

Abstract
Folate-mediated one-carbon metabolism (FOCM) is associated with risk for numerous pathological states including birth defects, cancers, and chronic diseases. Although the enzymes that constitute the biological pathways have been well described and their interdependency through the shared use of folate cofactors appreciated, the biological mechanisms underlying disease etiologies remain elusive. The FOCM network is highly sensitive to nutritional status of several B-vitamins and numerous penetrant gene variants that alter network outputs, but current computational approaches do not fully capture the dynamics and stochastic noise of the system. Combining the stochastic approach with a rule-based representation will help model the intrinsic noise displayed by FOCM, address the limited flexibility of standard simulation methods for coarse-graining the FOCM-associated biochemical processes, and manage the combinatorial complexity emerging from reactions within FOCM that would otherwise be intractable.
INTRODUCTION
Systems biology aims to develop a systemlevel description and understanding of biological phenomena.1–3 Advances in software and computational power, coupled with the availability of high-throughput data, have stimulated the application of simulation-based approaches that describe and predict the function and dynamics of biological systems, as well as their relationship to human physiology and pathophysiology (i.e., computational systems biology).1 Folate-mediated one-carbon metabolism (FOCM) has been an attractive network for systems modeling because: (1) the enzymes that constitute the biological pathways have been well described; (2) the metabolic pathways are interrelated through their shared use of folate cofactors, and therefore computational approaches enable detailed understand of the FOCM network and the interconnectedness of its pathways; (3) the FOCM network is highly sensitive to nutritional status of several vitamins (folate and vitamins B12, B6, and B2) and numerous penetrant gene variants that alter network outputs; and (4) numerous pathological states with unknown etiologies are associated with perturbations in this network. Although considerable research has elucidated biochemical details of FOCM, most studies have focused primarily on single reactions or pathways in isolation, failing to capture the overall functioning of the system. Mathematical modeling has proven to be a powerful tool for filling this gap. However, this approach can be limited by incomplete knowledge of the system that can impair its realistic description, and by drawbacks related to the coherence of the data used in the model, an issue that can affect the reliability of outcomes and predictions. These uncertainties include cell-type specific variations as well as the effect of multienzyme complex formation, referred to as metabolons, on substrate stability, metabolite channeling, and the regulation of pathway fluxes and efficiency within the FOCM network. Moreover, the almost exclusive use of a deterministic approach in modeling FOCM cannot capture the stochastic noise of the biological system.4 In this review, we highlight the major challenges to constructing models, using FOCM as an illustrative example.
OVERVIEW OF FOCM
Folate-mediated FOCM functions in the cytoplasm, mitochondria, and nucleus (Figure 1). In the cytoplasm, FOCM has been modeled as a network of three interdependent pathways involved in the de novo synthesis of purine nucleotides and thymidylate (dTMP), as well as the remethylation of homocysteine (HCY) to methionine (MET). The MET is an essential amino acid and is required for the initiation of protein synthesis. It can also be converted to S-adenosylmethionine (SAM), which functions as an intermediate for polyamine synthesis as well as a cofactor and methyl group donor for over 50 methylation reactions.5 These reactions include the methylation of chromatin (CpG islands in DNA and histone proteins), RNA, phospholipids, neurotransmitters and other small molecules, phosphatidylcholine, and numerous proteins. The products of these SAM-dependent methylations are involved in the regulation of fundamental biological processes including transcription, translation, signaling,6 protein localization,7 and metabolism.8 In the nucleus, folate cofactors are required for the synthesis of dTMP9 and may participate in KDM1-catalyzed histone demethylation.10 In mitochondria, formate is generated from the catabolism of the amino acids serine, glycine, sarcosine, and dimethylglycine. Mitochondrially-derived formate is the primary source of one-carbons for nuclear and cytoplasmic FOCM,11 although folate-activated one-carbons can be derived in the cytoplasm from the catabolism of histidine, purines, and serine (Figure 1).5
FIGURE 1.
