methionines were present exclusively in S1 from H2O2-treated fibers and were absent in S1 from unoxidized fibers, suggesting that these sites were most likely specific targets of H2O2 (Table 2). Five oxidized methionines found in S1 from unoxidized fibers were also found oxidized in S1 from H2O2-treated fibers, which indicates that accidental oxidation during preparations of peptides probably occurs only at the most reactive methionines, and these could be the first targets of H2O2. No oxidized methionine was found exclusively in S1 from unoxidized samples.
Effect of H2O2 on Cysteine Content in S1 Isolated from Oxidized Muscle Fibers
While sites of oxidative modifications detected by ESI-MS were limited to methionine residues, H2O2 is known to react also with cysteines (45, 84). Quantitation of reactive S1 cysteines by the DTNB assay showed 8.33 ± 0.65 (mean ± SD, n = 13) cysteines in S1 from unoxidized fibers, which is close to the 10 cysteines present in the amino acid sequence of skeletal muscle S1. Eight of these 10 cysteines are present in the S1 heavy chain, which includes the 1–809 NH2-terminal sequence of myosin heavy chain (88), and 2 remaining cysteines are present in the sequences of ELC1 and ELC2, each containing 1 cysteine (27). The experimentally determined slightly lower cysteine content in S1 is probably due to incomplete unfolding of S1 and/or formation of random disulfides. Initial oxidation of S1 is unlikely because the mass of its heavy chain, as determined by MS, is consistent among independent preparations, and masses of the ELCs are the same as predicted from the amino acid sequence (Figs. 7 and 8). Muscle fiber oxidation by 50 mM H2O2 decreased S1 cysteine content to 7.71 ± 0.64 (mean ± SD, n = 5), i.e., by ~0.6 cysteines/mol. Such a small difference is not statistically significant but could contribute to the functional effect of H2O2 if it represents oxidation of Cys707 (67) (8, 9, 22, 24), and such a small difference could have been missed by the previous peptide analyses. Therefore, we performed further experiments to determine the effect of 50 mM H2O2 on S1 cysteines with site-specific labeling and MALDI MS analysis.
Measurement of reactivity of Cys707 with the fluorescent derivative of iodoacetamide, IAF (81, 82), showed that the amount of IAF bound to native S1, as determined by absorbance at 496 nm, was 0.86 ± 0.04 IAF/mol S1 for S1 from unoxidized fibers and 0.88 ± 0.06 IAF/mol S1 for S1 from the oxidized fibers (means ± SD, n = 2). This result was directly confirmed by MALDI MS analysis, showing essentially the same relative content of the IAF-Cys707 peptide in S1 from oxidized and unoxidized fibers: 1.11 and 0.97 in trypsin and Glu-C/Lys-C digestions, respectively (Figs. 10 and 11). Thus we concluded that Cys707 of myosin is an unlikely target of H2O2.
Fig. 10.

Matrix-assisted laser desorption ionization (MALDI)-TOF spectra of the tryptic tripeptide IC707R of S1 from unoxidized (top) and 50 mM H2O2-oxidized (bottom) muscle fibers, which contains fluorescein iodoacetamide (IAF)-labeled Cys707 and a reference S1 peptide. The reference peptide GSSFQRVSALFR (mass 1,298.66 Da) in the trypsin digestion and GSSFQRVSALFRE (mass 1,427.71 Da) in the Glu-C/Lys-C digestion did not contain oxidizable residues (C, N, W, or Y). amu, Atomic mass units.
Fig. 11.

MS/MS of the tryptic peptide containing IAF-Cys707 of S1 (A) and of the reference peptide used for normalization (B). In B, the region from 650 to 1,250 m/z (indicated by arrow) is amplified 3 times. Peptide fragment ions are labeled according to Papayannopoulos (56).
The cysteine coverage of S1 was increased by analysis of Glu-C/Lys-C digests of S1 labeled by IAF after denaturation in 0.1% SDS and 10% acetonitrile. Detected peptides contained IAF label at Cys402, Cys674, Cys697, and also Cys707, and the relative content of these IAF peptides in S1 from 50 mM H2O2 oxidized and unoxidized fibers was 1.11 ± 0.30, 0.96 ± 0.20, 1.09 ± 0.30, and 1.10 ± 0.19 (means ± SD, n = 3 or 4), respectively, showing that none of these residues was oxidized.
The ICAT method (73) detected ICAT-labeled peptides with Cys402, Cys479, Cys697, and Cys794, and all of the cysteines showed a similar extent of reactivity, within the 30% error reported by the manufacturer. Thus MALDI analysis of IAF- and ICAT-labeled S1 peptides showed, consistently with the ESI results, that oxidation of muscle fibers by 50 mM H2O2 did not affect the content of cysteines in the myosin head.
DISCUSSION
We report that 1) oxidation-induced inhibition of muscle fiber contractility is associated with oxidative modifications of myosin when fibers are oxidized by high (50 mM) but not low (5 mM) concentrations of H2O2 and 2) the functional effects of oxidation result from multiple modifications of the heavy and essential light chains of myosin.
Effects of 5 mM H2O2
Five millimolar H2O2 significantly inhibited Fmax and V0 of fibers (Fig. 2), but these inhibitory effects were not associated with changes in myosin enzymatic activity (Figs. 5 and Fig. 6, Table 1), indicating that myosin heads were not modified. This conclusion is directly supported by our MS analyses, which did not detect changes in the masses of heavy or light chains of S1 from such muscle fibers. Inhibition of muscle fiber contractility by 5 mM H2O2 without changes in myosin head (Fig. 5) provides direct support for the conclusion from previous physiological studies on permeabilized muscle fibers that changes in contractile proteins cannot explain the effects of low (≤5 mM) concentrations of H2O2 (14, 61). Thus inhibition of fiber function under these conditions is probably due to oxidation in the “rod” regions of myosin, such as light meromyosin (LMM) and S2 (7, 54) and/or in other muscle proteins, e.g., proteins involved in regulation, maintaining structure, or transmitting force. The recovery of contractility in muscle fibers reduced with 100 mM DTT indicates H2O2-induced reversible oxidation of cysteine residues in affected muscle proteins (19). It is unlikely that our conditions of DTT treatment could have any reducing effect on muscle methionines (32).
