

Abstract
Cancer associated fibroblasts (CAFs), the most abundant cells in the tumor microenvironment (TME), are a key source of extracellular matrix (ECM) that constitutes the desmoplastic stroma. Through remodeling of the reactive tumor stroma and paracrine actions, CAFs regulate cancer initiation, progression, and metastasis, as well as tumor resistance to therapies. The CAFs found in stroma-rich primary hepatocellular carcinomas (HCCs) and liver metastases of primary cancers of other organs predominantly originate from hepatic stellate cells (HSTCs), which are pericytes associated with hepatic sinusoids. During tumor invasion, HSTCs transdifferentiate into myofibroblasts in response to paracrine signals emanating from either tumor cells or a heterogenous cell population within the hepatic tumor microenvironment. Mechanistically, HSTC-to-myofibroblast transdifferentiation, also known as, HSTC activation, requires cell surface receptor activation, intracellular signal transduction, gene transcription and epigenetic signals, which combined ultimately modulate distinct gene expression profiles that give rise to and maintain a new phenotype. The current review, defines a paradigm that explains how HSTCs are activated into CAFs to promote liver metastasis. Furthermore, focus on the most relevant intracellular signaling networks and epigenetic mechanisms that control HSTC activation is provided. Finally, we discuss the feasibility of targeting CAF/activated HSTCs, in isolation or in conjunction with targeting cancer cells, which constitutes a promising and viable therapeutic approach for the treatment of primary stroma-rich liver cancers and liver metastasis.
Keywords: Liver metastasis, cancer associated fibroblasts, hepatic stellate cells, signal transduction, transcription factors, epigenetics
Introduction
Tumor microenvironment as a key regulator of malignancy
Today solid tumors are no longer viewed as collections of homogeneous transformed epithelial cells. Tumors are organ-like, heterotypic tissues consisting of malignant cells embedded in a tumor microenvironment that is critical for either inhibiting or enhancing the efficiency of all the steps of neoplastic transformation including initiation, progression and metastasis. The tumor microenvironment is composed of heterogeneous stromal cells such as cancer associated fibroblasts (CAFs), immune cells, mastocytes, endothelial cells, pericytes, adipocytes, and a variable type of tissue-specific cell populations (e.g.: melanocytes in skin tumors). In addition, the three-dimensional structure of the niche in which the tumor develops and resides is maintained by a dynamic balance between the synthesis, degradation, assembly, disassembly and crosslinking of both soluble and fibril components of extracellular matrix (ECM). ECM not only provides tumor cells with a supporting scaffold but also acts as a reservoir for matrix metalloproteinases (MMPs), growth factors, cytokines, and chemokines, which provides combinatorial regulations for cancer-associated processes (1) (2) (3). Although the cancer-modifying functions of the tumor stroma have been ignored for several decades, it has recently become clear that the process of carcinogenesis requires an orchestrated interplay between cancer cells and all the stromal components. In fact, the tumor stroma contributes to seven of the eight Hanahan's hallmarks of cancer, namely the generation of factors that mediate tumor proliferation and escape growth suppression, resistance to cell death signals, induction of angiogenesis, modulation of invasion, evading immune surveillance and reprogramming of energy metabolism (1) (3). In addition, the composition and function of the stromal components that lead to aberrant tumor angiogenesis, distorted tissue architecture, and increased tumor stiffness, contribute to poor drug distribution and chemoresistance of cancer (4) (5). Therefore, the tumor microenvironment is of paramount importance in determining the ultimate fate of malignant tumors.
Cancer associated fibroblasts (CAFs)
CAFs are routinely identified by their expression of a myocyte marker, alpha- smooth muscle actin (α-SMA) (6) (7), and others such as fibroblast-specific protein (FSP-1, also called S100A4), fibroblast activating protein (FAP), vimentin, PDGF receptors and NG2 chondroitin sulfate proteoglycan (2). In certain cancers, such as pancreatic cancer, CAFs constitute up to 80% of cells of the tumor mass (7). CAFs are pivotal for tumor development, progression and metastasis (2) (6). However, CAFs are heterogeneous as defined by different expression patterns of specific markers. Sigimoto et al. found two distinct subsets of CAFs in mouse models of pancreatic and breast cancer; one subtype of CAFs coexpressed α-SMA, PDGFβ and NG2 proteoglycan and another expressed FSP-1 (8). In a recent review, F1- and F2- polarized fibroblasts have been assigned to CAFs based on the plasticity and emerging functional divergence of CAFs, although the marker/function relationship remains unknown (2). F1 subtype represents CAFs that inhibit tumors (7) (9) and F2 subtype represents CAFs with dominating tumor promoting effects. Thus, understanding of CAF biology and multifaceted role of CAFs in tumorigenesis is critical for development of therapeutics that selectively target against tumor promoting CAFs.
Our lab has been focused on biology and function of CAFs of liver metastases, which are predominantly activated from liver specific pericytes, hepatic stellate cells (HSTCs), through a process called myofibroblastic activation (10). In an experimental liver metastasis mouse model, we found that implantation of pancreatic cancer cells into mouse liver induced desmoplastic reaction, similar to the primary pancreatic cancer (11), and that higher CAF densities of liver metastases associated with larger tumor sizes in the liver of mice (10). Thus, to better illustrate these findings, we review clinical and laboratory evidence that supports this promoting role of CAFs/activated HSTCs for liver metastasis and mechanisms by which CAFs/activated HSTCs promote the development of these lesions and resistance to cancer therapy. Additionally, in this review we discuss mechanisms governing myfibroblastic activation of HSTCs with a focus on key receptor mediated intracellular signaling and downstream epigenetic regulation of gene transcription.
Cancer Associated Fibroblasts from Liver Metastases Originate Predominantly from Hepatic Stellate Cells
HSTCs are liver specific pericytes that reside in the space of Disse between sinusoidal endothelial cells and hepatocytes, where they are quiescent and store vitamin A. They control ECM turnover and release paracrine, autocrine, juxtacrine, and chemoattractant factors to regulate blood flow of hepatic vasculature and maintain homeostasis of the liver microenvironment (12). HSTCs are a key contributor to liver fibrosis in response to injury by activating into myofibroblasts responsible for the excessive ECM deposits, growth factors and cytokines (13) (14). Quiescent HSTCs express neuronal cell markers, including glial fibrillary acidic proteins (GFAP), and desmin (15). When cancer cells are colonized in sinusoidal area of liver lobules, they extravasate into the space of Disse to make a direct contact with desmin positive HSTCs to induce their transdifferentiation into CAFs (10) (16), which are phenotypically distinct from the lipid-droplet rich quiescent HSTCs. During HSTC activation, cells shed their lipid droplets, develop α-SMA positive stress fibers, and express additional mesenchymal markers including vimentin, PDGF receptor alpha (PDGFR-α) and beta (PDGFR-β). Activated HSTCs are proliferative, migratory, contractile and fibrogenic, contributing to the formation and remodeling of the desmoplastic stroma of liver metastases.
Cell lineage tracing in genetic modified mouse models demonstrate that HSTCs give rise to 82-96% of hepatic myofibroblasts under various liver injuries (14), and that mesothelial cells, which constitute a single layer of flat epithelial cells covering the liver, are another progenitor of hepatic myofibroblasts via mesothelial-mesenchymal transition (17). Portal fibroblasts may be another contributor to hepatic myofibroblasts (18). Although epithelial-to-mesenchymal transition (EMT) has been proposed as a mechanism for myofibroblastic activation in the liver, genetic labeling of hepatocytes and cholangiocytes in mice and tracing of the labeled cells in liver injury animal models failed to support this notion (19) (20). Bone marrow derived fibrocytes and mesenchymal stem cells can be activated into hepatic myofibroblasts (15). However, contribution of these cells to CAFs found in liver metastases remains to be determined and, therefore, this topic offers unique opportunities for further experimentations.