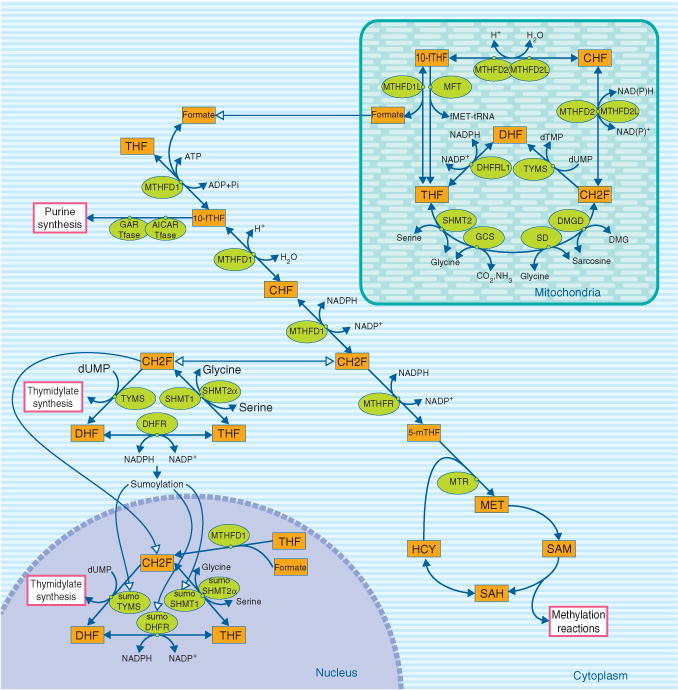
Compartmentation of folate-mediated one-carbon metabolism (FOCM) in the cytoplasm, mitochondrion, and nucleus. FOCM in the cytoplasm is required for the de novo synthesis of purines and thymidylate (dTMP), and for the remethylation of homocysteine to methionine. FOCM in the nucleus synthesizes dTMP from deoxyuridylate (dUMP) and serine. FOCM in mitochondrion is required to generate formate for FOCM in the cytoplasm, to generate and/or catabolize the amino acid glycine, and to synthesize dTMP. Enzyme abbreviations: AICAR Tfase, phosphoribosylaminoimidazolecarboxamide formyltransferase; CHF, 5,10-methenyltetrahydrofolate; DHFR, dihydrofolate reductase; GAR Tfase, phosphoribosylglycinamide formyltransferase; GCS, glycine cleavage system; 5-mTHF, 5-methyltetrahydrofolate; MTHFD, methylenetetrahydrofolate dehydrogenase which may contain up to three enzymatic activities depending on the specific isozyme (see Figure 4): methenyltetrahydrofolate cyclohydrolase, 10-formyltetrahydrofolate synthetase and methylenetetrahydrofolate dehydrogenase activities; MTHFR, methylenetetrahydrofolate reductase; MTR, methionine synthase; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine; SHMT, serine hydroxymethyltransferase; THF, tetrahydrofolate; TYMS, thymidylate synthase; 10-fTHF, 10-formyltetrahydrofolate.