Effects of 50 mM H2O2
In contrast to the effects of 5 mM H2O2, inhibition of fiber contractility by 50 mM H2O2 (Fig. 2) was associated with inhibition of the enzymatic function of myosin (Fig. 5, Table 1). Myofibrillar ATPase activities at pCa 4.5 and actin-activated ATPase of S1 both showed oxidation-induced inhibition; however, the extent of inhibition of actin-activated ATPase of S1 (~60%) was much more pronounced than that of myofibrillar ATPase (~30%). This difference could not be due to more selective extraction of oxidized myosin from the fibers, because the effect of oxidation on high-salt ATPases was essentially the same for the myofibrils and for isolated S1, i.e., isolated S1 accurately represents the catalytic properties of myofibrillar myosin (Fig. 6). A possible explanation for this difference is that cooperative interactions between active and inactive myosin heads within the myosin filament in muscle fibers compensated for the loss of activity, giving rise to the previously reported less pronounced inactivation in fibers than in solution (22, 70). However, inhibition of enzymatic function of myosin can provide only a partial explanation for the inhibition of fiber contractile properties, since reduction of 50 mM H2O2-oxidized fibers by 100 mM DTT partially restored Fmax without restoring V0 or myofibrillar ATPase at pCa 4.5. This suggests that 50 mM H2O2 not only affected the catalytic domain of myosin but also increased oxidation of noncatalytic regions of myosin and other muscle proteins, all of which were suggested targets of 5 mM H2O2.
Structural Basis of Oxidation-Induced Inhibition of Muscle Fiber Contractility
Analysis of EPR spectra from IASL-labeled fibers (Fig. 4) suggests a structural basis for the inhibitory effect of H2O2 on the contractile and enzymatic function of myosin (Fig. 2, Table 1). The increase in the fraction of myosin heads in the strongly bound structural state in relaxed fibers (XE) but not in contracting fibers (XC) implies a decreased fraction of myosin heads that undergoes the force-generating transition from weakly to strongly bound states during the actomyosin ATPase cycle (Fig. 12) (25, 83). This increased XE can also explain changes in Ca2+ regulation of fiber function, such as decreased pCa50 and Ca2+ threshold for tension development and increased resting tension and myofibrillar ATPase in the absence of calcium (Figs. 3 and 5) (8, 13, 47, 53). However, the molecular mechanism of changes in the structural states of myosin is probably dependent on the concentration of H2O2: low concentration induces changes indirectly, while high concentration induces changes both indirectly and directly, by oxidative modifications of the myosin head.
Fig. 12.

Model for the actomyosin ATPase cycle, showing the force-generating transition between weak-binding and strong-binding states in actin and myosin. AM, actomyosin; M, myosin.
Chemical Basis of Oxidation-Induced Inhibition of Muscle Fiber Contractility
ESI-MS analysis of intact S1 showed that oxidation of muscle fibers by 50 mM H2O2 increased masses of heavy and light chains of S1 compatible with addition of multiple oxygen atoms (Figs. 7 and 8). This suggests that functional and structural effects of H2O2 are not site specific but result from cumulative changes induced by oxidation at multiple sites. For example, oxidative modifications within the 23-kDa and 50-kDa domains of myosin heavy chain could affect, directly or allosterically, the nucleotide and actin binding regions (66), and oxidative modifications in the 20-kDa domain and the ELCs could affect force generation by changing the dynamics of the light chain domain (4, 10, 11, 36, 66).
ESI analysis of S1 peptides detected oxidative modifications exclusively at methionine residues (Table 2), and increases in the masses of S1 heavy and light chain calculated by adding masses of oxygen atoms to these specific sites were within increases experimentally detected in the whole protein (Figs. 7 and 8). For example, the single-oxygen adduct of the 23-kDa domain (Fig. 7) could represent a mixture of two species, one with oxidation at Met94 and another one with oxidation at Met166. The 4-oxygen adduct of the 50-kDa domain (Fig. 7) probably includes detected oxidations at Met496 and Met542, and oxidation of Met100, Met113, and Met189 in the common region of ELC1 and ELC2 (27) could explain ELC species with 1, 2, and 3 oxygens added (Fig. 8).
On the other hand, MS analysis of a protein digest can overestimate the extent of H2O2-induced oxidative modifications due to accidental oxidations introduced during the long processes of enzymatic digestion and LC separations. This probably explains oxidation of Met146 and Met160, which were detected in the peptides of ELCs from unoxidized fibers (Table 2) despite the lack of oxidation of the intact light chains from these fibers (Fig. 8). It is also probable that the single-oxygen adduct of the 23-kDa domain (Fig. 7) has only one H2O2-oxidized methionine and another one detected in the peptides results from artifact. Similarly, inconsistency between the singly oxidized 20-kDa domain (Fig. 7) and the double oxidation of one of its peptides containing Met779 (Table 2) suggests that one added oxygen is the product of the H2O2 reaction and another one, which was also found in sample from unoxidized fibers, is an artifact during sample processing. Thus while H2O2-induced mass increases on heavy and light chains (Figs. 7 and 8) can be partially explained by oxidation of methionines, we cannot eliminate the possibility that other (undetected) sites were also involved.