Bench and Bedside Evidence Supporting a Tumor Promoter Role of Cancer Associated Fibroblasts Activated from Hepatic Stellate Cells
Clinical studies demonstrate that gene expression signatures obtained by expression profiling of CAFs predict poor clinical outcome of patients with lung cancer, colon cancer, invasive ductal breast carcinoma, and esophageal squamous cell carcinoma (21) (22) (23) (24). In liver metastases of patients affected by colon, gastric or pancreas adenocarcinomas cancers, CAF/activated HSTCs are easily detected by α-SMA staining, which correlate with the degree of fibrous stroma (desmoplasia) (25). Studies, primarily performed during the last decade have found that, in patients with hepatocellular carcinoma (HCC) or intrahepatic cholangiocarcinoma (ICC), the presence of desmoplasia or, at least of intratumoral CAF/activated HSTCs, correlates with the occurrence of larger tumors sizes and poor patient survival (26) (27). Consequently, these discoveries have fuelled a large amount of studies, which seek to identify CAF associated molecules as either mechanistic regulators or biomarkers for these processes. For instance, Utispan K et al. compared gene expression profile of CAF/activated HSTCs of cholangiocarcinoma (CCA) patients with that of normal liver fibroblasts and they found that periostin was overexpressed in CAF/activated HSTCs of CCA patients. Furthermore, they found that patients with high levels of periostin in their cancer had significantly shorter survival time than those with low levels, indicating CAF/activated HSTCs derived ECM components such as periostin as prognostic markers for CCA patients (28). With a similar goal, Badioda et al., have implanted tumor cells into the liver of control and discoidin domain receptor 2 (DDR2) knockout mice via intrasplenic injection and they found that DDR2 mice developed three folds more liver metastases than control mice, which contained higher densities of CAF/activated HSTCs (29). Similarly, mice deficient for IQ motif containing GTPase activating protein 1 (IQGAP1) developed significantly more liver metastases than control mice, which associated with more CAFs/activated HSTCs (10). In vitro, conditioned medium of activated HSTCs stimulated tumor cell proliferation, migration and invasion (27) (30) and induced EMT phenotype of tumor cells (31). In a tumor/HSTC coimplantation model, HSTCs promoted tumor initiation and progress in mice (10) (27) (32), and in a rat ICC model, depletion of CAF/activated HSTCs by a cytotoxic drug navitoclax resulted in reduction of tumor size and metastasis secondarily (33). Thus, the desmoplastic reaction, which forms the tumor microenvironment of liver metastases, is not just a response or a barrier to invasion of tumor cells, but rather a niche for cancer cells to implant and progress (34). Consequently, the primary cell constituent of this tissue, CAFs, provide a large amount of currently known cellular mechanisms and molecular mediators that are essential for modulating cancer growth.
Cancer Associated Fibroblasts Derived from Activated Hepatic Stellate Cells Promote Liver Metastasis by Paracrine actions
Activated HSTCs promote liver metastases by multiple mechanisms (Fig. 1): (a) they are sources of PDGF, HGF, SDF-1/CXCL12, TGF-β and so on, which are potent mitogens and chemoattractants of cancer cells (10) (16) (35). Colorectal cancer cells express CXCR4, receptor of SDF-1/CXCL12, and this SDF-1/CXCL12-CXCR4 pathway has been shown to mediate liver specific metastasis of these tumors (35). Additionally, TGF-β promotes migration and invasion of tumor cells by inducing their EMT transition. (b) MMPs, released from CAFs/activated HSTCs, facilitate tumor cell migration and invasion by breaking down ECM and cleaving E-cadherin and disassembling adherens junctions of tumor cells (36). In addition, these proteases are involved in the processing of signaling molecules. For instance, TGF-β is usually anchored in the ECM by a covalently link to a large glycoprotein called latency associated protein (LAP) and it is released by MMPs through proteolytic cleavage of LAP from the ECM (12) (37). (c) TGF-β suppresses anti-tumor responses of a variety of inflammatory cells, including natural killer (NK) cells, dendritic cells, macrophages, neutrophils, CD8+ and CD4+ T cells and regulatory T cells. It has been shown that activated HSTCs inhibit T-cell responsiveness, T-cell mediated cytotoxicity and accelerate T-cell apoptosis by releasing B7-H1 (38) (39). (d) Activated HSTCs regulate tumor angiogenesis by releasing angiogenic factors such as VEGF, angiopoietin 1 or 2, and MMP9 (29) (30) (40). Additionally, ECM components, such as fibronectin, collagen and laminin, facilitate tumor angiogenesis by activating integrin mediated signaling in endothelial cells (41). (e) The desmoplastic and hypovascularized stroma formed by activated HSTCs and ECM acts as a physical barrier for drug delivery and confers chemo- and radio- resistance to cancer tissue. For example, activated HSTCs confer tumor resistance to cisplatin by releasing HGF which activates Met on HCCs to promote cancer cell survival (31). Activated HSTCs contribute to chemo resistance of cholangiocarcinoma as well by simulating production of IL-1α, C5α, IL-1β and G-CSF cytokines in a HSTC/tumor cell coculture (27). Furthermore, ECM and inflammatory cytokines in the tumor stroma protect tumor cells from radiation therapy-induced death in part by activating integrin mediated intracellular signaling of tumor cells (42). Thus, CAFs/activated HSTCs are emerging as a bonafide target of therapies that seek to improve the efficacy of both chemotherapy and radiation therapy (5) (34).
Figure 1. Paracrine mechanisms by which CAF/activated HSCs promote liver metastasis.
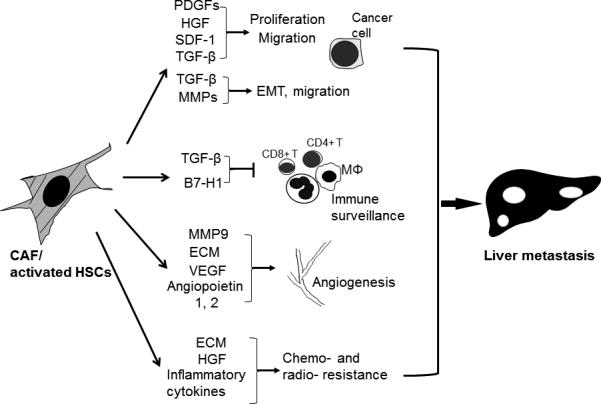
CAF/activated HSCs release excessive growth factors, cytokines and chemokines to promote tumor cell proliferation and migration. TGF-β derived from CAF/activated HSCs promotes tumor cell migration and invasion by inducing EMT of cancer cells. CAF/activated HSCs also produce MMPs to breakdown ECM and cadherin based adherens junctions of cancer to further potentiate tumor cell migration. Furthermore, TGF-β suppresses anti-tumor responses of a variety of inflammatory cells. CAF/activated HSCs also promote tumor angiogenesis by releasing MMP9 and angiogenic factors. Excessive ECM and inflammatory cytokines within the desmoplastic stroma impair drug delivery, contributing to tumor resistance to conventional chemotherapy and radiation therapy. EMT: epithelial to mesenchymal transition; MΦ: macrophage.