PATHWAYS AND THEIR SUBCELLULAR LOCALIZATION
FOCM in the Cytoplasm and Nucleus
The enzymes that constitute three folate-dependent biosynthestic pathways of de novo purine biosynthesis, de novo dTMP biosynthesis, and HCY remethylation have been described as an interconnected FOCM network.5,11,12 These enzymes are present in the cytoplasm and are assumed to compete for a limiting pool of folate cofactors within the network, as the concentration of folate enzymes exceeds intracellular folate levels.5,13 However, more recent studies have demonstrated the formation of multienzyme complexes by enzymes that constitute individual FOCM pathways and undergo dynamic physical compartmentation away from other folate-dependent enzymes.14 Complex formation may be required for pathway function.14 Furthermore, the formation of multienzyme complexes and their physical compartmentation exhibit cell-cycle dependence, indicating that pathways within the network may be both spatially and temporally isolated from each other.15,16 These newer studies call into question an equilibrium model whereby individual pathways are tightly interconnected through direct competition for a limiting pool of folate cofactors. The dynamic assembly of FOCM pathways into compartmentalized metabolic complexes adds additional dimensions and complexity to regulation of these pathways, including the necessity to regulate the trafficking of folate cofactors among compartmentalized pathways and within multienzyme complexes.14
Purine Biosynthesis
Folate-dependent de novo purine nucleotide biosynthesis involves a 10-step pathway (Figure 2). Two of these reactions are dependent on the folate cofactors 10-formylTHF, which donate the #2 and #8 carbon to the purine ring in reactions catalyzed by glycinamide ribonucleotide transformylase (GAR Tfase) and aminoimidazolecarboxamide ribonucleotide transformylase (AICAR Tfase), respectively.17 The 10 reactions are catalyzed by six proteins that form a multienzyme complex, or metabolon, in the cytoplasm that is referred to as the purinosome (Figure 1).17 The formation of the purinosome is dynamic: it disassembles when exposed to exogenous sources of purines nucleosides or casein kinase II (CK2) inhibitors.18 Moreover, the complex is present only during G1, indicating that de novo purine biosynthesis is cell cycle-dependent.16 Although it has not been definitively established if de novo purine nucleotide biosynthesis is completely dependent on the formation of the purinosome, the essentiality of complex formation for pathway function is consistent with established knowledge of the regulation of the folate-dependent de novo pathway by the folate-independent salvage pathway.19 Therefore, de novo purine biosynthesis may not be as tightly connected to other pathways in the FOCM metabolic network as previously thought, as it exhibits both physical and temporal isolation from the other folate-dependent pathways.
FIGURE 2.
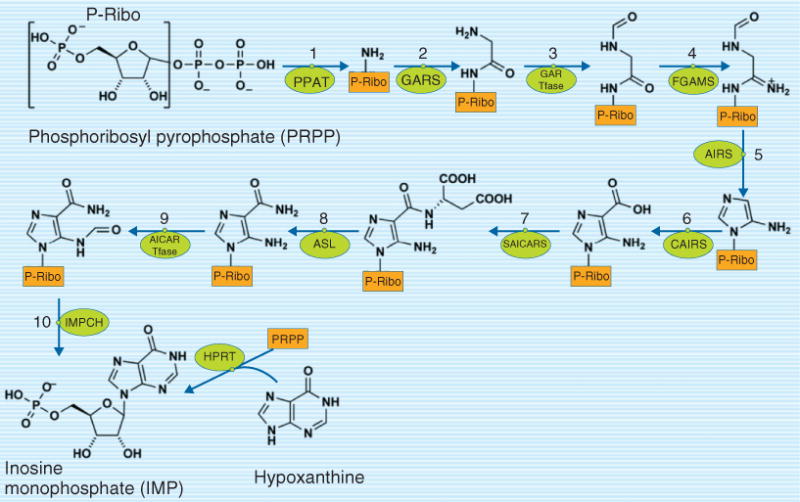
De novo purine nucleotide biosynthesis is a 10-step pathway in the cytoplasm that functions as a multienzyme complex referred to as a purinosome. Reactions 3 and 9 require 10-formyltetrahydrofolate as a cofactor. Enzyme abbreviations: AICAR Tfase, phosphoribosylaminoimidazole carboxamide formyltransferase; AIRS, aminoimidazole ribonucleotide synthetase; ASL, adenylosuccinate lyase; CAIRS, carboxyaminoimidazole ribonucleotide synthase; FGAMS, phosphoribosylformylglycinamidine synthase; GARS, glycinamide ribonucleotide synthetase; GAR Tfase, phosphoribosylglycinamide formyltransferase; HPRT, hypoxanthine phosphoribosyl transferase; IMPCH, IMP cyclohydrolase; PPAT, PRPP amidotransferase; SAICARS, succinylaminoimidazolecarboxamide ribonucleotide synthetase.