Comparison With Previous Studies of H2O2 Treatment of Muscle Proteins
Ours is the first study to investigate specific protein modifications due to treatment of muscle fibers with H2O2, but several studies have investigated the oxidation of isolated muscle proteins. Oxidation of methionines by peroxide has been previously detected in isolated skeletal muscle actin (49, 50), smooth muscle myosin subfragments (59), and calmodulin (26). Oxidation of cysteine residues has been detected in isolated myosin fragments treated in solution with peroxide (49, 50, 59) or peroxynitrite (86). In contrast, our study clearly showed that H2O2 treatment of myosin in the muscle filament lattice does not affect myosin cysteines, particularly the most reactive, Cys707 (Fig. 10). It is not surprising that myosin cysteine reactivity is less in the muscle fiber lattice than in solution. The protective effect of actin binding on the reactivity of Cys707 has been previously documented (24) and is supported by the results of control experiments showing that the 50 mM H2O2-induced decrease in the K-ATPase activity of myosin was more pronounced when oxidation was performed on isolated myosin (4.4-fold) than when myosin was isolated from oxidized muscle fibers (2.2-fold). Thus our results indicate that oxidation-induced molecular changes in purified myosin do not necessarily mimic molecular changes that occur after oxidation of myosin in the muscle filament lattice.
Comparison of Changes in Muscle Function Associated With In Vitro Oxidation and In Vivo Muscle Aging
Our interest in the physiological relevance of H2O2 was based in part on muscle aging, since age-related deterioration of muscle function was the subject of previous studies from our laboratories (43, 44, 64, 85) and it has been suggested that oxidation of the contractile proteins contributes to age-related deterioration of muscle contractility (31, 40, 43, 64, 85). Furthermore, a possible role of H2O2 in age-related deterioration of function is supported by its ability to produce a senescent phenotype in cell metabolism (18). The inhibitory effects of 5 mM H2O2 on muscle fiber contractility, as characterized by Fmax or V0 (Fig. 2), were similar to those observed for age-related effects reported previously (23, 40, 43, 44, 64, 85).
However, when we explored the physiological and biochemical effects in more detail, some important qualitative differences between H2O2-induced and age-related effects emerged. In aging rats, the major functional effect is the decline in Fmax (active isometric tension), which agrees quantitatively with the decline in XC (the fraction of myosin heads in the strong-binding structural state) (43) and in the actin-activated ATPase activity of isolated myosin S1 (64). In contrast, peroxide treatment decreases Fmax (Fig. 2A) without a detectable change in XC (Fig. 4) or acto-S1 ATPase activity (Table 1), showing clearly that peroxide and aging must affect muscle function by different molecular mechanisms. The clearest difference was in Ca2+ regulation: the decrease in Ca2+ regulation of force and myofibrillar ATPase was much more pronounced in peroxide-treated (Figs. 3 and 5) than aged (42, 44) muscle. EPR studies revealed the structural basis of this difference: the fraction of myosin heads in the strongly bound structural state in relaxed muscle significantly increased after H2O2 treatment (Fig. 4) but not in aged muscle (43).
It is not surprising that the effects of peroxide and aging are in some aspects similar and in other aspects different. One issue is the concentration of H2O2 in the in vitro and in vivo systems. In healthy living cells H2O2 is present at submicro-molar concentrations (28, 29), but its concentrations are much less defined in pathological states that result in cell damage. This probably explains why low concentrations of H2O2 have been used in most in vitro studies focused on its role in healthy cells, but a much wider range of concentrations have been used in studies on the mechanism of oxidative damage. For example, oxidation of permeabilized skeletal muscle fibers was performed with 10 μM to 10 mM H2O2 (14, 38, 61), but up to 280 mM H2O2 was used to demonstrate higher sensitivity of bladder muscle from aged than young rats to oxidative damage (1) and up to 100 mM H2O2 was used in studies on the effects of oxidation-induced damage to the epithelium (33) and myometrium (95). Another issue in comparing the in vivo and in vitro effects of an oxidant is the time and temperature of exposure. Our H2O2 treatment time was only 30 min at 25°C, while the in vivo systems are exposed to oxidants for much longer periods of time and at higher temperatures, and the effects may be amplified by age-related, slower myosin turnover rates (5, 90). On the molecular level, the similarities and differences between the in vitro oxidation-induced and age-related changes are dependent on the targeted proteins, amino acids, and products of oxidation, which in turn are determined by concentration and type of oxidant. Most relevant to our results is the age-related oxidation of methionine detected in various rat and mice tissues (78, 79), although the state of methionine oxidation in individual muscle proteins and the corresponding oxidation-related functional changes have not been investigated. Thus, while our observed effects of H2O2 provide some support for the “oxidative stress” hypothesis of age-related muscle weakening, the understanding of in vivo oxidation requires more studies on the targets of oxidation by a wider range of biological oxidants.
Summary
We used physiological, biochemical, structural, and chemical analysis of the effects of H2O2 on the properties of muscle fibers and myosin from those fibers. Isolation of myosin from oxidized fibers allowed us to show directly that inhibition of muscle contractility is associated with oxidative modification of myosin at high, but not low, concentrations of H2O2 and the inhibitory effects result from oxidations of multiple methionines in myosin heavy and essential light chains. The presented experimental approach is applicable in other studies on the role of oxidation in loss of muscle function and can be extended to studies on the mechanism of muscle recovery.
ACKNOWLEDGMENTS
The authors express their thanks to Kari Pedersen for preparation of glycerinated muscle fibers, Christina Karim for discussions about protein chemistry, Octavian Cornea for assistance with preparation of the manuscript, Lori Anderson for peptide separation, and the Minnesota Supercomputing Institute for computational resources. Mass spectrometric measurements were performed at the Center for Mass Spectroscopy and Proteomics, Department of Biochemistry, Molecular Biology, and Biophysics, University of Minnesota.
