

Abstract
We previously found that neurons are able to affect the ability of brain capillary endothelial cells to form in vitro a monolayer with properties resembling the blood-brain barrier. We then looked, by immunofluorescence and western analysis, for factors, produced by neurons, with the potential to influence growth and differentiation of endothelial cells. In the present paper, we report that neurons produce both vascular endothelial growth factor and fibroblast growth factor 2, two well-known angiogenic factors. More interestingly, we gained evidence that both factors are released by neurons, at least in part, by shedding of extracellular vesicles, that contain β1 integrin, a membrane protein already known to be part of extracellular vesicles released by tumour cells. Shedding of extracellular vesicles by neurons was also confirmed by scanner electron microscopy.
Keywords: neurons, extracellular vesicles, shedding, VEGF, FGF-2
Introduction
We previously found, by using a transwell co-culture system, that neurons and astrocytes have synergistic effects on reduction of brain capillary endothelial cell permeability to radioactive sucrose, used as a marker of paracellular flux [1–5].
A further point was that in the three-cell type system of culture, neurons, astrocytes and endothelial cells were not in physical contact. Therefore, the effects discovered should be due, at least in part, to molecular mechanisms not based on cell–cell contacts. As independent experiments showed that transformed glial cells (oligodendroglioma cells) are able to shed membrane vesicles [6], we wondered whether extracellular vesicles could be also the way adopted by neurons (this paper) and/or astrocytes (manuscript in preparation) to address inductive signals to endothelial cells.
Many eukaryotic cell types have been indeed shown to release into the extracellular environment vesicles of different sizes and composition which have been differently defined [for recent reviews, see: 7–9]. The first population of exovesiscles to be described seemed to originate from the cell plasma membrane through a mechanism similar to virus budding [10]. By electron microscopic analyses, vesicles shed by circulating human lymphocytes [10] and by in vitro cultured human breast carcinoma cells [11] were found to be large and heterogeneous in size (100–1000 nm). Extracellular vesicles of this kind, called microvesicles (MVs), are released by several kinds of viable cells through an active process that require RNA and protein synthesis [10, 12] and seem to originate from domains of the plasma membrane selectively enriched in specific membrane components, such as β1 integrin and membrane-bound gelatinase B/MMP-9 [see, e.g. 13]. Other vesicles, known as exosomes, are smaller (30–90 nm) and seem to originate from the endosomal system by exocytosis of cell structures, called multivesicular bodies [MVBs; 14, 15]; exosomes are released by most cells of the haemopoietic lineage and contain several cytosolic proteins (including heterotrimeric G protein Gi2α, annexin II and heat shock cognate protein Hsc73), as well as different integral and peripheral membrane proteins [15]. Both MVs and exosomes are involved in cell-to-cell communications. The phenomenon of shedding was first demonstrated in tumour cells, where shedding was related to the invasive potential of the tumour [16–19]. The presence in shed vesicles of numerous extracellular matrix (ECM)-degrading proteolytic enzymes can also have an effect on angiogenesis [20]. However, vesicles promote angiogenesis [21, 22] also because they vehicle numerous pro-angiogenic signalling molecules. For example, fibroblast growth factor 2 (FGF-2) was demonstrated to be externalized from the cells as a component of shed membrane vesicles [12]; and vascular endothelial growth factor (VEGF), perhaps the most effective angiogenic factor known at present, also appears to be present in vesicles [23].
FGF-2, also known as basic FGF, is a non-glycosylated 18 kD polypeptide with very high affinity for heparin, that is secreted in spite of lacking a conventional secretory peptide and being independent of the classical endoplasmic reticulum-Golgi pathway [12]. It is a potent inducer of blood vessel formation, involved in the pathogenesis of several diseases characterized by abnormal neovascularization [24] and in a cancer [25, 26], as well as in development and differentiation of various tissues [27, 28], including the nervous system [28]. In addition to the 18-kD peptide, larger isoforms of FGF-2, ranging in size from 22 to 34 kD, and with different sub-cellular localization have been described [28]. While the 18-kD isoform is cytoplasmic and secreted, the larger isoforms, that derive from unusual translation initiation at CUG codons, have been reported to be predominantly intracellular and, in particular, nuclear [28].
Like FGF-2, VEGF, also known as vascular permeability factor (VPF), is a potent mitogen of endothelial cells, involved both in pathological angiogenic processes and in the embryonic development and differentiation of the vascular system [29]. VEGF expression is regulated not only at the transcriptional level but also at a post-transcriptional level, by alternative splicing, which generates in human beings 5 isoforms (of 121, 145, 165, 189 and 206 residues, respectively). The major isoform is VEGF165, a basic, heparin-binding glycoprotein, existing as a homodimer of 45 kD. It has been reported that VEGF121 is a freely soluble protein, while VEGF165, even if also secreted, tends to remain “bound” to the ECM [for review, see 29]. Interestingly, VEGF189 and VEGF206 seem to be almost completely sequestered in the ECM [30]. In addition to these shorter isoforms, a further high molecular weight isoform (large VEGF), containing an N-terminal extension of about 200 residues and with a monomeric mass of about 45 kD, has been demonstrated in human beings and mouse [31, 32]. This isoform is generated by an unusual translation initiation process, which, like in the case of FGF-2 larger isoforms, occurs at one of four potential CUG translation initiation codons, positioned in between two ribosome internal entry sites (IRES), and in frame with the classical AUG start codon [31, 32]. Complexity of this family of growth factors is further increased by proteolitic cleavage of the longer isoform into N-terminal and C-terminal fragments [31, 32] and glycosylation [31, 33].