dTMP Biosynthesis
Folate-dependent de novo dTMP synthesis involves the methylation of deoxyuridylate (dUMP) catalyzed by enzymes thymidylate synthase (TYMS), dihydrofolate reductase (DHFR), two serine hydoxymethyltransferase isozymes (SHMT1 and SHMT2α),20 and trifunctional enzyme methyleneTHF dehydrogenase (MTHFD1). 5,10-MethyleneTHF is the required one-carbon donor for the TYMS-catalyzed conversion of dUMP to dTMP and DHF; in this reaction 5,10-methyleneTHF serves as both a one-carbon donor and source of reducing equivalents.5 THF is regenerated from DHF through the activity of the NADPH-dependent enzyme DHFR, and the pathway is completed by the generation of 5,10-methyleneTHF from THF, a reaction catalyzed by the enzyme serine hydoxymethyltransferase (SHMT1 and SHMT2α). Stable isotope tracer studies indicate that the one-carbon of 5,10-methyleneTHF is derived either from formate, ATP, NADPH, and THF through the three enzymatic activities of MTHFD1 or, alternatively, from serine through the vitamin B6-dependent activity of SHMT, which catalyzes the transfer of the hydroxymethyl group of serine to THF to generate glycine and 5,10-methyleneTHF (Figure 1).9,21 The de novo dTMP biosynthesis is known to be cell cycle regulated,22 but the mechanisms have only been recently understood.15 SHMT, DHFR, and TYMS are present in the cytoplasm, but not in the nucleus at G1. At the transition to S-phase,23 or as a result of UV exposure,24 a significant percentage of these enzymes undergo modification by the small ubiquitin modifier (SUMO), which enables their translocation to the nucleus where they form a multienzyme complex and synthesize dTMP from dUMP at the replication fork.9 MTHFD1 is also translocated to the nucleus at G1.9 Both SHMT and MTHFD1 contribute one-carbons for dTMP synthesis in the nucleus, but tracer studies indicate that MTHFD1 is the primary source of 5,10-methyleneTHF for dTMP synthesis.21 SHMT plays an essential, noncatalytic role as a scaffold protein that enables assembly of the enzymes involved in the dTMP synthesis pathway at the replication fork.9 Nuclear localization of the dTMP synthesis pathway is essential to prevent uracil incorporation into nuclear DNA, but it is not clear if de novo dTMP synthesis occurs exclusively in the nucleus or if there is residual activity in the cytoplasm.25 Nonetheless, it is apparent that de novo purine and de novo dTMP biosynthesis do not directly compete within the one-carbon metabolic network as they function in different subcellular compartments and during different stages of the cell cycle.
HCY Remethylation
The remethylation of HCY to MET is catalyzed by the folate- and vitamin B12-dependent enzyme methionine synthase (MTR). The one-carbon donor of the reaction is 5-methylTHF (5mTHF), which is generated by the reduction of 5,10-methyleneTHF in an NADPH-dependent reaction catalyzed by methyleneTHF dehydrogenase (MTHFR; Figure 1). Formate is the primary source of one-carbons for MET synthesis.12 This is the only FOCM biosynthetic pathway that has not been reported to form a multienzyme complex or show cell-cycle dependence, but requires the activities of MTHFD1, MTHFR, and MTR to convert formate and HCY to MET.14
The MTR-catalyzed reaction plays two other important functions in addition to generating MET; it consumes: (1) HCY, a toxic intermediate in the HCY remethylation cycle and (2) 5mTHF, a folate cofactor which can accumulate at the expense of other one-carbon forms of folate and thereby impair the reaction velocities throughout the folate cycle in case of its accumulation. Because the MTHFR-catalyzed reaction is essentially irreversible in vivo, loss of MTR activity, which can occur in vitamin B12 deficiency, results in the accumulation of cellular folate as 5mTHF, otherwise referred to as a folate methyl trap. The methyl trap impacts all cellular folate-dependent pathways and enzymes by removing other one-carbon folate forms from the cofactor pool.26
Other folate interconverting and binding enzymes
The cytoplasm also contains enzymes that intercovert the formyl folate forms and may function to balance the distribution of the one-carbon forms of folate (Figure 3). 10-FormylTHF dehydrogenase (FDH) catalyzes the removal of the formyl group from 10-formylTHF to generate THF and CO2. SHMT and methenylTHF synthetase (MTHFS) constitute a futile cycle that interconverts 5-formylTHF and 10-formylTHF. 5-FormylTHF is not a cofactor (one-carbon donor) but rather serves as a storage form of THF and is an inhibitor of folate-dependent enzymes.27,28
FIGURE 3.