GRANTS This work was supported by grants to D. D. Thomas from the National Institutes of Health (NIH) (AR-32961 and AG-26160) and the University of Minnesota Biomedical Genomics Center, to D. A. Lowe from NIH (AG-20990 and AG-25861), and to L. V. Thompson from NIH (AG-17768 and AG-21626).
REFERENCES
- 1.Aikawa K, Leggett RE, Levin RM. Effect of age on hydrogen peroxide mediated contraction damage in the male rat bladder. J Urol. 2003;170:2082–2085. doi: 10.1097/01.ju.0000081461.73156.48. [DOI] [PubMed] [Google Scholar]
- 2.Andrade FH, Reid MB, Allen DG, Westerblad H. Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J Physiol. 1998;509:565–575. doi: 10.1111/j.1469-7793.1998.565bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andrade FH, Reid MB, Allen DG, Westerblad H. Effect of nitric oxide on single skeletal muscle fibres from the mouse. J Physiol. 1998;509:577–586. doi: 10.1111/j.1469-7793.1998.577bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baker JE, Brust-Mascher I, Ramachandran S, LaConte LE, Thomas DD. A large and distinct rotation of the myosin light chain domain occurs upon muscle contraction. Proc Natl Acad Sci USA. 1998;95:2944–2949. doi: 10.1073/pnas.95.6.2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Balagopal P, Rooyackers OE, Adey DB, Ades PA, Nair KS. Effects of aging on in vivo synthesis of skeletal muscle myosin heavy-chain and sarcoplasmic protein in humans. Am J Physiol Endocrinol Metab. 1997;273:E790–E800. doi: 10.1152/ajpendo.1997.273.4.E790. [DOI] [PubMed] [Google Scholar]
- 6.Barnett VA, Thomas DD. Resolution of conformational states of spin-labeled myosin during steady-state ATP hydrolysis. Biochemistry. 1987;26:314–323. doi: 10.1021/bi00375a044. [DOI] [PubMed] [Google Scholar]
- 7.Bhoite-Solomon V, Kessler-Icekson G, Shaklai N. Peroxidative crosslinking of myosins. Biochem Int. 1992;26:181–189. [PubMed] [Google Scholar]
- 8.Bobkov AA, Bobkova EA, Homsher E, Reisler E. Activation of regulated actin by SH1-modified myosin subfragment 1. Biochemistry. 1997;36:7733–7738. doi: 10.1021/bi963185o. [DOI] [PubMed] [Google Scholar]
- 9.Bobkova EA, Bobkov AA, Levitsky DI, Reisler E. Effects of SH1 and SH2 modifications on myosin: similarities and differences. Biophys J. 1999;76:1001–1007. doi: 10.1016/S0006-3495(99)77264-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Borejdo J, Ushakov DS, Akopova I. Regulatory and essential light chains of myosin rotate equally during contraction of skeletal muscle. Biophys J. 2002;82:3150–3159. doi: 10.1016/S0006-3495(02)75657-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Borejdo J, Ushakov DS, Moreland R, Akopova I, Reshetnyak Y, Saraswat LD, Kamm K, Lowey S. The power stroke causes changes in the orientation and mobility of the termini of essential light chain 1 of myosin. Biochemistry. 2001;40:3796–3803. doi: 10.1021/bi002527u. [DOI] [PubMed] [Google Scholar]
- 12.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 13.Brandt PW, Roemer D, Schachat FH. Co-operative activation of skeletal muscle thin filaments by rigor crossbridges. The effect of troponin C extraction. J Mol Biol. 1990;212:473–480. doi: 10.1016/0022-2836(90)90326-H. [DOI] [PubMed] [Google Scholar]
- 14.Callahan LA, She ZW, Nosek TM. Superoxide, hydroxyl radical, and hydrogen peroxide effects on single-diaphragm fiber contractile apparatus. J Appl Physiol. 2001;90:45–54. doi: 10.1152/jappl.2001.90.1.45. [DOI] [PubMed] [Google Scholar]
- 15.Chalovich JM, Eisenberg E. Inhibition of actomyosin ATPase activity by troponin-tropomyosin without blocking the binding of myosin to actin. J Biol Chem. 1982;257:2432–2437. [PMC free article] [PubMed] [Google Scholar]
- 16.Chalovich JM, Greene LE, Eisenberg E. Crosslinked myosin subfragment 1: a stable analogue of the subfragment-1.ATP complex. Proc Natl Acad Sci USA. 1983;80:4909–4913. doi: 10.1073/pnas.80.16.4909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chase PB, Denkinger TM, Kushmerick MJ. Effect of viscosity on mechanics of single, skinned fibers from rabbit psoas muscle. Biophys J. 1998;74:1428–1438. doi: 10.1016/S0006-3495(98)77855-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen QM, Tu VC, Catania J, Burton M, Toussaint O, Dilley T. Involvement of Rb family proteins, focal adhesion proteins and protein synthesis in senescent morphogenesis induced by hydrogen peroxide. J Cell Sci. 2000;113:4087–4097. doi: 10.1242/jcs.113.22.4087. [DOI] [PubMed] [Google Scholar]
- 19.Claiborne A, Yeh JI, Mallett TC, Luba J, Crane EJ, 3rd, Charrier V, Parsonage D. Protein-sulfenic acids: diverse roles for an unlikely player in enzyme catalysis and redox regulation. Biochemistry. 1999;38:15407–15416. doi: 10.1021/bi992025k. [DOI] [PubMed] [Google Scholar]