Key Intracellular Signaling Pathways for Hepatic Stellate Cells Activation
Myofibroblastic activation of HSTCs requires paracrine signals from cancer cells and other cell populations within the hepatic microenvironment to initiate intracellular signaling of HSTCs (Fig. 2). Below we list key signaling cascades governing HSTC activation. We need to point out that HSTC activation under tumor invasion is a relatively new and understudied topic as compared to HSTC activation under fibrotic stimuli. Some mechanisms presented here are therefore adapted from those in liver fibrosis studies, which we think are applicable to both liver diseases. Furthermore, each receptor mediated intracellular signaling does not act alone. In fact, it often cross-talks, regulates or converges with others to form complexes which function as an interconnected signaling network for HSTC activation (Fig. 2).
Figure 2. HSC activation is regulated by receptor mediated an intracellular signaling network and downstream orchestrated gene transcriptional events.

Only key receptor mediated signaling cascades and the downstream effectors are shown. In the nucleus, transcription of fibrogenic genes is turned on by recruitment of transcription factors, posttranscriptional modifications of transcription factors, and histone modifications to form an active chromatin structure, whereas transcription of adipogenic genes is turned off by epigenetic mechanisms such as DNA methylation, histone modifications to form a repressed chromatin structure and miRNAs. TFs: transcription factors; miRNAs: microRNAs; Ac: acetyl group; PM: plasma membrane; Nu: nucleus.
TGF-β signaling
TGF-β ligands consist of TGF-β1, TGF-β2 and TGF-β3 and activated HSTCs express TGF-β receptor I (TβRI) and II (TβRII) (10). They also express TGF-β receptor III (endoglin and betaglycan) although their functions for HSTC activation remain undefined. TGF-β induces heterodimerization of TβRII/TβRI, which results in the activation of the Serine/Threonine kinase domain of TβRI. The activated TβRI then phosphorylates SMAD family proteins to stimulate the nuclear translocation of SMAD2/3/4 complexes that are transcription factors for TGF-β target genes (43). Treatment of HSTCs with TGF-β1 induces expression of mesenchymal markers such as α-SMA and fibronectin, as well as the development of stress fibers, hallmarks of HSTC activation (10). Smad anchor for receptor activation (SARA), which localizes at early endosomes, is required for TβRI mediated phosphorylation of SMADs (44). TGF-β receptors undergo ligand induced endocytosis, late endosome/lysosomes targeting for degradation, and recycling (45) (10) (32). Additionally, TGF-β stimulation also activates non-canonical pathways by phosphorylating AKT and ERK to regulate fibroblast cell migration and proliferation (46). Furthermore, TGF-β also activates the p38 MAPK pathway, which leads to further SMAD3 phosphorylation and downstream fibrogenic responses (47). Therefore, both the TGF-β mediated canonical and noncanonical pathways are the most potent factors for myofibroblastic activation of HSTCs, and they present important targets for reducing CAFs and ECM accumulation within the tumor microenvironment.
PDGF signaling
PDGF ligands include PDGF-A, B, C and D (48). In response to liver injury, HSTCs express PDGFRα and PDGFRβ (49) (50) (51). PDGF-A binds to PDGFRα, PDGF-B binds to both PDGFRα and PDGFRβ, PDGF-C predominantly binds to PDGFRα, and PDGF-D binds to PDGFRβ (48). PDGFs are secreted as homo- or heterodimers, PDGF-AA, PDGF-BB, PDGF-AB, PDGF-CC, PDGF-DD. Binding of PDGFs with the receptors induces formation of PDGFRαα, PDGFRββ homodimers, or PDGFRαβ heterodimers, leading to activation of downstream Ras-MAPK, PI3K, and PLC-γ signaling pathways (52). PDGF/PDGFR binding and downstream signaling are regulated by neuropilin-1 (NRP-1) in HSTCs (50). PDGFRs contain multiple major autophosphorylation sites, Tyr-740, Tyr-751, Tyr-771, Tyr-857, which define their binding to specific downstream adaptor proteins for signal transduction. Similar to TGF-β receptors, PDGFRs also undergo endocytosis and ubiquitination mediated lysosomal targeting for degradation (53). Taken together, PDGF signaling is a major mitogen and chemoattractant for HSTCs and, consequently, an important target of efforts that seek to inhibit CAF proliferation and migration. In a later section of this review, targeting the tumor microenvironment by pharmacologic inhibition of PDGF signaling will be further discussed.
Integrin, Hedgehog (Hh) and Wnt signaling
HSTCs express three kinds of ECM receptors: integrins, disintegrin and metalloproteinase domain (ADAM) molecules, and DDR (54). Integrins are heterodimeric transmembrane proteins consisting of α and β subunit. Combinations of one of 8 β with one of 15 α subunits can potentially give rise to 21 different integrin receptors, which bind to collagen, fibronectin or laminin (55). Integrins activate FAK, which cross-talks with PI3K, Ras-MAPK and PLC-γ signaling pathways to promote survival and migration of HSTCs (55). Recent studies suggest that integrins are mechanosensors for ECM mediated mechanical signals. Integrins initiate mechanotransduction within the cell by recruiting numerous signaling molecules at focal adhesion sites, where the mechano signals from the ECM are converted into biochemical signals within the cells (56) (57). In addition to integrin signaling, Hh signaling promotes HSTC activation by regulating glycolysis (58). Consequently, pharmacologic inhibition of Hh signaling in mice blocks HSTC activation in the liver (59). Similarly, activation of Wnt signaling by ligands, Wnt3a and Wnt5a, results in enhanced myofibroblastic activation and survival of HSTCs (60). In summary, integrin, Hh and Wnt mediated signaling pathways are important players for HSTC activation. In this regard, we underscore the fact that IPI-926 is an Hh inhibitor that is currently under investigation in phase I/II trials to target the desmoplastic stroma of pancreatic cancer patients. Thus, it is likely that IPI-926 may be a potential therapeutic agent to reduce HSTC activation and thus impact on the growth of liver metastasis.
Signaling by Inflammatory Cytokines
HSTCs express CCR5 which is the receptor for RANTES and together they promote cell migration and proliferation (61). MCP-1 has also been shown to promote migration of activated, not quiescent, HSTCs in a dose-dependent manner (62). Additional cytokines including TNF-α, IFN-α, IFN-γ, IL-8, IL-6 and IL-10 may regulate HSTC activation by modulating NF-κB activity or JAK-STATs signaling pathway of HSTCs (54). Furthermore, activated HSTCs express leptin, an adipokine encoded by the obese gene, and its receptor (63). Leptin stimulates HSTC proliferation and potentiates TGF-β mediated collagen production (64), which is counteracted by adiponectin (65). Interestingly, a recent study demonstrates that an adiponectin-like small synthetic peptide agonist (ADP355: H-DAsn-Ile-Pro-Nva-Leu-Tyr-DSer-Phe-Ala-DSer-NH2) significantly inhibits both HSTC activation and fibrosis in mice (66), indicating that cytokine mediated signaling may also serve as an additional therapeutic target for antagonizing HSTC activation and liver metastasis.