Regulation of cytoplasmic folate-activated one-carbon pools. The relative distribution of one-carbon activated forms of THF is regulated by folate-binding proteins and one-carbon depleting reactions. FDH, 10-formyltetrahydofolate dehydrogenase ; MTHFS, methenyltetrahydrofolate synthetase; SHMT, serine hydroxymethyltransferase; GNMT, glycine N-methyltransferase. THF, tetrahydrofolate; 10-fTHF, 10-formyltetrahydrofolate; CHF, 5,10-methenyltetrahydrofolate; 5-mTHF, 5-methyltetrahydrofolate.
There are many enzymes that bind tightly but do not metabolize folate cofactors, such as MTHFS with 10-formylTHF, SHMT with 5-formylTHF, FDH with THF and glycine-N-methyltransferase (GNMT), an enzyme that catalyzes the SAM-dependent methylation of glycine to sarcosine, with 5mTHF. In all cases, the bound folate inhibits the activity of these proteins. However, these folate tight-binding proteins may serve other functions, including sequestering folate to regulate flux through the pathways and/or traffic folate cofactors among the folate-dependent pathways.14
FOCM in Mitochondria
The FOCM in mitochondria functions to generate formate for cytoplasmic one-carbon metabolism from the catabolism of the amino acids serine, glycine, dimethylglycine, and sarcosine; to synthesize glycine from serine; to generate fMet-tRNA for mitochondrial protein synthesis initiation; and to conduct de novo dTMP synthesis from dUMP (Figure 1).11,29 Formate synthesized in mitochondria is subsequently transported to the cytoplasm and nucleus. Unlike nuclear de novo dTMP synthesis which is cell cycle regulated,15 de novo dTMP synthesis in mitochondria and mitochondrial DNA replication is neither linked to cell cycle nor nuclear DNA replication, and occurs in both proliferating and nonproliferating cells.30
The potential sources of one-carbons generated in mitochondria in the form of formate differ among cell types. With the exception of red blood cells, all cells can convert serine to glycine and formate, whereas the generation of formate from glycine, sarcosine, and dimethylglycine is cell-type restricted and may be limited to liver, kidney, stem cells, and other undifferentiated cell types. All THF-dependent catabolism of amino acids generates 5,10-methyleneTHF, which is oxidized to 10-formylTHF by the bifunctional enzymes MTHFD231,32 and MTHFD2L.33 MTHFD1L hydrolyzes 10-formylTHF to form THF and formate in an ATP-generating reaction.12,34 Formate exits the mitochondria by an unknown mechanism to the cytoplasm to support nucleotide biosynthesis and HCY remethylation (Figure 1).11
SUBCELLULAR DISTRIBUTION OF FOLATE COFACTORS
A few studies have examined the subcellular distribution of total folate and individual folate one-carbon forms. A study of isolated rat liver indicated that the nucleus contains about 10% of total cellular folate,35 although the one-carbon distribution of folate cofactor forms has not been investigated. Nothing is known about the consequences of nuclear dTMP synthesis metabolon formation on: (1) transport, processing, and accumulation of folates into the nucleus; (2) the forms of folates localized in the nucleus, and (3) the regulation of dTMP biosynthesis by folate availability throughout the cell cycle.14 To elucidate whether an active transport is required for accumulating folates into the nucleus, and investigate the possible role of SUMOylation, we represent some of the next challenges to understanding FOCM. Mitochondria constitute as much as 40% of total cellular folate,35,36 with 10-formylTHF as the major form. This indicates that the generation of formate from 10-formylTHF is the limiting step in mitochondrial FOCM. THF monoglutamates are transported into mitochondria by SLC25A3237 and must be converted to THF polyglutamates to be retained within mitochondria; they form a distinct cofactor pool that is not in equilibrium with THF polyglutamates in the cytoplasm.36
CHANNELING IN FOCM