- 20.Cooke R, Crowder MS, Thomas DD. Orientation of spin labels attached to cross-bridges in contracting muscle fibres. Nature. 1982;300:776–778. doi: 10.1038/300776a0. [DOI] [PubMed] [Google Scholar]
- 21.Crosbie RH, Miller C, Cheung P, Goodnight T, Muhlrad A, Reisler E. Structural connectivity in actin: effect of C-terminal modifications on the properties of actin. Biophys J. 1994;67:1957–1964. doi: 10.1016/S0006-3495(94)80678-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crowder MS, Cooke R. The effect of myosin sulphydryl modification on the mechanics of fibre contraction. J Muscle Res Cell Motil. 1984;5:131–146. doi: 10.1007/BF00712152. [DOI] [PubMed] [Google Scholar]
- 23.D'Antona G, Pellegrino MA, Adami R, Rossi R, Carlizzi CN, Cane-pari M, Saltin B, Bottinelli R. The effect of ageing and immobilization on structure and function of human skeletal muscle fibres. J Physiol. 2003;552:499–511. doi: 10.1113/jphysiol.2003.046276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duke J, Takashi R, Ue K, Morales MF. Reciprocal reactivities of specific thiols when actin binds to myosin. Proc Natl Acad Sci USA. 1976;73:302–306. doi: 10.1073/pnas.73.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eisenberg E, Hill TL. Muscle contraction and free energy transduction in biological systems. Science. 1985;227:999–1006. doi: 10.1126/science.3156404. [DOI] [PubMed] [Google Scholar]
- 26.Ferrington DA, Sun H, Murray KK, Costa J, Williams TD, Bigelow DJ, Squier TC. Selective degradation of oxidized calmodulin by the 20 S proteasome. J Biol Chem. 2001;276:937–943. doi: 10.1074/jbc.M005356200. [DOI] [PubMed] [Google Scholar]
- 27.Frank G, Weeds AG. The amino-acid sequence of the alkali light chains of rabbit skeletal-muscle myosin. Eur J Biochem. 1974;44:317–334. doi: 10.1111/j.1432-1033.1974.tb03489.x. [DOI] [PubMed] [Google Scholar]
- 28.Gonzalez-Flecha B, Evelson P, Sterin-Speziale N, Boveris A. Hydrogen peroxide metabolism and oxidative stress in cortical, medullary and papillary zones of rat kidney. Biochim Biophys Acta. 1993;1157:155–161. doi: 10.1016/0304-4165(93)90059-h. [DOI] [PubMed] [Google Scholar]
- 29.Halliwell B, Clement MV, Long LH. Hydrogen peroxide in the human body. FEBS Lett. 2000;486:10–13. doi: 10.1016/s0014-5793(00)02197-9. [DOI] [PubMed] [Google Scholar]
- 30.Heunks LM, Cody MJ, Geiger PC, Dekhuijzen PN, Sieck GC. Nitric oxide impairs Ca2+ activation and slows cross-bridge cycling kinetics in skeletal muscle. J Appl Physiol. 2001;91:2233–2239. doi: 10.1152/jappl.2001.91.5.2233. [DOI] [PubMed] [Google Scholar]
- 31.Hook P, Sriramoju V, Larsson L. Effects of aging on actin sliding speed on myosin from single skeletal muscle cells of mice, rats, and humans. Am J Physiol Cell Physiol. 2001;280:C782–C788. doi: 10.1152/ajpcell.2001.280.4.C782. [DOI] [PubMed] [Google Scholar]
- 32.Houghten RA, Li CH. Reduction of sulfoxides in peptides and proteins. Anal Biochem. 1979;98:36–46. doi: 10.1016/0003-2697(79)90702-4. [DOI] [PubMed] [Google Scholar]
- 33.Jeppsson AB, Sundler F, Luts A, Waldeck B, Widmark E. Hydrogen peroxide-induced epithelial damage increases terbutaline transport in guinea-pig tracheal wall: implications for drug delivery. Pulm Pharmacol. 1991;4:73–79. doi: 10.1016/0952-0600(91)90055-8. [DOI] [PubMed] [Google Scholar]
- 34.Kanski J, Hong SJ, Schoneich C. Proteomic analysis of protein nitration in aging skeletal muscle and identification of nitrotyrosine-containing sequences in vivo by nanoelectrospray ionization tandem mass spectrometry. J Biol Chem. 2005;280:24261–24266. doi: 10.1074/jbc.M501773200. [DOI] [PubMed] [Google Scholar]
- 35.Kapphahn RJ, Ethen CM, Peters EA, Higgins L, Ferrington DA. Modified alpha A crystallin in the retina: altered expression and truncation with aging. Biochemistry. 2003;42:15310–15325. doi: 10.1021/bi034774e. [DOI] [PubMed] [Google Scholar]
- 36.LaConte LE, Baker JE, Thomas DD. Transient kinetics and mechanics of myosin's force-generating rotation in muscle: resolution of millisecond rotational transitions in the spin-labeled myosin light-chain domain. Biochemistry. 2003;42:9797–9803. doi: 10.1021/bi034288r. [DOI] [PubMed] [Google Scholar]
- 37.Lafoux A, Divet A, Gervier P, Huchet-Cadiou C. Greater susceptibility of the sarcoplasmic reticulum to H2O2 injuries in diaphragm muscle from mdx mice. J Pharmacol Exp Ther. 2006;318:1359–1367. doi: 10.1124/jpet.106.103291. [DOI] [PubMed] [Google Scholar]
- 38.Lamb GD, Posterino GS. Effects of oxidation and reduction on contrac-tile function in skeletal muscle fibres of the rat. J Physiol. 2003;546:149–163. doi: 10.1113/jphysiol.2002.027896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lanzetta PA, Alvarez LJ, Reinach PS, Candia OA. An improved assay for nanomole amounts of inorganic phosphate. Anal Biochem. 1979;100:95–97. doi: 10.1016/0003-2697(79)90115-5. [DOI] [PubMed] [Google Scholar]