Extracellular Matrix initiated Mechanotransduction for HSTC Activation
Progression of cancer and fibrosis caused by overproduction, deposition and cross-linking of ECM proteins leads to a stiff microenvironment. In this regard, transglutaminases, lysyl oxidases and prolyl hydroxylases are three examples of enzymes that mediate ECM cross-linking (56). Cells respond to physical signals of the ECM by forming integrin- and actomyosin-linked focal adhesions that sense mechanical force and stiffness of the ECM and initiate mechanotransduction of the cell. Thus, in addition to soluble biochemical factors, mechanotransduction induced by changes of the stiffness of the surrounding ECM has been proposed as another key factor for myofibroblastic activation of cells. Indeed, Olsen et al. have shown that when cultured in a stiff environment, freshly isolated rat HSTCs differentiated into myofibroblasts without TGF-β stimulation (67). In fact, TGF-β stimulation potentiated HSTC activation in response to a stiff environment (67). In the original study, in which this phenomenon was described, HSTCs were cultured on insert polyacrylamide supports of variable but precisely defined shear modulus (stiffness) ranging from 0.4 to 12 kPa, and cell morphology and α-SMA IF were used to assess HSTC activation. HSTCs cultured on a support of 0.4-1 kPa, representing the stiffness of normal liver (0.3-0.6 kPa), failed to differentiate into myofibroblasts. In contrast, cells on a support of 8-12 kPa showed myofibroblastic morphology and α-SMA positive stress fibers. Interestingly, when plated on Teflon, which is 4-5 orders of magnitude stiffer than cirrhotic liver, HSTCs failed to differentiate into myofibroblasts because they were not able to generate an effective mechanical tension on the polymer Teflon (67). Together, these data support the notion that HSTC activation requires mechanical tension generated within the cell in response to a stiff environment.
It appears that increased matrix stiffness leads to increased cell-generated tension, which ultimately leads to α-SMA expression and ECM deposition of activated HSTCs. Thus, it is critical to define the signaling pathways that are activated by mechanical signals and, which in turn, transduce these signals into defined gene expression patterns within the nucleus. In this regard, recent studies have begun to identify several signaling pathways as mediators of mechanotransduction of the cell. For example, as noted earlier, TGF-β is secreted as a latent form and anchored into ECM by LAP which directly binds to certain integrins. Single molecule analysis demonstrates that release of the active form of TGF-β is mechanosensitive; if cells are attached to a soft ECM, LAP-TGF-β complexes remain intact. On the other hand, if cells are attached to a stiff ECM, the resistance of ECM against cell-generated tension leads to LAP-TGF-β breakdown and the consequent release of active TGF-β (57). Of the numerous proteins within the focal adhesions, FAK is a key molecule mediating mechanotransduction. Besides the capability of FAK to crosstalk with both the PI3K and MAPK pathways, its activation also couples to RhoA/ROCK signaling (68), a step which is essential for generating actomyosin-dependent tension and reestablishment of force equilibrium within a cell. Samuel et al. demonstrated that overexpression of ROCK2 in skin of mice activated RhoA/ROCK signaling and led to tissue stiffening, intracellular tension, β-catenin nuclear translocation and β-catenin mediated hyperproliferation of epidermal cells in mice, all effects which were abrogated by inhibitors of actomyosin contractility (69). Although these findings were originally reported in epidermal cells, we believe that it is likely that FAK serves as an important mechanotransducer for HSTC activation. Complementing this observation, Yes-associated protein (YAP) and TAZ (WW domain-containing transcription regulator protein 1, also called WWTR1) have been recently identified as novel mechanosensors and mechanomediators in response to cues from the cellular microenvironment (70). YAP/TAZ become oncogenic through their ability to interact with the transcription factors TEAD/TEF in the nucleus to regulate cancer-associated gene expression networks (71). YAP/TAZ are downstream effectors of Hippo signaling, a pathway that controls cell proliferation and organ size. In the Hippo signaling cascade, cell-cell contacts activate LATS1/2 serine/threonine kinases, which induces phosphorylation and sequestration of YAP/TAZ in the cytoplasm and their subsequent ubiquitin-mediated degradation (72). Dupont et al. found that ECM stiffness and cell spreading increased nuclear accumulation and function of YAP/TAZ, which was abrogated by Rho inhibitor C3 or actin depolymerization agent latrunculin A (70). Additionally, Calvo et al. found that YAP of CAFs isolated from mouse mammary carcinomas was nuclear as compared to cytoplasmic YAP found in normal fibroblasts used as control (73). Furthermore, YAP in the nucleus of CAFs promoted the expression of ANLN and DIAPH3 which stabilized actomyosin fibers and led to further ECM stiffening required for maintenance of CAF phenotypes (73). Thus, YAP/TAZ, which are novel mechano sensors and transducers of the cell, may play important roles in the process of myofibroblastic activation of HSTCs.
Epigenetic Mechanisms for HSTC Activation
The receptor mediated intracellular signaling cascades and ECM medicated mechanotransduction, as mentioned above, eventually converge in the nucleus to regulate gene transcription. Indeed, the downstream effectors of these signaling pathways are often transcription factors and chromatin modification proteins that together constitute epigenetic mechanisms to induce the expression of gene networks responsible for the activated HSTC phenotype (Fig. 2). Thus, the epigenetic mechanisms are an obliged requisite for myofibroblastic activation of HSTCs.
Transcription factors
Numerous transcription factors have been identified during past decades as mediators for HSTC activation. For example, PPAR-γ is a transcription factor responsible for HSTC quiescence phenotype, which is repressed during HSTC activation (54). KLF11, which is both a binding partner and a target of PPAR-γ, also regulates stellate cell activation and live fibrosis (74). IκBα, an inhibitor of NFκB activity, is also silenced during HSTC activation, resulting in increased NFκB activity for both the survival and fibrogenic effects of HSTCs (75). SMADs are required for TGF-β1 mediated upregulation of α-SMA and fibronectin in HSTCs and STATs are downstream transcription factors mediating the effect of IFN-γ or leptin on proliferation of HSTCs (54). Other transcription factors characterized for HSTC activation include AP-1, AP-2, NF-1, C/EBP, Lhx2, KLF6, Sp1, Sp3, Foxf1 and FoxO1, and so on (for detailed information, see reviews (54) (76)). In addition, the effects of these transcription factors on bridging the upstream signaling to gene transcription are made possible by posttranscriptional modifications on them, such as phosphorylation, glycosylation, acetylation, ubiquitination, sumoylation and others. These posttranscriptional modifications regulate nuclear translocation, DNA binding capability, and protein stability of the transcription factors, adding an additional layer of complexity into gene transcription of HSTCs during myofibroblastic activation (54) (76). Thus, the complexity of transcription factors is one of important components of the epigenetic mechanisms that mediate HSTC activation.
DNA modifications, histone modifications and microRNAs in the epigenetic regulation of gene expression
Epigenetic regulation of gene expression is stable, heritable and without alterations of DNA sequence, which can be induced by tissue or tumor microenvironmental cues. Epigenetic regulation of gene expression includes DNA methylation, hydroximethylation, formilation, histone modifications, nucleosome remodeling machines, long noncoding RNAs (lncRNAs), and small noncoding RNAs such as microRNAs (miRNAs) and small interfering RNAs (siRNAs). However, only DNA methylation, histone modifications and miRNAs have been studied and characterized to date in activated HSTCs.
DNA methylation mediated epigenetic silencing is an important mechanism for turning off gene expression of proteins associated with quiescent HSTC phenotype. For example, epigenetic silencing of IκBα during HSTC activation was prevented by DNA methylation inhibitor 5-aza-2’-deoxycytidine (5-azadC) (75). Similarly, epigenetic silencing of both PTEN and Ras GTPase activating-like protein 1 (RASAL1), a Ras inhibitor, was reversed by 5-azadC (77) (78). Although histone modifications such as histone methylation and histone acetylation are less investigated, the data thus far available indicate that these marks are implicated in myofibroblastic activation of HSTCs. For example, histone methyltransferase ASH1 is recruited to the promoter region of genes including TIMP-1, α-SMA, TGFβ1 and collagen 1 to induce histone methylation (H3K4me3) to increase expression of these genes during HSTC activation (79), and ethanol treatment of HSTCs induces a dose and time dependent increase of acetylation of histone 3 at lysine 9 (80).