Substrate channeling refers to the direct transfer of substrates and products between sequential enzymes in a pathway without complete mixing with the bulk solvent.38 Channeling can confer unique properties to a metabolic pathway or network, including stabilization of labile metabolic intermediates, regulation of metabolite partitioning at branch points, driving of unfavorable equilibrium and kinetics associated with a particular enzyme, and acceleration of overall flux through a pathway.39 Formation of a metabolon, a multienzyme complex involving enzyme components of a metabolic pathway, is generally considered necessary but not sufficient for metabolic channeling to occur. Channeling can be ‘perfect’, meaning that pathway intermediates are transferred directly between a donor and acceptor enzyme without its diffusion into the bulk phase, or can be ‘leaky’, in which case the donor and acceptor enzymes are located in close proximity to one another, allowing local concentrations of intermediates to accumulate with high probability that the acceptor enzyme will bind the metabolite generated by the donor. For an excellent review on this subject, see Spivey and Ovadi.39
As detailed above, the enzymes involved in folate-dependent de novo purine and de novo dTMP biosynthesis form metabolons in the cytoplasm16,17 and nucleus,20 respectively. Evidence for metabolic channeling of folate cofactors has been observed between active sites of multifunctional enzymes involved in FOCM, as well as between active sites of monofunctional enzymes that function sequentially in a pathway. Transfer of 5,10-methenylTHF between the active sites of the bifunctional methyleneTHF dehydrogenase/cyclohydrolase enzyme occurs with 50% efficiency when the enzyme reaction proceeds in the forward direction. This leaky channeling has been attributed to movement of the folate cofactor between two proximal and overlapping active sites that have different affinities for the methenylTHF cofactor that drives the channeling.40 The bifunctional 5-amino-4-imidazolecarboxamide ribonucleotide transformylase/IMP cyclohydrolase (ATIC) enzyme, which sequentially catalyzes the final two steps in IMP biosynthesis, does not exhibit metabolic channeling between the two active sites (Figure 2). However, the rate-limiting step in the transformylase reaction is the release of THF from the enzyme,41 and this off rate could be enhanced by channeling the THF product to a donor tetrahydrofolate-binding protein within the purinosome, as has been observed for other folate-dependent enzymes.42 Such kinetic enhancement has been demonstrated in vitro by the reconstitution of the folate-dependent formate to serine pathway, which involves two enzymes and four reactions catalyzed by the activities of SHMT and MTHFD1. In one study, the actual rate of the in vitro reconstituted pathway was shown to be five times faster than predicted from deterministic modeling based on the Michaelis–Menten kinetic parameters.43
BIOMARKERS OF IMPAIRED FOCM
There are only a limited number of biomarkers that report on impairments in FOCM. There are no established biomarkers of impaired de novo purine biosynthesis. Z-nucleotides appear in the plasma when 10-formylTHF-dependent formylation of AICAR is incomplete, which occurs as a result of increased rates of de novo purine biosynthesis as observed in Lesch-Nyhan syndrome, but this biomarker is likely not relevant for healthy populations.44
An unfavorable dUTP/dTMP ratio can lead to uracil misincorporation and subsequent accumulation in DNA has been suggested to be a biomarker of impaired de novo dTMP biosynthesis.45 Thymidine is unique from other deoxyribonucleotide precursors for DNA replication because its metabolic precursor, deoxyuridine nucleotides, can be incorporated into DNA during replication and repair.45 Impairments in dTMP synthesis cause uracil accumulation in both nuclear and mitochondrial DNA by affecting the dUTP/dTTP ratio.46–49 Uracil content in DNA is not a specific biomarker of vitamin status, but is responsive to both folate48 and vitamin B12 status.50 However, high levels of folic acid (5 mg/day) and vitamin B12 (1.25 mg/day) supplementation has been shown to increase uracil DNA levels in the rectal mucosa,51 and levels of uracil accumulation in DNA differ by tissue in mice,52 raising concerns about the utility of this biomarker as both an indicator of vitamin status and de novo dTMP synthesis functionality.