- 40.Larsson L, Li X, Frontera WR. Effects of aging on shortening velocity and myosin isoform composition in single human skeletal muscle cells. Am J Physiol Cell Physiol. 1997;272:C638–C649. doi: 10.1152/ajpcell.1997.272.2.C638. [DOI] [PubMed] [Google Scholar]
- 41.Levine RL, Berlett BS, Moskovitz J, Mosoni L, Stadtman ER. Methionine residues may protect proteins from critical oxidative damage. Mech Ageing Dev. 1999;107:323–332. doi: 10.1016/s0047-6374(98)00152-3. [DOI] [PubMed] [Google Scholar]
- 42.Lowe DA, Husom AD, Ferrington DA, Thompson LV. Myofibrillar myosin ATPase activity in hindlimb muscles from young and aged rats. Mech Ageing Dev. 2004;125:619–627. doi: 10.1016/j.mad.2004.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lowe DA, Surek JT, Thomas DD, Thompson LV. Electron paramagnetic resonance reveals age-related myosin structural changes in rat skeletal muscle fibers. Am J Physiol Cell Physiol. 2001;280:C540–C547. doi: 10.1152/ajpcell.2001.280.3.C540. [DOI] [PubMed] [Google Scholar]
- 44.Lowe DA, Thomas DD, Thompson LV. Force generation, but not myosin ATPase activity, declines with age in rat muscle fibers. Am J Physiol Cell Physiol. 2002;283:C187–C192. doi: 10.1152/ajpcell.00008.2002. [DOI] [PubMed] [Google Scholar]
- 45.Luo D, Smith SW, Anderson BD. Kinetics and mechanism of the reaction of cysteine and hydrogen peroxide in aqueous solution. J Pharm Sci. 2005;94:304–316. doi: 10.1002/jps.20253. [DOI] [PubMed] [Google Scholar]
- 46.Metzger JM. Effects of phosphate and ADP on shortening velocity during maximal and submaximal calcium activation of the thin filament in skeletal muscle fibers. Biophys J. 1996;70:409–417. doi: 10.1016/S0006-3495(96)79584-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Metzger JM. Myosin binding-induced cooperative activation of the thin filament in cardiac myocytes and skeletal muscle fibers. Biophys J. 1995;68:1430–1442. doi: 10.1016/S0006-3495(95)80316-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Metzger JM, Moss RL. Greater hydrogen ion-induced depression of tension and velocity in skinned single fibres of rat fast than slow muscles. J Physiol. 1987;393:727–742. doi: 10.1113/jphysiol.1987.sp016850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Milzani A, DalleDonne I, Colombo R. Prolonged oxidative stress on actin. Arch Biochem Biophys. 1997;339:267–274. doi: 10.1006/abbi.1996.9847. [DOI] [PubMed] [Google Scholar]
- 50.Milzani A, Rossi R, Di Simplicio P, Giustarini D, Colombo R, Dalle-Donne I. The oxidation produced by hydrogen peroxide on Ca-ATP-G-actin. Protein Sci. 2000;9:1774–1782. doi: 10.1110/ps.9.9.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mornet D, Pantel P, Bertrand R, Audemard E, Kassab R. Isolation and characterization of the trypsin-modified myosin-S1 derivatives. FEBS Lett. 1981;123:54–58. doi: 10.1016/0014-5793(81)80018-x. [DOI] [PubMed] [Google Scholar]
- 52.Mornet D, Ue K, Morales MF. Proteolysis and the domain organization of myosin subfragment 1. Proc Natl Acad Sci USA. 1984;81:736–739. doi: 10.1073/pnas.81.3.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nagashima H, Asakura S. Studies on co-operative properties of tropomyosin-actin and tropomyosin-troponin-actin complexes by the use of N-ethylmaleimide-treated and untreated species of myosin subfragment 1. J Mol Biol. 1982;155:409–428. doi: 10.1016/0022-2836(82)90479-x. [DOI] [PubMed] [Google Scholar]
- 54.Ooizumi T, Xiong YL. Biochemical susceptibility of myosin in chicken myofibrils subjected to hydroxyl radical oxidizing systems. J Agric Food Chem. 2004;52:4303–4307. doi: 10.1021/jf035521v. [DOI] [PubMed] [Google Scholar]
- 55.Ostap EM, Barnett VA, Thomas DD. Resolution of three structural states of spin-labeled myosin in contracting muscle. Biophys J. 1995;69:177–188. doi: 10.1016/S0006-3495(95)79888-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Papayannopoulos I. The interpretation of collision-induced dissociation tandem mass spectra of peptides. Mass Spectrometry Rev. 1995;14:49–73. [Google Scholar]
- 57.Park C, So HS, Kim SJ, Youn MJ, Moon BS, Shin SH, Lee I, Moon SK, Park R. Samul extract protects against the H2O2-induced apoptosis of H9c2 cardiomyoblasts via activation of extracellular regulated kinases (Erk) 1/2. Am J Chin Med. 2006;34:695–706. doi: 10.1142/S0192415X06004211. [DOI] [PubMed] [Google Scholar]
- 58.Pearson AR, Jones LH, Higgins L, Ashcroft AE, Wilmot CM, Davidson VL. Understanding quinone cofactor biogenesis in methylamine dehydrogenase through novel cofactor generation. Biochemistry. 2003;42:3224–3230. doi: 10.1021/bi027073a. [DOI] [PubMed] [Google Scholar]