Several lines of evidence indicate that VEGF influences early stages of vascularization of the developing brain and that VEGF inactivation in the nervous system results in abnormalities of vessel density and cell number in the cerebral cortex. Moreover, it was reported that reduced levels of VEGF caused ALSlike motoneuron degeneration in mice [reviewed in 34]. This latter finding suggested that, in addition to its specific angiogenic action, VEGF might also have neuroprotective functions.
FGF-2 was also demonstrated to play a role in hippocampal neurogenesis [35] and to have neurotrophic functions both in vitro and in vivo[see, e.g. 36–38]. Interestingly, there are also evidence of a cross-talk between FGF-2 and different members of the VEGF family during angiogenesis and vasculogenesis [39].
In this paper, we report that neurons synthesize growth factors FGF-2 and VEGF and release them at least in part via extracellular vesicles.
Materials and methods
Animals
Wistar rats (Stefano Morini, San Polo d’Enza, Italy) were housed in the institutional animal care facility of the Department of “Biologia Cellulare e Sviluppo,” (University of Palermo, Palermo, Italy), under direction of a licensed veterinary who approved protocols. Procedures involving animals were conducted according to the European Community Council Directive 86/609, OJL 358 1, December 12, 1987. The number of animals used has been minimized as well as their suffering.
Cell cultures
Neurons were purified from fetal rat cortices at the 16th day of gestation and cultured in serum-free Maat medium (MM) [40], on laminin (2.5 μg/cm2), as already described [41]. In order to induce cell death, some neuronal cultures were incubated for 24 hrs with camptothecin (Sigma-Aldrich, MO, USA), at a final concentration of 10 μM [42]. Cell apoptosis was observed by staining the cells with a combination of the fluorescent DNA-binding dyes, acridine orange (AO) and ethidium bromide (EB), 100 μg/ml in PBS, as previously described [6]. The differential uptake of these two dyes allowed the identification of viable and non-viable cells by fluorescence microscopy: normal nuclei in alive cells should appear bright green, while apoptotic nuclei in dead cells should appear bright orange/red. In many cases, nuclei of dead cells are fragmented into apoptotic bodies.
Methionine 35S labelling of neurons
Neurons, previously cultured for 15 days in Maat medium (MM), were fed with methionine-free MM containing 0.4 μl of 35S-methionine (10 μCi/μl)/ml of medium for 24 hrs. At the end of treatment, the 35S-methionine-containing medium was washed away and neurons were cultured for 24 or 48 hrs in unlabelled MM. Conditioned medium was finally collected and centrifuged to obtain labelled vesicles. When the last conditioned medium was collected, cells were also lysed to obtain total labelled cell proteins.
Vesicle purification from conditioned medium
Vesicles were purified from conditioned media as described elsewhere [11]. Briefly, media conditioned for 3–24 hrs by neurons were centrifuged at 2,000 ×g for 10 min and 4,000 ×g for 15 min. The first centrifugation step allows pelletting a few entire cells, still present in the medium; the second centrifugation step also removes big cell fragments and nuclei deriving from broken cells. The supernatant was finally centrifuged at 105,000 ×g for 90 min. Pelleted vesicles were resuspended in PBS.
Western analyses
Neurons were homogenized in homogenization buffer (0.32 M sucrose; 50 mM sodium phosphate buffer, pH 6.5; 50 mM KCl, 0.5 mM spermine; 0.15 mM spermidine; 2 mM EDTA, and 0.15 mM EGTA), containing the protease inhibitors aprotinin (2 μg/ml), antipain (2 μg/ml), leupeptin (2 μg/ml), pepstatin A (2 μg/ml), benzamidine (1.0 mM) and phenylmethylsulfonyl fluoride (1.0 mM), all purchased from Sigma-Aldrich. Proteins (20 μg of total cell extracts) were separated by electrophoresis on denaturing 10% polyacrylamide slab gels (SDS-PAGE) and immunoblotted as described elsewhere [1]. Immunostaining of membranes was done with rabbit polyclonal anti-human VEGF165 antibodies (Calbiochem, Darmstadt, Germany) that recognize all the known VEGF isoforms, and mouse monoclonal antibovine purified FGF-2 antobodies (Upstate, Charlottesville, VA, USA) that recognize all the known isoforms of FGF-2.
Scanner electron microscopy (SEM)
Neurons were cultured on coverslips for 15 days. Cells were rinsed with PBS at 37°C and fixed with 4% glutaraldehyde in MM for 10 min. After rinsing with MM, cells were dehydrated by a series of washes in aceton/water at increasing percentage of aceton (15%, 30%, 50% 75% and 100%; 3 min each). Coverslips were finally dried in air, coated with gold and observed by a Philips 505 scanner electron microscope.