There are several robust biomarkers for HCY remethylation function. These include plasma levels of SAM, HCY and S-adenosylhomocysteine (SAH).21,53 However, these biomarkers are sensitive to both folate and vitamin B12 levels as well as genetic variation. A common genetic variant in the MTHFR gene (677 C→T) encodes an enzyme that exhibits less MTHFR enzymatic activity due to thermolability of the enzyme,54 resulting in lower 5mTHF pools,55 lower cellular folate concentrations,55 and elevated plasma total HCY.56 Other biomarkers of HCY remethylation function include CpG DNA methylation levels and protein methylation (including histones), which affect gene expression and DNA stability.55,57–59
There are no established biomarkers that report on the activity of mitochondrial FOCM. Plasma formate levels may report on mitochondrial formate production60 but its utility as a functional biomarker has not been extensively investigated. Impairment in de novo dTMP synthesis in mitochondria do result in elevated uracil levels in mitochondrial DNA,29 but the utility of uracil in mitochondrial DNA as a robust biomarker of folate nutritional status or de novo dTMP synthesis has yet to be established. Impairment in folate-dependent glycine cleavage activity, as observed in patients with nonketotic hyperglycinemia, results in elevated glycine and HCY levels in cerebral spinal fluid, but these elevations are likely restricted to severe disruptions associated with that inborn error in metabolism.61
DETERMINISTIC APPROACHES TO MODELING FOCM
Deterministic mathematical models of FOCM based on enzyme kinetics and regulatory mechanisms have been developed to describe the network and predict the impact of genetic and nutritional variation on network outputs.62,63 The deterministic approach relies on ordinary differential equations (ODEs, reaction rate equations) where metabolite concentrations are represented in terms of continuous variables.64 These models represent well-defined sets of reactions, with system architecture determined during the phase of model construction (i.e., reaction-based models).65–67 They aim to capture system behavior from reaction velocities, usually described in terms of Michaelis–Menten kinetics (parameters used in these equations take into account the affinities between enzymes and substrates).
The construction of a simulated model of metabolic pathways requires knowledge of the main actors involved (e.g., enzymes and metabolites), their patterns of interaction (e.g., metabolic reactions, inhibition, and activation processes), and their range of concentration. Also an explicit description concerning the spatial distribution of interactions (i.e., to know whether they occur in the nucleus, mitochondria, and/or cytoplasm), the mechanisms of transport and substrate channeling, and the formation of complexes (i.e., enzymes and metabolites composing the complex) further enhance the realism and predictive power of the model. System-level dynamics (e.g., metabolite concentration in a time series) emerge from the interplay between basic components of the system (i.e., reaction velocities), which have been modeled through ODEs. Adoption of a reaction-based approach requires complete knowledge of the system, well-grounded and testable hypotheses regarding the reactions and their elementary mechanism (i.e., mathematical equations describing the velocity for each reaction step), or availability of data describing time-courses of reactants in wet-lab experiments, in order to derive the metabolic model and infer admissible parameter sets.68,69 Therefore, the validity of the model depends on the completeness of existing knowledge, whereas its long-term utility depends on its adaptability in response to new knowledge. Current mathematical models describing the folate and MET metabolism62,70,71 are deterministic. They simulate elementary reaction steps by means of ODEs62,63,67,72 founded on the reaction-based approach. Reaction velocities have been modeled assuming the underlying interactions are adherent to mass-action kinetics (monomolecular, bimolecular) or using higher-level approximations to the elementary interactions (e.g., Michaelis–Menten of first and second order for enzymatic reactions; Hill equation for cooperative binding). Below are several examples of reaction-based equations that have been built into existing models of FOCM and the underlying assumptions upon which the model is constructed.