- 59.Penheiter AR, Bogoger M, Ellison PA, Oswald B, Perkins WJ, Jones KA, Cremo CR. H2O2-induced kinetic and chemical modifications of smooth muscle myosin: correlation to effects of H2O2 on airway smooth muscle. J Biol Chem. 2007;282:4336–4344. doi: 10.1074/jbc.M609499200. [DOI] [PubMed] [Google Scholar]
- 60.Perkins WJ, Han YS, Sieck GC. Skeletal muscle force and actomyosin ATPase activity reduced by nitric oxide donor. J Appl Physiol. 1997;83:1326–1332. doi: 10.1152/jappl.1997.83.4.1326. [DOI] [PubMed] [Google Scholar]
- 61.Plant DR, Lynch GS, Williams DA. Hydrogen peroxide modulates Ca2+-activation of single permeabilized fibres from fast- and slow-twitch skeletal muscles of rats. J Muscle Res Cell Motil. 2000;21:747–752. doi: 10.1023/a:1010344008224. [DOI] [PubMed] [Google Scholar]
- 62.Prochniewicz E, Thomas DD. Differences in structural dynamics of muscle and yeast actin accompany differences in functional interactions with myosin. Biochemistry. 1999;38:14860–14867. doi: 10.1021/bi991343g. [DOI] [PubMed] [Google Scholar]
- 63.Prochniewicz E, Thomas DD. Site-specific mutations in the myosin binding sites of actin affect structural transitions that control myosin binding. Biochemistry. 2001;40:13933–13940. doi: 10.1021/bi010893n. [DOI] [PubMed] [Google Scholar]
- 64.Prochniewicz E, Thomas DD, Thompson LV. Age-related decline in actomyosin function. J Gerontol A Biol Sci Med Sci. 2005;60:425–431. doi: 10.1093/gerona/60.4.425. [DOI] [PubMed] [Google Scholar]
- 65.Prochniewicz E, Walseth TF, Thomas DD. Structural dynamics of actin during active interaction with myosin: different effects of weakly and strongly bound myosin heads. Biochemistry. 2004;43:10642–10652. doi: 10.1021/bi049914e. [DOI] [PubMed] [Google Scholar]
- 66.Rayment I, Holden HM, Whittaker M, Yohn CB, Lorenz M, Holmes KC, Milligan RA. Structure of the actin-myosin complex and its implications for muscle contraction. Science. 1993;261:58–65. doi: 10.1126/science.8316858. [DOI] [PubMed] [Google Scholar]
- 67.Reisler E. Sulfhydryl modification and labeling of myosin. Methods Enzymol. 1982;85:84–93. doi: 10.1016/0076-6879(82)85012-x. [DOI] [PubMed] [Google Scholar]
- 68.Riddles PW, Blakeley RL, Zerner B. Reassessment of Ellman's reagent. Methods Enzymol. 1983;91:49–60. doi: 10.1016/s0076-6879(83)91010-8. [DOI] [PubMed] [Google Scholar]
- 69.Rojkind M, Dominguez-Rosales JA, Nieto N, Greenwel P. Role of hydrogen peroxide and oxidative stress in healing responses. Cell Mol Life Sci. 2002;59:1872–1891. doi: 10.1007/PL00012511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Root DD, Cheung P, Reisler E. Catalytic cooperativity induced by SH1 labeling of myosin filaments. Biochemistry. 1991;30:286–294. doi: 10.1021/bi00215a039. [DOI] [PubMed] [Google Scholar]
- 71.Schwyter D, Phillips M, Reisler E. Subtilisin-cleaved actin: polymerization and interaction with myosin subfragment 1. Biochemistry. 1989;28:5889–5895. doi: 10.1021/bi00440a027. [DOI] [PubMed] [Google Scholar]
- 72.Sekine T, Kielly WW. The enzymic properties of N-ethylmaleimide modified myosin. Biochim Biophys Acta. 1964;81:336–345. [Google Scholar]
- 73.Sethuraman M, McComb ME, Heibeck T, Costello CE, Cohen RA. Isotope-coded affinity tag approach to identify and quantify oxidant-sensitive protein thiols. Mol Cell Proteomics. 2004;3:273–278. doi: 10.1074/mcp.T300011-MCP200. [DOI] [PubMed] [Google Scholar]
- 74.Sharov VS, Dremina ES, Galeva NA, Williams TD, Schoneich C. Quantitative mapping of oxidation-sensitive cysteine residues in SERCA in vivo and in vitro by HPLC-electrospray-tandem MS: selective protein oxidation during biological aging. Biochem J. 2006;394:605–615. doi: 10.1042/BJ20051214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sherwood JJ, Waller GS, Warshaw DM, Lowey S. A point mutation in the regulatory light chain reduces the step size of skeletal muscle myosin. Proc Natl Acad Sci USA. 2004;101:10973–10978. doi: 10.1073/pnas.0401699101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Silveira LR, Pereira-Da-Silva L, Juel C, Hellsten Y. Formation of hydrogen peroxide and nitric oxide in rat skeletal muscle cells during contractions. Free Radic Biol Med. 2003;35:455–464. doi: 10.1016/s0891-5849(03)00271-5. [DOI] [PubMed] [Google Scholar]
- 77.Stadtman ER, Berlett BS. Reactive oxygen-mediated protein oxidation in aging and disease. Chem Res Toxicol. 1997;10:485–494. doi: 10.1021/tx960133r. [DOI] [PubMed] [Google Scholar]
- 78.Stadtman ER, Moskovitz J, Berlett BS, Levine RL. Cyclic oxidation and reduction of protein methionine residues is an important antioxidant mechanism. Mol Cell Biochem. 2002;234–235:3–9. [PubMed] [Google Scholar]