MiRNAs suppress gene expression by binding to 3’-untranslated region (3’ -UTR) of mRNAs to induce their degradation. Using miRNA arrays, Roderburg et al. have identified 10 miRNAs upregulated and 21 miRNAs downregulated in fibrotic mouse liver, supporting that HSTC activation requires cooperation of divergent effects of different miRNAs (81). Consistently, miR-29 is downregulated in activated HSTCs and restoration of miR-29 inhibits collagen expression (81); miR-19 is downregulated and restoration of miR-19 represses TβRII and SMAD3 thereby inhibiting TGF-β signaling (82). Similarly, miR-146a inhibits TGF-β signaling in HSTCs by targeting SMAD4 for degradation (83). Furthermore, miR-15, miR-16 and miR-335 target proteins or signaling pathways important for proliferation, apoptosis and migration of HSTCs respectively (84) (85). These studies underscore the importance of this type of posttranscriptional regulation and the fact that understanding the role of miRNAs for HSTC activation will be important for developing diagnostic markers or novel therapeutic strategy for clinical application.
In summary, HSTC activation in response to tumor derived stimuli can be explained by a paradigm that involves two steps: (1) initiation of signals at the cell membrane through the activation of various types of receptors to either activate or inhibit intracellular signaling cascades, and (2) convergence of the intracellular signaling cascades at the nucleus for gene expression networks that define phenotypes of active HSTCs. While initiation and cytoplasmic transduction of signals are important, the final fate of HSTCs is determined by the inherited pattern of gene expression which occurs via epigenetic mechanisms. Of the numerous epigenetic mechanisms, chromatin remodeling is a dominant effect that dictates which regions of the genome are read for transcription of mRNAs, lncRNAs, miRNAs, or siRNAs. Although significant advances have been made in the area of receptor biology and intracellular signal transduction, gene transcription regulated by chromatin remodeling and epigenetic inheritance of gene expression patterns remains an under developed area of study, which we predict will significantly grow in the near future. Therefore, filling this gap in knowledge may constitute potentially one of the most promising area of investigation in this field.
Conclusions and Perspectives on Therapeutic Targeting of CAF/activated HSTCs
Liver metastasis is dependent on bidirectional interactions between cancer cells and the microenvironment of the liver. HSTCs are one of important components of prometastatic liver microenvironment for their transdifferentiation into CAFs critical for tumor implantation, propagation and invasion in the liver. CAFs/activated HSTCs also confer chemo and radio resistance of cancer. Activation of HSTCs into CAFs is regulated by a complex interconnected intracellular signaling network and downstream orchestrated gene transcriptional events to induce genes responsible for activated HSTC phenotype and repress genes responsible for quiescent HSTC phenotype. CAF/activated HSTCs thus present a therapeutic target for preventing or reducing liver metastasis, improving drug delivery, and reducing tumor resistance to anti-cancer therapy.
Therapeutic targeting of cancer cells specifically has progressed slowly for decades because each patient's cancer cells harbor individual genetic mutations and display unique biological behaviors, making them difficult to target. CAF/activated HSTCs are targetable because they are significantly more sensitive than tumor cells to cytotoxic drugs such as navitoclax (ABT-263), a BH3 mimetic (33). In rats orthotopically transplanted with ICC tumor cells, navitoclax treatment increased apoptosis of CAF/activated HSTCs, which resulted in reduced tumor size and metastasis, providing a “proof-of-principle” for depletion of CAF/activated HSTCs as an anticancer strategy for tumor growth in the liver (33) (34). In a mouse model of pancreatic ductal adenocarcinoma, combined therapy with gemcitabine to target tumor cells and Hh inhibitor IPI-926 to target desmoplastic stroma, improved delivery and efficacy of gemcitabine, and extended median survival of mice as compared with either drug alone (4). Congruently, in phase I/II and phase III clinical studies, combination of nab-paclitaxel (Abraxane) to cause depletion of tumor stroma and Gemcitabine significantly extended survival of patients with advanced/metastatic pancreatic cancer as compared to Gemcitabine alone (86) (87). Based upon these findings, nab-paclitaxel plus gemcitabine has received FDA approval in 2013 as a first-line treatment for patients with metastatic pancreatic adenocarcinoma. Interestingly, however, using the same experimental paradigm (4), Rhim at al. recently demonstrated that IPI-926 alone or combined with gemcitabine, both are known to target CAFs, shortened survival of mice (88). Additionally, CAF depletion in a genetic modified pancreatic cancer mouse model shortened mouse survival but enhanced the efficacy of anti-CTLA4 (Ipilimumab) immunotherapy (7). Together, these data reveal that, though it is still poorly understood, targeting CAFs is beneficial for ameliorating pancreatic cancer in experimental animal models. Thus, more in depth investigations are needed to more solidly establish the beneficial outcomes of combinational therapies that target both CAFs and tumor cells for the treatment of liver metastasis.
PDGF and TGF-β signaling are two major intracellular signaling pathways that control HSTC activation. We recently found that activated HSTCs express a high level of PDGFRα in addition to PDGFRβ and that PDGFRα is required for TGF-β signaling in HSTCs, highlighting PDGFRα as a target for inhibiting both PDGF and TGF-β signaling of HSTCs (51). Crenolanib besylate, a drug currently under investigation (89), and approved anti-cancer drug Imatinib (Gleeve, ST1-571) and Sunitinib, are designed to inhibit both PDGFR receptors. It is worth investigating if these drugs inhibit both PDGF and TGF-β signaling of HSTCs and suppress both tumor cells and the tumor microenvironment. Targeting TGF-β signaling of the tumor microenvironment is a promising direction in cancer therapy for tumors that lost TGF-β mediated tumor suppression pathways (90). Drugs targeting TGF-β signaling are still under investigation with some of them showing encouraging results in clinical trials. Besides IPI-926, GDC-0449 is another promising Hh inhibitor currently being tested in clinical trials. GDC-0449 has been shown to inhibit accumulation of liver fibroblasts and induce HCC regression in aged mice deficient for multidrug resistant gene 2 (Mdr2−/− mice) (59). Furthermore, continuously elucidating epigenetic regulation of gene expression for HSTC activation may help lead to new directions for drug development, for instance, delivering miRNAs or screening for novel compounds that target histone methylation or acetylation to inhibit myofibroblastic activation of HSTCs and the tumor microenvironment.
Acknowledgments
The authors wish to thank M. Gyarmaty, J. Horner, R. Williamson, C. Drefcinski, J. Lintz, S. Friedman and A. Quinn at Hormel Foods for their strong support and inspiration. The authors also apologize to the authors for their work not being cited owing to space limitations.
Grants: This work is supported by NIH grants R01 CA160069 to N. Kang, R01 DK059615 and AA021171 to V. H. Shah, and R01 DK052913 to R. Urrutia. The authors also wish to acknowledge a startup fund to N. Kang at the Hormel Institute.
Footnotes
The authors have declared that no conflict of interest exists.