Immunofluorescence
Neurons were cultured on coverslips for 15 days. Cells were fixed with 96% ethanol, on ice, for 10 min and permeabilized for 5 min with 0.1% Triton X-100, in PBS. Cells were finally incubated with rabbit polyclonal anti-human VEGF165 antibodies (Calbiochem) that recognize all the known VEGF isoforms, mouse monoclonal anti-bovine purified FGF-2 antobodies (Upstate) that recognize all the known isoforms of FGF-2, and goat anti-integrin β-1 (Santa Cruz, CA, USA). In some control experiments, we also used mouse monoclonal anti-neurofilament 68 (NF68)- (Sigma-Aldrich), policlonal rabbit anti-glial fibrillary acidic protein (GFAP)- (Sigma-Aldrich), and mouse monoclonal anti-microtubule accociated protein (MAP)-2 (Sigma- Aldrich) primary antibodies. The secondary antibodies were anti-rabbit-, anti-goat- or antimouse-IgGs, conjugated to rodhamine or to fluoresceine (Promega Corporation, WI, USA). Cells were observed in an Olympus fluoview FV300 confocal microscope, equipped with two laser multipliers. In some cases, cells prepared as described, were finally stained for DNA by treating with Vectashield mounting medium for fluorescence, containing 4′-,6-diamino-2- phenylindole (DAPI; Vector Laboratories, Youngstown, OH, USA) and observed in an Olympus BX-50 microscope (Olympus Italia s.r.l., Segrate, Italy) equipped with Vario Cam B/W camera (Nikon Instruments s.p.a., Calenzano, Italy).
Results
We previously found that, in a three-cell type system of culture, containing neurons, astrocytes and endothelial cells, both neurons and astrocytes influence the ability of endothelial cells to form a barrier with properties resembling those of BBB [3, 4]. As in that system cells were not in physical contact, we deduced that the effects discovered should be due, at least in part, to molecular mechanisms not based on cell-to-cell contacts. Moreover, independent experiments showed that oligodendroglioma cells shed membrane vesicles [6]. Thus, we asked whether extracellular vesicles could be also produced by neurons to address inductive signals to endothelial cells. A further question was which kind of signals neurons deliver to them. As two of the major factors acting on endothelial cells (i.e. FGF-2 and VEGF) have been reported to be released through extracellular vesicles in other cell systems, we looked for the possible existence of extracellular structures, produced by neurons, carrying the two factors. As shown in Fig. 1A, there is evidence that neuronal cultures contain vesicular structures with sizes ranging from 400 to even 2000 nm. Most of these vesicles are in the range of size (100–1000 nm) already reported for the extracellular structures produced by several kinds of viable cells [10–12] and called MVs. As some of the structures observed are very large, we also looked for the presence of whole nuclei or nuclei fragments in them. We found that only intact nuclei, but not the putative MVs, were stained by DAPI (Fig. 1Ab).
1.
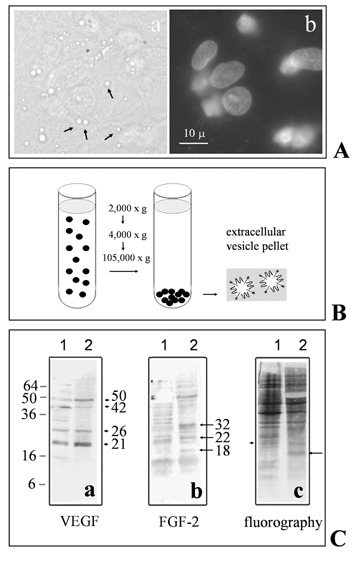
Neurons cultured for 15 days in serum-free Maat Medium release extracellular vesicles that contain both VEGF and FGF-2. A(a): Typical neuronal culture, observed in bright field, showing many extracellular vesicles of different sizes, some of which are indicated by arrows for reference; A(b) immunofluorescent staining of nuclei with DAPI; same field as in A(a). B: Schematic drawing of the protocol used to prepare an extracellular vesicle fraction (according to Dolo et al., 1994). C: Western analysis of total cell lysates (lane 1) and vesicles (lane 2) prepared, as outlined in B, from neurons cultured for 15 days in Maat medium and then labelled with 35S-methionine for 24 hrs. Proteins were immunostained with either rabbit polyclonal anti-human VEGF antibodies (a) or mouse monoclonal anti-FGF-2 (b). After electrophoresis and western blot, the same membranes were also exposed to X-ray sensitive films for 3–5 days: an example of the radioactive protein pattern, 48 hrs after metabolic labelling and medium replacement, is shown in (c). Arrowheads in (a) and arrows in (b) indicate VEGF isoforms and FGF-2 isoforms, respectively. The arrow in (c) indicates the only band that could be interpreted as an FGF-2 isoform. The arrowhead in (c) indicates the only band that should represent a radioactive VEGF isoform.