- 79.Stadtman ER, Van Remmen H, Richardson A, Wehr NB, Levine RL. Methionine oxidation and aging. Biochim Biophys Acta. 2005;1703:135–140. doi: 10.1016/j.bbapap.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 80.Sweeney HL, Kushmerick MJ, Mabuchi K, Sreter FA, Gergely J. Myosin alkali light chain and heavy chain variations correlate with altered shortening velocity of isolated skeletal muscle fibers. J Biol Chem. 1988;263:9034–9039. [PubMed] [Google Scholar]
- 81.Takashi R. Fluorescence energy transfer between subfragment-1 and actin points in the rigor complex of actosubfragment-1. Biochemistry. 1979;18:5164–5169. doi: 10.1021/bi00590a021. [DOI] [PubMed] [Google Scholar]
- 82.Takashi R, Duke J, Ue K, Morales MF. Defining the “fast-reacting” thiols of myosin by reaction with 1,5 IAEDANS. Arch Biochem Biophys. 1976;175:279–283. doi: 10.1016/0003-9861(76)90509-9. [DOI] [PubMed] [Google Scholar]
- 83.Thomas DD, Prochniewicz E, Roopnarine O. Changes in actin and myosin structural dynamics due to their weak and strong interactions. Results Probl Cell Differ. 2002;36:7–19. doi: 10.1007/978-3-540-46558-4_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Thomas JA, Mallis RJ. Aging and oxidation of reactive protein sulfhydryls. Exp Gerontol. 2001;36:1519–1526. doi: 10.1016/s0531-5565(01)00137-1. [DOI] [PubMed] [Google Scholar]
- 85.Thompson LV, Brown M. Age-related changes in contractile properties of single skeletal fibers from the soleus muscle. J Appl Physiol. 1999;86:881–886. doi: 10.1152/jappl.1999.86.3.881. [DOI] [PubMed] [Google Scholar]
- 86.Tiago T, Simao S, Aureliano M, Martin-Romero FJ, Gutierrez-Merino C. Inhibition of skeletal muscle S1-myosin ATPase by peroxynitrite. Biochemistry. 2006;45:3794–3804. doi: 10.1021/bi0518500. [DOI] [PubMed] [Google Scholar]
- 87.Timson DJ. Fine tuning the myosin motor: the role of the essential light chain in striated muscle myosin. Biochimie. 2003;85:639–645. doi: 10.1016/s0300-9084(03)00131-7. [DOI] [PubMed] [Google Scholar]
- 88.Tong SW, Elzinga M. Amino acid sequence of rabbit skeletal muscle myosin. 50-kDa fragment of the heavy chain. J Biol Chem. 1990;265:4893–4901. [PubMed] [Google Scholar]
- 89.Tong SW, Elzinga M. The sequence of the NH2-terminal 204-residue fragment of the heavy chain of rabbit skeletal muscle myosin. J Biol Chem. 1983;258:13100–13110. [PubMed] [Google Scholar]
- 90.Toth MJ, Matthews DE, Tracy RP, Previs MJ. Age-related differences in skeletal muscle protein synthesis: relation to markers of immune activation. Am J Physiol Endocrinol Metab. 2005;288:E883–E891. doi: 10.1152/ajpendo.00353.2004. [DOI] [PubMed] [Google Scholar]
- 91.Trayer HR, Trayer IP. Fluorescence resonance energy transfer within the complex formed by actin and myosin subfragment 1. Comparison between weakly and strongly attached states. Biochemistry. 1988;27:5718–5727. doi: 10.1021/bi00415a049. [DOI] [PubMed] [Google Scholar]
- 92.Viner RI, Ferrington DA, Aced GI, Miller-Schlyer M, Bigelow DJ, Schoneich C. In vivo aging of rat skeletal muscle sarcoplasmic reticulum Ca-ATPase. Chemical analysis and quantitative simulation by exposure to low levels of peroxyl radicals. Biochim Biophys Acta. 1997;1329:321–335. doi: 10.1016/s0005-2736(97)00125-9. [DOI] [PubMed] [Google Scholar]
- 93.Wagner PD, Slater CS, Pope B, Weeds AG. Studies on the actin activation of myosin subfragment-1 isoezymes and the role of myosin light chains. Eur J Biochem. 1979;99:385–394. doi: 10.1111/j.1432-1033.1979.tb13267.x. [DOI] [PubMed] [Google Scholar]
- 94.Wagner PD, Weeds AG. Determination of the association of myosin subfragment 1 with actin in the presence of ATP. Biochemistry. 1979;18:2260–2266. doi: 10.1021/bi00578a020. [DOI] [PubMed] [Google Scholar]
- 95.Warren AY, Matharoo-Ball B, Shaw RW, Khan RN. Hydrogen peroxide and superoxide anion modulate pregnant human myometrial contractility. Reproduction. 2005;130:539–544. doi: 10.1530/rep.1.00437. [DOI] [PubMed] [Google Scholar]
- 96.Weeds AG, Pope B. Studies on the chymotryptic digestion of myosin. Effects of divalent cations on proteolytic susceptibility. J Mol Biol. 1977;111:129–157. doi: 10.1016/s0022-2836(77)80119-8. [DOI] [PubMed] [Google Scholar]
- 97.White HD, Ashcroft AE. Real-time measurement of myosin-nucleotide noncovalent complexes by electrospray ionization mass spectrometry. Biophys J. 2007;93:914–919. doi: 10.1529/biophysj.106.101618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wolkart G, Kaber G, Kojda G, Brunner F. Role of endogenous hydrogen peroxide in cardiovascular ischaemia/reperfusion function: studies in mouse hearts with catalase-overexpression in the vascular endothelium. Pharmacol Res. 2006;54:50–56. doi: 10.1016/j.phrs.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 99.Yates LD, Greaser ML. Quantitative determination of myosin and actin in rabbit skeletal muscle. J Mol Biol. 1983;168:123–141. doi: 10.1016/s0022-2836(83)80326-x. [DOI] [PubMed] [Google Scholar]