References
- 1.Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer cell. 2012;21:309–22. doi: 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 2.Augsten M. Cancer-Associated Fibroblasts as Another Polarized Cell Type of the Tumor Microenvironment. Frontiers in oncology. 2014;4:62. doi: 10.3389/fonc.2014.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iovanna JL, Marks DL, Fernandez-Zapico ME, Urrutia R. Mechanistic insights into self-reinforcing processes driving abnormal histogenesis during the development of pancreatic cancer. The American journal of pathology. 2013;182:1078–86. doi: 10.1016/j.ajpath.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olive KP, Jacobetz MA, Davidson CJ, Gopinathan A, McIntyre D, Honess D, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–61. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sebens S, Schafer H. The tumor stroma as mediator of drug resistance--a potential target to improve cancer therapy? Current pharmaceutical biotechnology. 2012;13:2259–72. doi: 10.2174/138920112802501999. [DOI] [PubMed] [Google Scholar]
- 6.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nature reviews Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 7.Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer cell. 2014;25:719–34. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sugimoto H, Mundel TM, Kieran MW, Kalluri R. Identification of fibroblast heterogeneity in the tumor microenvironment. Cancer biology & therapy. 2006;5:1640–6. doi: 10.4161/cbt.5.12.3354. [DOI] [PubMed] [Google Scholar]
- 9.Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, et al. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science. 2004;303:848–51. doi: 10.1126/science.1090922. [DOI] [PubMed] [Google Scholar]
- 10.Liu C, Billadeau DD, Abdelhakim H, Leof E, Kaibuchi K, Bernabeu C, et al. IQGAP1 suppresses TbetaRII-mediated myofibroblastic activation and metastatic growth in liver. The Journal of clinical investigation. 2013;123:1138–56. doi: 10.1172/JCI63836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Decker NK, Abdelmoneim SS, Yaqoob U, Hendrickson H, Hormes J, Bentley M, et al. Nitric oxide regulates tumor cell cross-talk with stromal cells in the tumor microenvironment of the liver. The American journal of pathology. 2008;173:1002–12. doi: 10.2353/ajpath.2008.080158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geerts A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Seminars in liver disease. 2001;21:311–35. doi: 10.1055/s-2001-17550. [DOI] [PubMed] [Google Scholar]
- 13.Friedman SL. Mechanisms of disease: Mechanisms of hepatic fibrosis and therapeutic implications. Nature clinical practice Gastroenterology & hepatology. 2004;1:98–105. doi: 10.1038/ncpgasthep0055. [DOI] [PubMed] [Google Scholar]
- 14.Mederacke I, Hsu CC, Troeger JS, Huebener P, Mu X, Dapito DH, et al. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nature communications. 2013;4:2823. doi: 10.1038/ncomms3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fausther M, Lavoie EG, Dranoff JA. Contribution of Myofibroblasts of Different Origins to Liver Fibrosis. Current pathobiology reports. 2013;1:225–30. doi: 10.1007/s40139-013-0020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimizu S, Yamada N, Sawada T, Ikeda K, Kawada N, Seki S, et al. In vivo and in vitro interactions between human colon carcinoma cells and hepatic stellate cells. Japanese journal of cancer research : Gann. 2000;91:1285–95. doi: 10.1111/j.1349-7006.2000.tb00916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Wang J, Asahina K. Mesothelial cells give rise to hepatic stellate cells and myofibroblasts via mesothelial-mesenchymal transition in liver injury. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:2324–9. doi: 10.1073/pnas.1214136110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iwaisako K, Jiang C, Zhang M, Cong M, Moore-Morris TJ, Park TJ, et al. Origin of myofibroblasts in the fibrotic liver in mice. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E3297–305. doi: 10.1073/pnas.1400062111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taura K, De Minicis S, Seki E, Hatano E, Iwaisako K, Osterreicher CH, et al. Hepatic stellate cells secrete angiopoietin 1 that induces angiogenesis in liver fibrosis. Gastroenterology. 2008;135:1729–38. doi: 10.1053/j.gastro.2008.07.065. [DOI] [PubMed] [Google Scholar]
- 20.Scholten D, Osterreicher CH, Scholten A, Iwaisako K, Gu G, Brenner DA, et al. Genetic labeling does not detect epithelial-to-mesenchymal transition of cholangiocytes in liver fibrosis in mice. Gastroenterology. 2010;139:987–98. doi: 10.1053/j.gastro.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Navab R, Strumpf D, Bandarchi B, Zhu CQ, Pintilie M, Ramnarine VR, et al. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:7160–5. doi: 10.1073/pnas.1014506108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herrera M, Islam AB, Herrera A, Martin P, Garcia V, Silva J, et al. Functional heterogeneity of cancer-associated fibroblasts from human colon tumors shows specific prognostic gene expression signature. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:5914–26. doi: 10.1158/1078-0432.CCR-13-0694. [DOI] [PubMed] [Google Scholar]
- 23.Farmer P, Bonnefoi H, Anderle P, Cameron D, Wirapati P, Becette V, et al. A stroma-related gene signature predicts resistance to neoadjuvant chemotherapy in breast cancer. Nature medicine. 2009;15:68–74. doi: 10.1038/nm.1908. [DOI] [PubMed] [Google Scholar]
- 24.Ha SY, Yeo SY, Xuan YH, Kim SH. The prognostic significance of cancer-associated fibroblasts in esophageal squamous cell carcinoma. PloS one. 2014;9:e99955. doi: 10.1371/journal.pone.0099955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Terada T, Makimoto K, Terayama N, Suzuki Y, Nakanuma Y. Alpha-smooth muscle actin-positive stromal cells in cholangiocarcinomas, hepatocellular carcinomas and metastatic liver carcinomas. Journal of hepatology. 1996;24:706–12. doi: 10.1016/s0168-8278(96)80267-4. [DOI] [PubMed] [Google Scholar]
- 26.Ju MJ, Qiu SJ, Fan J, Xiao YS, Gao Q, Zhou J, et al. Peritumoral activated hepatic stellate cells predict poor clinical outcome in hepatocellular carcinoma after curative resection. American journal of clinical pathology. 2009;131:498–510. doi: 10.1309/AJCP86PPBNGOHNNL. [DOI] [PubMed] [Google Scholar]
- 27.Okabe H, Beppu T, Hayashi H, Ishiko T, Masuda T, Otao R, et al. Hepatic stellate cells accelerate the malignant behavior of cholangiocarcinoma cells. Annals of surgical oncology. 2011;18:1175–84. doi: 10.1245/s10434-010-1391-7. [DOI] [PubMed] [Google Scholar]
- 28.Utispan K, Thuwajit P, Abiko Y, Charngkaew K, Paupairoj A, Chau-in S, et al. Gene expression profiling of cholangiocarcinoma-derived fibroblast reveals alterations related to tumor progression and indicates periostin as a poor prognostic marker. Molecular cancer. 2010;9:13. doi: 10.1186/1476-4598-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Badiola I, Olaso E, Crende O, Friedman SL, Vidal-Vanaclocha F. Discoidin domain receptor 2 deficiency predisposes hepatic tissue to colon carcinoma metastasis. Gut. 2012;61:1465–72. doi: 10.1136/gutjnl-2011-300810. [DOI] [PubMed] [Google Scholar]