We then purified the vesicular fraction (Fig. 1B) from the medium conditioned by neurons, according to the protocol used by Dolo et al. (1994) for vesicles released by tumour cells. This fraction, together with total neuronal lysates, was analyzed by Western blot. One example of this analysis is reported in Fig. 1C, which shows that bands of the expected range of size for both VEGF- (Ca: arrowheads) and FGF-2- isoforms (Cb: arrows) are present both in cell lysates and vesicles. Interestingly, the patterns of proteins are not exactly identical when cell lysates and vesicles are compared. In more detail, among the main bands immunostained by anti-VEGF antibodies, two bands of about 21 and 26 kD, respectively, together with one of about 50 kD, are more concentrated in vesicles (lane 2) than in lysates (lane 1), while, on the contrary, a protein with apparent mass of 42 kD is more concentrated in total cell lysates (lane 1) than in vesicles (lane 2). On the other hand, anti-FGF-2 antibodies recognize three main proteins, with apparent masses of about 18, 22 and 32 kD. Among these proteins, two (18 kD and 32 kD) are apparently present only in vesicles (lane 2), while one (22 kD) is present both in cell lysates and vesicles. In order to investigate the fate of vesicle proteins from biosynthesis to appearance in vesicles, neurons at the 15th day of culture were also metabolically labelled with 35S-methionine. After labelling for 24 hrs, the 35S methionine- containing medium was washed away and neurons were cultured for further 24 or 48 hrs, in unlabelled complete Maat Medium. Conditioned medium was finally collected and centrifuged to obtain labelled vesicles. When the last conditioned medium was collected, cells were also lysed to obtain in parallel total labelled cell proteins. As we did not find any difference in the pattern of radioactive proteins, either in cell lysates or in vesicles, at the different times at which media had been collected, only radioactive proteins recovered 48 hrs after incubation with 35S-methionine are shown (Fig. 1Cc). Interestingly, at least one of the putative VEGF- (arrowhead) and one of the putative FGF-2- (arrow) isoforms, respectively, seem to be radioactive (Fig 1Cc).
The fact that neurons produce structures resembling MVs has been also confirmed by scanner electron microscopy (Fig. 2). As shown in Fig. 2B, some of these structures are very large, around 2000 nm (see below).
2.

Analysis by scanner electron microscopy of neuronal cultures. Cells produce and release extracellular vesicular structures (indicated by arrows). A detail (in the white box) of part A was observed at higher magnification (B).
MVs have been reported to originate from domains of the plasma membrane selectively enriched in specific membrane components, such as β1 integrin and membrane-bound gelatinase B/MMP-9 [13]. Therefore we investigated, by double immunofluorescence and confocal microscopy, the possible colocalization of both FGF-2 and VEGF with β1 integrin. The results confirmed, first of all, that neurons produce both FGF-2 and VEGF. As shown in Fig. 3, indeed, neurons were immunostained by both anti-FGF-2 antibodies (b, b*) and anti-VEGF antibodies (e, e*). Second, we found that both molecules are at least in part present in round extracellular structures, emanating from neurons, also stained by anti-β1 integrin antibodies (a, a*; d, d*). The sizes of the extracellular structures, visible in Fig. 3, are comparable with the sizes of those visible in Figs. 1 and 2. We noticed in some cases a tendency of vesicles to cluster around neurites (see, e.g.Fig. 3f*, structures indicated by arrows).
3.

Colocalization of either FGF-2 or VEGF with β1 integrin in extracellular vesicles. Double immunofluorescence was used to co-localize either FGF-2 (b, b*: red) or VEGF (e, e*: red) and β1-integrin (a, a*; d, d*: green) immunoreactivity in neurons cultured for 15 days in Maat Medium. Overlay of FGF-2 and b1-integrin fluorescence is shown in c and c*. Overlay of VEGF and β1-integrin fluorescence is shown in f and f*. a*–f*, enlarged views of the fields boxed in the figures a–f. Cells were observed in an Olympus fluoview FV300 confocal microscope, equipped with two laser multipliers.
As we know that neuronal cultures are contaminated by some rare astrocytes (Fig. 4), we also looked by immunofluorescence at colocalization in the same cell of different neuron-specific cytoskeletal proteins such as NF68 and microtubule-associated protein 2, MAP2) and either FGF-2- or VEGF- immunoreactivity. One example of this kind of analysis was reported in Fig. 5, that shows neurons immunostained for both MAP2 (A: red fluorescence) and VEGF (B: green fluorescence). Extracellular vesicles are clearly seen, attached to MAP2-positive cells.
4.

Different localization of the astrocyte-specific glial fibrillary acidic protein (GFAP, red fluorescence), and neuronspecific neurofilament protein, 68-kD component (NF68, green fluorescence), in cultures of rat cortical neurons.
5.

Localization of MAP-2 (A: red) and VEGF (B: green) immunoreactivity in neurons cultured for 15 days in Maat Medium. Extracellular vesicles are indicated by arrows. Cells were observed in an Olympus BX-50 microscope, equipped with Vario Cam B/W camera.