- 30.Coulouarn C, Corlu A, Glaise D, Guenon I, Thorgeirsson SS, Clement B. Hepatocytestellate cell cross-talk in the liver engenders a permissive inflammatory microenvironment that drives progression in hepatocellular carcinoma. Cancer research. 2012;72:2533–42. doi: 10.1158/0008-5472.CAN-11-3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu G, Jing Y, Kou X, Ye F, Gao L, Fan Q, et al. Hepatic stellate cells secreted hepatocyte growth factor contributes to the chemoresistance of hepatocellular carcinoma. PloS one. 2013;8:e73312. doi: 10.1371/journal.pone.0073312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tu K, Li J, Verma VK, Liu C, Billadeau DD, Lamprecht G, et al. VASP promotes TGF-beta activation of hepatic stellate cells by regulating Rab11 dependent plasma membrane targeting of TGF-beta receptors. Hepatology. 2014 doi: 10.1002/hep.27251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mertens JC, Fingas CD, Christensen JD, Smoot RL, Bronk SF, Werneburg NW, et al. Therapeutic effects of deleting cancer-associated fibroblasts in cholangiocarcinoma. Cancer research. 2013;73:897–907. doi: 10.1158/0008-5472.CAN-12-2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sirica AE, Gores GJ. Desmoplastic stroma and cholangiocarcinoma: Clinical implications and therapeutic targeting. Hepatology. 2014;59:2397–402. doi: 10.1002/hep.26762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsusue R, Kubo H, Hisamori S, Okoshi K, Takagi H, Hida K, et al. Hepatic stellate cells promote liver metastasis of colon cancer cells by the action of SDF-1/CXCR4 axis. Annals of surgical oncology. 2009;16:2645–53. doi: 10.1245/s10434-009-0599-x. [DOI] [PubMed] [Google Scholar]
- 36.Cirri P, Chiarugi P. Cancer-associated-fibroblasts and tumour cells: a diabolic liaison driving cancer progression. Cancer metastasis reviews. 2012;31:195–208. doi: 10.1007/s10555-011-9340-x. [DOI] [PubMed] [Google Scholar]
- 37.Artacho-Cordon A, Artacho-Cordon F, Rios-Arrabal S, Calvente I, Nunez MI. Tumor microenvironment and breast cancer progression: a complex scenario. Cancer biology & therapy. 2012;13:14–24. doi: 10.4161/cbt.13.1.18869. [DOI] [PubMed] [Google Scholar]
- 38.Yu MC, Chen CH, Liang X, Wang L, Gandhi CR, Fung JJ, et al. Inhibition of T-cell responses by hepatic stellate cells via B7-H1-mediated T-cell apoptosis in mice. Hepatology. 2004;40:1312–21. doi: 10.1002/hep.20488. [DOI] [PubMed] [Google Scholar]
- 39.Zhao W, Su W, Kuang P, Zhang L, Liu J, Yin Z, et al. The role of hepatic stellate cells in the regulation of T-cell function and the promotion of hepatocellular carcinoma. International journal of oncology. 2012;41:457–64. doi: 10.3892/ijo.2012.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olaso E, Salado C, Egilegor E, Gutierrez V, Santisteban A, Sancho-Bru P, et al. Proangiogenic role of tumor-activated hepatic stellate cells in experimental melanoma metastasis. Hepatology. 2003;37:674–85. doi: 10.1053/jhep.2003.50068. [DOI] [PubMed] [Google Scholar]
- 41.Hodkinson PS, Mackinnon AC, Sethi T. Extracellular matrix regulation of drug resistance in small-cell lung cancer. International journal of radiation biology. 2007;83:733–41. doi: 10.1080/09553000701570204. [DOI] [PubMed] [Google Scholar]
- 42.Mantoni TS, Lunardi S, Al-Assar O, Masamune A, Brunner TB. Pancreatic stellate cells radioprotect pancreatic cancer cells through beta1-integrin signaling. Cancer research. 2011;71:3453–8. doi: 10.1158/0008-5472.CAN-10-1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 44.Truty MJ, Urrutia R. Basics of TGF-beta and pancreatic cancer. Pancreatology : official journal of the International Association of Pancreatology. 2007;7:423–35. doi: 10.1159/000108959. [DOI] [PubMed] [Google Scholar]
- 45.Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nature cell biology. 2003;5:410–21. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- 46.Rahimi RA, Leof EB. TGF-beta signaling: a tale of two responses. Journal of cellular biochemistry. 2007;102:593–608. doi: 10.1002/jcb.21501. [DOI] [PubMed] [Google Scholar]
- 47.Furukawa F, Matsuzaki K, Mori S, Tahashi Y, Yoshida K, Sugano Y, et al. p38 MAPK mediates fibrogenic signal through Smad3 phosphorylation in rat myofibroblasts. Hepatology. 2003;38:879–89. doi: 10.1053/jhep.2003.50384. [DOI] [PubMed] [Google Scholar]
- 48.Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes & development. 2008;22:1276–312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wong L, Yamasaki G, Johnson RJ, Friedman SL. Induction of beta-platelet-derived growth factor receptor in rat hepatic lipocytes during cellular activation in vivo and in culture. The Journal of clinical investigation. 1994;94:1563–9. doi: 10.1172/JCI117497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cao S, Yaqoob U, Das A, Shergill U, Jagavelu K, Huebert RC, et al. Neuropilin-1 promotes cirrhosis of the rodent and human liver by enhancing PDGF/TGF-beta signaling in hepatic stellate cells. The Journal of clinical investigation. 2010;120:2379–94. doi: 10.1172/JCI41203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu C, Li J, Xiang X, Guo L, Tu K, Liu Q, et al. PDGF receptor alpha promotes TGF-beta signaling in hepatic stellate cells via transcriptional and post transcriptional regulation of TGF-beta receptors. American journal of physiology Gastrointestinal and liver physiology. 2014 doi: 10.1152/ajpgi.00138.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pinzani M. PDGF and signal transduction in hepatic stellate cells. Frontiers in bioscience : a journal and virtual library. 2002;7:d1720–6. doi: 10.2741/A875. [DOI] [PubMed] [Google Scholar]
- 53.Mori S, Heldin CH, Claesson-Welsh L. Ligand-induced ubiquitination of the platelet-derived growth factor beta-receptor plays a negative regulatory role in its mitogenic signaling. The Journal of biological chemistry. 1993;268:577–83. [PubMed] [Google Scholar]
- 54.Hernandez-Gea V, Friedman SL. Pathogenesis of liver fibrosis. Annual review of pathology. 2011;6:425–56. doi: 10.1146/annurev-pathol-011110-130246. [DOI] [PubMed] [Google Scholar]
- 55.Pinzani M, Marra F, Carloni V. Signal transduction in hepatic stellate cells. Liver. 1998;18:2–13. doi: 10.1111/j.1600-0676.1998.tb00120.x. [DOI] [PubMed] [Google Scholar]
- 56.Tschumperlin DJ, Liu F, Tager AM. Biomechanical regulation of mesenchymal cell function. Curr Opin Rheumatol. 2013;25:92–100. doi: 10.1097/BOR.0b013e32835b13cd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Janmey PA, Wells RG, Assoian RK, McCulloch CA. From tissue mechanics to transcription factors. Differentiation. 2013;86:112–20. doi: 10.1016/j.diff.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen Y, Choi SS, Michelotti GA, Chan IS, Swiderska-Syn M, Karaca GF, et al. Hedgehog controls hepatic stellate cell fate by regulating metabolism. Gastroenterology. 2012;143:1319–29. e1–11. doi: 10.1053/j.gastro.2012.07.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Philips GM, Chan IS, Swiderska M, Schroder VT, Guy C, Karaca GF, et al. Hedgehog signaling antagonist promotes regression of both liver fibrosis and hepatocellular carcinoma in a murine model of primary liver cancer. PloS one. 2011;6:e23943. doi: 10.1371/journal.pone.0023943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miao CG, Yang YY, He X, Huang C, Huang Y, Zhang L, et al. Wnt signaling in liver fibrosis: progress, challenges and potential directions. Biochimie. 2013;95:2326–35. doi: 10.1016/j.biochi.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 61.Lee UE, Friedman SL. Mechanisms of hepatic fibrogenesis. Best practice & research Clinical gastroenterology. 2011;25:195–206. doi: 10.1016/j.bpg.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harada K, Chiba M, Okamura A, Hsu M, Sato Y, Igarashi S, et al. Monocyte chemoattractant protein-1 derived from biliary innate immunity contributes to hepatic fibrogenesis. Journal of clinical pathology. 2011;64:660–5. doi: 10.1136/jclinpath-2011-200040. [DOI] [PubMed] [Google Scholar]
- 63.Ikejima K, Takei Y, Honda H, Hirose M, Yoshikawa M, Zhang YJ, et al. Leptin receptor-mediated signaling regulates hepatic fibrogenesis and remodeling of extracellular matrix in the rat. Gastroenterology. 2002;122:1399–410. doi: 10.1053/gast.2002.32995. [DOI] [PubMed] [Google Scholar]
- 64.Tang M, Potter JJ, Mezey E. Leptin enhances the effect of transforming growth factor beta in increasing type I collagen formation. Biochemical and biophysical research communications. 2002;297:906–11. doi: 10.1016/s0006-291x(02)02300-8. [DOI] [PubMed] [Google Scholar]
- 65.Handy JA, Fu PP, Kumar P, Mells JE, Sharma S, Saxena NK, et al. Adiponectin inhibits leptin signalling via multiple mechanisms to exert protective effects against hepatic fibrosis. The Biochemical journal. 2011;440:385–95. doi: 10.1042/BJ20102148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kumar P, Smith T, Rahman K, Thorn NE, Anania FA. Adiponectin Agonist ADP355 Attenuates CCl4-Induced Liver Fibrosis in Mice. PloS one. 2014;9:e110405. doi: 10.1371/journal.pone.0110405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Olsen AL, Bloomer SA, Chan EP, Gaca MD, Georges PC, Sackey B, et al. Hepatic stellate cells require a stiff environment for myofibroblastic differentiation. American journal of physiology Gastrointestinal and liver physiology. 2011;301:G110–8. doi: 10.1152/ajpgi.00412.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schaller MD. Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. Journal of cell science. 2010;123:1007–13. doi: 10.1242/jcs.045112. [DOI] [PubMed] [Google Scholar]
- 69.Samuel MS, Lopez JI, McGhee EJ, Croft DR, Strachan D, Timpson P, et al. Actomyosin-mediated cellular tension drives increased tissue stiffness and beta-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer cell. 2011;19:776–91. doi: 10.1016/j.ccr.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–83. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- 71.Vassilev A, Kaneko KJ, Shu H, Zhao Y, DePamphilis ML. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes & development. 2001;15:1229–41. doi: 10.1101/gad.888601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Piccolo S, Dupont S, Cordenonsi M. The Biology of YAP/TAZ: Hippo Signaling and Beyond. Physiol Rev. 2014;94:1287–312. doi: 10.1152/physrev.00005.2014. [DOI] [PubMed] [Google Scholar]
- 73.Calvo F, Ege N, Grande-Garcia A, Hooper S, Jenkins RP, Chaudhry SI, et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nature cell biology. 2013;15:637–46. doi: 10.1038/ncb2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mathison A, Grzenda A, Lomberk G, Velez G, Buttar N, Tietz P, et al. Role for Kruppel-like transcription factor 11 in mesenchymal cell function and fibrosis. PloS one. 2013;8:e75311. doi: 10.1371/journal.pone.0075311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mann J, Oakley F, Akiboye F, Elsharkawy A, Thorne AW, Mann DA. Regulation of myofibroblast transdifferentiation by DNA methylation and MeCP2: implications for wound healing and fibrogenesis. Cell death and differentiation. 2007;14:275–85. doi: 10.1038/sj.cdd.4401979. [DOI] [PubMed] [Google Scholar]
- 76.Friedman SL. Transcriptional regulation of stellate cell activation. Journal of gastroenterology and hepatology. 2006;21(Suppl 3):S79–83. doi: 10.1111/j.1440-1746.2006.04585.x. [DOI] [PubMed] [Google Scholar]
- 77.Tao H, Huang C, Yang JJ, Ma TT, Bian EB, Zhang L, et al. MeCP2 controls the expression of RASAL1 in the hepatic fibrosis in rats. Toxicology. 2011;290:327–33. doi: 10.1016/j.tox.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 78.Bian EB, Huang C, Ma TT, Tao H, Zhang H, Cheng C, et al. DNMT1-mediated PTEN hypermethylation confers hepatic stellate cell activation and liver fibrogenesis in rats. Toxicology and applied pharmacology. 2012;264:13–22. doi: 10.1016/j.taap.2012.06.022. [DOI] [PubMed] [Google Scholar]
- 79.Perugorria MJ, Wilson CL, Zeybel M, Walsh M, Amin S, Robinson S, et al. Histone methyltransferase ASH1 orchestrates fibrogenic gene transcription during myofibroblast transdifferentiation. Hepatology. 2012;56:1129–39. doi: 10.1002/hep.25754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim JS, Shukla SD. Histone h3 modifications in rat hepatic stellate cells by ethanol. Alcohol and alcoholism. 2005;40:367–72. doi: 10.1093/alcalc/agh170. [DOI] [PubMed] [Google Scholar]
- 81.Roderburg C, Urban GW, Bettermann K, Vucur M, Zimmermann H, Schmidt S, et al. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology. 2011;53:209–18. doi: 10.1002/hep.23922. [DOI] [PubMed] [Google Scholar]
- 82.Lakner AM, Steuerwald NM, Walling TL, Ghosh S, Li T, McKillop IH, et al. Inhibitory effects of microRNA 19b in hepatic stellate cell-mediated fibrogenesis. Hepatology. 2012;56:300–10. doi: 10.1002/hep.25613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.He Y, Huang C, Zhang SP, Sun X, Long XR, Li J. The potential of microRNAs in liver fibrosis. Cellular signalling. 2012;24:2268–72. doi: 10.1016/j.cellsig.2012.07.023. [DOI] [PubMed] [Google Scholar]
- 84.Guo CJ, Pan Q, Li DG, Sun H, Liu BW. miR-15b and miR-16 are implicated in activation of the rat hepatic stellate cell: An essential role for apoptosis. Journal of hepatology. 2009;50:766–78. doi: 10.1016/j.jhep.2008.11.025. [DOI] [PubMed] [Google Scholar]
- 85.Chen C, Wu CQ, Zhang ZQ, Yao DK, Zhu L. Loss of expression of miR-335 is implicated in hepatic stellate cell migration and activation. Experimental cell research. 2011;317:1714–25. doi: 10.1016/j.yexcr.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 86.Heinemann V, Reni M, Ychou M, Richel DJ, Macarulla T, Ducreux M. Tumour-stroma interactions in pancreatic ductal adenocarcinoma: rationale and current evidence for new therapeutic strategies. Cancer treatment reviews. 2014;40:118–28. doi: 10.1016/j.ctrv.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 87.Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. The New England journal of medicine. 2013;369:1691–703. doi: 10.1056/NEJMoa1304369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer cell. 2014;25:735–47. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Heinrich MC, Griffith D, McKinley A, Patterson J, Presnell A, Ramachandran A, et al. Crenolanib inhibits the drug-resistant PDGFRA D842V mutation associated with imatinib-resistant gastrointestinal stromal tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:4375–84. doi: 10.1158/1078-0432.CCR-12-0625. [DOI] [PubMed] [Google Scholar]
- 90.Katz LH, Li Y, Chen JS, Munoz NM, Majumdar A, Chen J, et al. Targeting TGF-beta signaling in cancer. Expert opinion on therapeutic targets. 2013;17:743–60. doi: 10.1517/14728222.2013.782287. [DOI] [PMC free article] [PubMed] [Google Scholar]