Finally, as neurons in our cell culture system are post-mitotic and are fed with a serum-free medium, the possibility existed that vesicle production was the effect of cell death. This possibility was also suggested by the large size of vesicles. In order to compare vesicles produced by vital cells with vesicular structures (such as apoptotic bodies), produced by dying cells, we treated neurons with campothecin, an S-phase-specific topoisomerase I inhibitor that has been shown to induce significant, dose-dependent cell death of post-mitotic rat cortical neurons in vitro[42]. Camptothecin-induced neuronal death was already shown to be apoptotic, as characterized by chromatin condensation, cytoplasmic shrinking, plasma membrane blebbing, and fragmentation of neurites [42]. Camptothecin induced massive cell death also in our cell culture system (Fig. 6): in most fields, neurons were fragmented into putative apoptotic bodies (indicated by arrows in Fig. 6B and D). These cell fragments appeared orange/red when cells were stained with a mixture of the fluorescent DNA-binding dyes AO and EO (Fig. 6B). On the contrary, nuclei of control neurons (Fig. 6A) appeared intact and bright green (as expected for alive cells). More important, vesicular structures deriving from cell death were also stained by DAPI (Fig. 6D: structures indicated by arrows), while the putative extracellular vesicles identified by us in normal cultures were not stained (Figs 1A and 6C).
6.
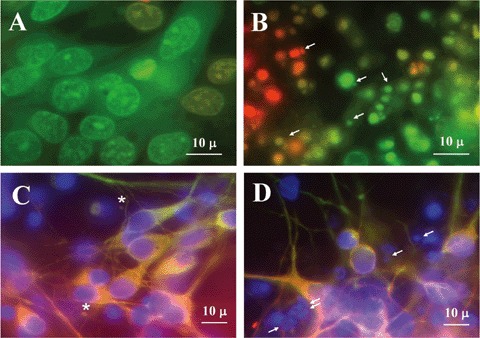
Control- (A and C) and camptothecin-treated (B and D) neurons cultured in Maat Medium for 15 days. Cells were incubated for 24 hrs with 10-mM camptothecin. As camptothecin was dissolved in dimethyl sulfoxide (DMSO), control cells were treated for 24 hrs with an equivalent amount of DMSO alone. A, B: Control cells (A) and cells treated with camptothecin (B), stained with a combination of the fluorescent DNA-binding dyes acridine orange and ethidium bromide, 100 mg each/ml in PBS. C, D: Control cells (C) and cells treated with campthothecin (D), immunostained with anti- MAP2- (green fluorescence) and anti-FGF-2 antibodies (red fluorescence). In addition, all cells were also stained with DAPI. The figure shows the overlay of the three fluorescences. Putative apoptotic bodies in treated cells are indicated by arrows, while putative extracellular vesicles in untreated cells are indicated by an asterisk. Cells were observed in an Olympus BX-50 microscope, equipped with Vario Cam B/W camera.
Discussion
Many independent lines of evidence suggest that shedding of extracellular vesicles is a general route, largely adopted to mediate cell-to-cell communications [7–9]. Although the biochemical mechanisms underlying the phenomenon are largely unknown, some proteins, among which β1 integrin, have been consistently localized to extracellular vesicles [13]. Moreover, it has been suggested that extracellular vesicles can mediate secretion of proteins that lack standard signal sequence and cannot be sorted the endoplamic reticulum. One of the secreted factors that lack a signal sequence is FGF-2, which has been reported to be included into extracellular vesicles shed by tumour cells [12].
We previously reported that RBE4.B brain capillary endothelial cells, cultured on collagen IV, form a barrier with BBB permeability properties, if co-cultured cultured with astrocytes and/or neurons [2–4]. In the co-culture systems, neurons, astrocytes and endothelial cells were not in physical contact. Therefore, we assumed that the effects discovered were due, at least in part, to molecular mechanisms not based on cell–cell contacts.
All of these considerations prompted us to investigate: (i) the possibility that neurons produce extracellular factors responsible for addressing signals to endothelial cells; (ii) the putative presence, among these factors, of angiogenic proteins, such as FGF-2 and VEGF and (iii) possible release of these factors through extracellular vesicles. The study described in this paper demonstrates by different techniques that neurons do shed extracellular vesicles of different sizes, from about 400 nm to over 2000 nm. Because of their dimensions, but also because they contain β1 integrin, these structures should be ranked as MVs.
Their very large size, however, induced us to investigate possible relationships between these structures and structures arising from dead cell fragmentation (e.g. apoptotic bodies). In order to directly compare extracellular vesicles and dead cell fragments, we treated neurons with campothecin, a topisomerase I inhibitor already known to induce apoptosis in postmitotic rat cortical neurons in culture [42]. We then looked by immunofluorescence at both morphological integrity of cell structures (first of all, nuclei and neurites) and the availability of nuclear DNA to be stained by different dyes. We did not find any evidence of DNA presence in extracellular vesicles observed in control cultures, not treated with camptothecin. Moreover, these cells appeared quite intact, with unfragmented nuclei and intricate branches of neurites. Extracellular vesicles shed from these wellshaped, alive cells, were not found to be stained by DAPI (Fig. 1). On the contrary, apoptotic and possibly necrotic cells entirely dissolve into structures that are stained by DAPI. In other words, in this case, there is no evidence of extracellular structures pinching off from otherwise intact cells. These findings corroborate the idea that the phenomenon of shedding associates with viable cells, in accord with previous reports demonstrating that cells are more active in producing extracellular vesicles when they are fed with serum-rich media [7].
We also demonstrate that the extracellular vesicles shed by neurons contain proteins of different sizes which react with either anti-VEGF- or anti- FGF-2-antibodies. Unfortunately, most commercially available antibodies are rinsed against portions of the two growth factors that are present in all the putative isoforms of them; this fact, together with the generally low concentration of the factors and their great variability, results in a complex pattern in the western analyses. Importantly, however, even if some bands of unknown origin are present, the main observable bands show apparent molecular masses compatible with those of VEGF and FGF-2, respectively. Moreover, some of these isoforms are clearly enriched in vesicles respect to cell lysates; curiously, we found in vesicles not only isoforms, such as the 18 kD FGF-2, normally secreted [30] but also isoforms that have been reported to be retained in the cells.
Vesicles released by neurons could interact and/or fuse with other vesicles and/or dock to specific components of ECM, thus releasing the factors to target cells only slowly. Actually, the fate of vesicles in the extracellular environment is not yet clear: they might fuse with target cells, thus directly delivering their contents into the cells or, alternatively, bind to specific membrane receptors or co-receptors, thus eliciting a signal transduction pathway. In any case, tendency of neurons to release extracellular vesicles add new strategies to their communication skills. One could indeed suppose that, beside influencing target cells through traditional transmission by neurotransmitters and neuromodulators at the level of synapses, neurons can also modulate their own survival and functioning by releasing growth factors, that can either directly act on neurons themselves or influence differentiation and functioning of other cell types, first of all astrocytes and endothelial cells. Shedding of growth factor-containing extracellular vesicles might allow protection of the factors in the ECM and/or their modulated release to target cells. In contrast with traditional localized neurotransmission, in the course of our analyses, we observed vesicles pinching off from different sites of neurons, from the cell body to different levels of neurites. It is important to underline that neurons analyzed in this paper are fetal and it cannot be excluded at the moment that their ability to shed extracellular vesicles is bound to their developmental stage. In the future, it will be of great interest to investigate the phenomenon in vivo both in developing and in mature brain, under physiological as well as pathological conditions, such as those that normally involve BBB modifications and neurodegeneration. In order to face these more complex problems, we think that the first step is to understand the fate of secreted VEGF and FGF-2 when vesicles shed by neurons are added to other cell cultures: neurons themselves, astrocytes and/or endothelial cells. An important second step will be to study the effect of extracellular vesicles released by neurons in a model of bloodbrain barrier.
Aknowledgements
The first two authors equally contributed to this paper. We wish to thank Dr. F Carfi Pavia for helpful discussions, advices and assistance with scanner electron microscopy, and Mr. S Agnello for technical assistance with confocal microscopy.
This work was supported by a special grant by Serono S.p.A., by the Italian Ministero dell’Università e della Ricerca (PRIN 2004) and by the University of Palermo (fondi di Ateneo, Università degli Studi di Palermo). P. Proia was supported by a Ph.D. fellowship of the Università degli Studi di Palermo, Palermo, Italy. G. Schiera was supported by a post-doctoral fellowship by Università degli Studi di Palermo, Palermo, Italy.
References
- 1.Savettieri G, Di Liegro I, Catania C, Licata L, Pitarresi GL, D'Agostino S, Schiera G, De Caro V, Giandalia G, Giannola LI, Cestelli A. Neurons and ECM regulate occludin localization in brain endothelial cells. Neuroreport. 2000;11:1081–4. doi: 10.1097/00001756-200004070-00035. [DOI] [PubMed] [Google Scholar]
- 2.Cestelli A, Catania C, D'Agostino S, Di Liegro I, Licata L, Schiera G, Pitarresi GL, Savettieri G, De Caro V, Giandalia G, Giannola LI. Functional feature of a novel model of blood brain barrier: studies on permeation of test compounds. J Control Release. 2001;76:139–47. doi: 10.1016/s0168-3659(01)00431-x. [DOI] [PubMed] [Google Scholar]
- 3.Schiera G, Bono E, Raffa MP, Gallo A, Pitarresi GL, Di Liegro I, Savettieri G. Synergistic effects of neurons and astrocytes on the differentiation of brain capillary endothelial cells in culture. J Cell Mol Med. 2003;7:165–70. doi: 10.1111/j.1582-4934.2003.tb00215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schiera G, Sala S, Gallo A, Raffa MP, Pitarresi GL, Savettieri G, Di Liegro I. Permeability properties of a three-cell type in vitro model of blood-brain barrier. J Cell Mol Med. 2005;9:373–9. doi: 10.1111/j.1582-4934.2005.tb00362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schiera G, Proia P. In vitro models of blood-brain barrier formation and functioning. In: Di Liegro I, Savettieri G, editors. Molecular Bases of Neurodegeneration. KERALA: Research Signpost; 2005. pp. 183–97. [Google Scholar]
- 6.D'Agostino S, Salomone M, Di Liegro I, Vittorelli ML. Membrane vesicles shed by oligodendroglioma cells induce neuronal apoptosis. Int J Oncol. 2006;29:1075–85. [PubMed] [Google Scholar]
- 7.Vittorelli ML. Shed membrane vesicles and clustering of membrane-bound proteolytic enzymes. Curr Top Dev Biol. 2003;54:411–32. doi: 10.1016/s0070-2153(03)54017-0. [DOI] [PubMed] [Google Scholar]
- 8.Pilzer D, Gasser O, Moskovich O, Schifferli JA, Fishelson Z. Emission of membrane vesicles: roles in complement resistance, immunity and cancer. Springer Semin Immunopathol. 2005;27:375–87. doi: 10.1007/s00281-005-0004-1. [DOI] [PubMed] [Google Scholar]
- 9.Whiteside TL. Tumour-derived exosomes or microvesicles: another mechanism of tumour escape from the host immune system. Br J Cancer. 2005;92:209–11. doi: 10.1038/sj.bjc.6602360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dainiak N. Surface membrane-associated regulation of cell assembly, differentiation, and growth. Blood. 1991;78:264–76. [PubMed] [Google Scholar]
- 11.Dolo V, Ginestra A, Ghersi G, Nagase H, Vittorelli ML. Human breast carcinoma cells cultured in the presence of serum shed membrane vesicles rich in gelatinolytic activities. J Submicrosc Cytol Pathol. 1994;26:173–80. [PubMed] [Google Scholar]
- 12.Taverna S, Ghersi G, Ginestra A, Rigogliuso S, Pecorella S, Alaimo G, Saladino F, Dolo V, Dell'Era P, Pavan A, Pizzolanti G, Mignatti P, Presta M, Vittorelli ML. Shedding of membrane vesicles mediates fibroblast growth factor-2 release from cells. J Biol Chem. 2003;278:51911–9. doi: 10.1074/jbc.M304192200. [DOI] [PubMed] [Google Scholar]
- 13.Dolo V, Ginestra A, Cassara D, Violini S, Lucania G, Torrisi MR, Nagase H, Canevari S, Pavan A, Vittorelli ML. Selective localization of matrix metalloproteinase 9, beta1 integrins, and human lymphocyte antigen class I molecules on membrane vesicles shed by 8701-BC breast carcinoma cells. Cancer Res. 1998;58:4468–74. [PubMed] [Google Scholar]
- 14.Denzer K, Kleijmeer MJ, Heijnen HF, Stoorvogel W, Geuze HJ. Exosome: from internal vesicle of the multivesicular body to intercellular signaling device. J Cell Sci. 2000;113:3365–74. doi: 10.1242/jcs.113.19.3365. [DOI] [PubMed] [Google Scholar]
- 15.Thery C, Boussac M, Veron P, Ricciardi-Castagnoli P, Raposo G, Garin J, Amigorena S. Proteomic analysis of dendritic cell-derived exosomes: a secreted subcellular compartment distinct from apoptotic vesicles. J Immunol. 2001;166:7309–18. doi: 10.4049/jimmunol.166.12.7309. [DOI] [PubMed] [Google Scholar]
- 16.Poste G, Nicolson GL. Arrest and metastasis of blood-borne tumor cells are modified by fusion of plasma membrane vesicles from highly metastatic cells. Proc Natl Acad Sci USA. 1980;77:399–403. doi: 10.1073/pnas.77.1.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ginestra A, La Placa MD, Saladino F, Cassara D, Nagase H, Vittorelli ML. The amount and proteolytic content of vesicles shed by human cancer cell lines correlates with their in vitro invasiveness. Anticancer Res. 1998;18:3433–7. [PubMed] [Google Scholar]
- 18.Taylor DD, Taylor CG, Jiang CG, Black PH. Characterization of plasma membrane shedding from murine melanoma cells. Int J Cancer. 1988;41:629–35. doi: 10.1002/ijc.2910410425. [DOI] [PubMed] [Google Scholar]
- 19.Angelucci A, D'Ascenzo S, Festuccia C, Gravina GL, Bologna M, Dolo V, Pavan A. Vesicle-associated urokinase plasminogen activator promotes invasion in prostate cancer cell lines. Clin Exp Metastasis. 2000;18:163–70. doi: 10.1023/a:1006778000173. [DOI] [PubMed] [Google Scholar]
- 20.Taraboletti G, D'Ascenzo S, Borsotti P, Giavazzi R, Pavan A, Dolo V. Shedding of the matrix metalloproteinases MMP-2, MMP-9, and MT1-MMP as membrane vesicle-associated componente by endothelial cells. Am J Pathol. 2002;160:673–80. doi: 10.1016/S0002-9440(10)64887-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ancellin N, Colmont C, Su J, Mittereder N, Chae SS, Stefansson S, Liau G, Hla T. Extracellular export of sphingosine kinase-1 enzyme. Sphingosine 1-phosphate generation and the induction of angiogenic vascular maturation. J Biol Chem. 2002;22:6667–75. doi: 10.1074/jbc.M102841200. [DOI] [PubMed] [Google Scholar]
- 22.Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4:397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- 23.Taraboletti G, D'Ascenzo S, Giusti I, Marchetti D, Borsotti P, Millimaggi D, Giavazzi R, Pavan A, Dolo V. Bioavailability of VEGF in tumor-shed vesicles depends on vesicle burst induced by acidic pH. Neoplasia. 2006;8:96–103. doi: 10.1593/neo.05583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Slavin J. Fibroblast growth factors: at the heart of angiogenesis. Cell Biol Int. 1995;19:431–44. doi: 10.1006/cbir.1995.1087. [DOI] [PubMed] [Google Scholar]
- 25.Folkman J, Shing Y. Angiogenesis. J Biol Chem. 1992;267:10931–4. [PubMed] [Google Scholar]
- 26.Nguyen M, Watanabe H, Budson AE, Richie JP, Hayes DF, Folkman J. Elevated levels of an angiogenic peptide, basic fibroblast growth factor, in the urine of patients with a wide spectrum of cancers. J Natl Cancer Inst. 1994;86:356–61. doi: 10.1093/jnci/86.5.356. [DOI] [PubMed] [Google Scholar]
- 27.Basilico C, Moscatelli D. The FGF family of growth factors and oncogenes. Adv Cancer Res. 1992;59:115–65. doi: 10.1016/s0065-230x(08)60305-x. [DOI] [PubMed] [Google Scholar]
- 28.Bikfalvi A, Klein S, Pintucci G, Rifkin DB. Biological roles of fibroblast growth factor-2. Endocr Rev. 1997;18:26–45. doi: 10.1210/edrv.18.1.0292. [DOI] [PubMed] [Google Scholar]
- 29.Ferrara N, Davis-Smith T. The biology of Vascular Endothelial Growth Factor. Endocr Rev. 1997;18:4–25. doi: 10.1210/edrv.18.1.0287. [DOI] [PubMed] [Google Scholar]
- 30.Park JE, Keller GA, Ferrara N. The vascular endothelial growth factor (VEGF) isoforms: differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol Biol Cell. 1993;4:1317–26. doi: 10.1091/mbc.4.12.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huez I, Bornes S, Bresson D, Créancier L, Prats H. New vascular endothelial isoform generated by internal ribosomal entry site-driven CUG translation initiation. Mol Endocrinol. 2001;15:2197–210. doi: 10.1210/mend.15.12.0738. [DOI] [PubMed] [Google Scholar]
- 32.Tee MK, Jaffe RB. A precursor form of vascular endothelial growth factor arises by initiation from an upstream in-frame CUG codon. Biochem J. 2001;359:219–26. doi: 10.1042/0264-6021:3590219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brandner B, Kurkela R, Vihko P, Kungl AJ. Investigating the effect of VEGF glycosylation on glycosaminoglycan binding and protein unfolding. Biochem Biophys Res Commun. 2006;340:836–9. doi: 10.1016/j.bbrc.2005.12.079. [DOI] [PubMed] [Google Scholar]
- 34.Iambrechts D, Storkebaum E, Carmeliet P. VEGF: necessary to prevent motoneuron degeneration, sufficient to treat ALS. Trends Mol Med. 2004;10:275–82. doi: 10.1016/j.molmed.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 35.Palmer TD, Markakis EA, Willhoite AR, Safar F, Gage FH. Fibroblast growth factor-2 activates a latent neurogenic program in neural stem cells from diverse regions of the adult CNS. J Neurosci. 1999;19:8487–97. doi: 10.1523/JNEUROSCI.19-19-08487.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lenhard T, Schober A, Suter-Crazzolara C, Unsicker K. Fibroblast growth factor-2 requires glial-cell-line derived neurotrophic factor for exerting its neuroprotective actions on glutamate-lesioned hippocampal neurons. Mol Cell Neurosci. 2002;20:181–97. doi: 10.1006/mcne.2002.1134. [DOI] [PubMed] [Google Scholar]
- 37.Rao MS, Hattiangady B, Shetty AK. Fetal hippocampal CA3 cell grafts enriched with FGF-2 and BDNF exhibit robust long-term survival and integration and suppress aberrant mossy fiber sprouting in the injured middle-aged hippocampus. Neurobiol Dis. 2006;21:276–90. doi: 10.1016/j.nbd.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 38.Johnson-Farley NN, Patel K, Kim D, Cowen DS. Interaction of FGF-2 with IGF-1 and BDNF in stimulating Akt, ERK, and neuronal survival in hippocampal cultures. Brain Res. 2007;1154:40–9. doi: 10.1016/j.brainres.2007.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Presta M, Dell'Era P, Mitola S, Moroni E, Ronca R, Rusnati M. Fribloblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev. 2005;16:159–78. doi: 10.1016/j.cytogfr.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 40.Cestelli A, Savettieri G, Ferraro D, Vitale F. Formulation of a novel synthetic medium for selectively culturing rat CNS neurons. Dev Brain Res. 1987;22:219–27. doi: 10.1016/0165-3806(85)90173-7. [DOI] [PubMed] [Google Scholar]
- 41.Savettieri G, Licata L, Catania C, Raneri R, Di Liegro I, Cestelli A. Synergistic effects of laminin and thyroid hormones on neuron polarity in culture. NeuroReport. 1999;10:1269–72. doi: 10.1097/00001756-199904260-00021. [DOI] [PubMed] [Google Scholar]
- 42.Morris EJ, Geller HM. Induction of neuronal apoptosis by camptothecin, an inhibitor of DNA topoisomerase- I: evidence for cell cycle-independent toxicity. J Cell Biol. 1996;134:757–70. doi: 10.1083/jcb.134.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]