Example 1: Modeling the Generation of Folate-Activated Formaldehyde as 5,10-MethyleneTHF
The 5,10-MethyleneTHF represents a key substrate in the process of HCY remethylation to MET, de novo synthesis of purine nucleotides and dTMP. It carries a one-carbon at the oxidation level of formaldehyde (Figure 1) and can be generated reversibly through both spontaneous and enzyme-catalyzed reactions in the cytoplasm, nucleus, and mitochondria. In solution, 5,10-methyleneTHF exists in equilibrium with formaldehyde and THF. Through enzyme-mediated catalysis, 5,10-methyleneTHF forms reversibly from THF with amino acids serving as the one-carbon donors (serine, glycine, dimethylglycine, and sarcosine), or can be produced reversibly from THF, ATP, formate, and NADPH (Figure 1). Michaelis–Menten equations of first and second order (one or two reactants, respectively) are the most extensively used functions for modeling enzymatic reactions in FOCM65,67,71 including the generation of 5,10-methyleneTHF by SHMT, dimethylglycine dehydrogenase (DMGD), sarcosine dehydrogenase (SDH), glycine decarboxylase complex (GDC) or MTHFD1. MTHFD1 and SHMT are the two primary enzymes responsible for generating folate-activated one-carbons for purine, dTMP, and HCY remethylation to MET.5,11 MTHFD1 activity produces 5,10-methyleneTHF from formate, ATP, and NADPH through a three-step pathway (Figure 1); the conversion of THF and formate into 10-formylTHF is described by a second-order Michaelis–Menten function, whereas the subsequent reaction velocities are modeled as first-order Michaelis–Menten equations.66 The generation of 5,10-methyleneTHF (CH2F) through MTHFD1 (last step only) and SHMT are described by first- and second-order Michaelis–Menten functions, respectively.66
where Vmax,MTHFD1 = 594,000 μM h−1, Km,CHF = 10 μM73 (CHF = 5,10-methenylTHF); parameters of the second equation are: Vmax,SHMT = 5200 μM h−1, Km,THF = 50μM and Km,Serine = 600 μM.66,73–80 Also described is the nonenzymatic generation of 5,10-methyleneTHF by the condensation of formaldehyde (HCHO) and THF (K1 = 0.03 μM−1 h−1),75,81 adopting a bimolecular kinetics (i.e., two reactants yielding to a singular product).
However, the physiological significance of this spontaneous reaction is probably negligible, as free formaldehyde concentrations are low in the bulk solvent (reaction velocity is proportional to the reactant concentrations), and there are numerous biological amines other than THF that can be modified by formaldehyde. The binding affinities of formaldehyde with THF and other biological amines have not been explicitly modeled and the propensity of the nonenzymatic reaction is simply described by the kinetic parameter. Despite this simplification, outcomes of the Reed et al.66 model indicate that reaction velocities mediated by MTHFD1 and SHMT are much faster than the nonenzymatic reaction, reflecting its trivial contribution to the formation of 5,10-methyleneTHF.
Example 2: Modeling Mitochondrial Deoxyribonucleotide Biosynthesis
Deterministic models of mitochondrial deoxyribonucleotide metabolism and mitochondrial DNA (mtDNA) replication have been valuable in identifying gaps in knowledge of nucleotide and DNA synthesis and stimulating research to address these gaps.82–84 Poovathingal et al.85 estimated through a stochastic model that mtDNA turnover rates in the order of months are the most consistent with published mtDNA mutation levels. Similarly, in silico experiments revealed a gap between the contributions of deoxyribonucleoside synthesis from salvage pathway relative to the levels of deoxyribonucleotides required for mtDNA replication.82 Computational models have revealed that the activities of mitochondrial nucleotide salvage enzymes are inadequate to support mitochondrial DNA replication and that instead, other sources of deoxyribonucleosides are essential. These findings have been partially corroborated by the recent identification of a dTMP biosynthesis pathway in mammalian mitochondria29 and other studies identifying mitochondrial nucleotide transport proteins. PNC1 (pyrimidine nucleotide carrier) transports a variety of metabolites, with a preference for UTP.86,87 Import of radioactively labeled dTMP from cytoplasm to mitochondria88 and a preference for the active transport of dCTP89 have also been observed. However, tissue specificity of dNTP pools,90 deoxynucleotide transporters,87 and enzymes involved in the salvage pathway90 have been revealed, which limits the ability to generalize the model to describe mtDNA turnover and regulation of deoxyribonucleoside pools in different organs. For example, the model proposed by Gandhi and Samuels assumes equal concentrations for all four deoxyribonucleotides, failing to capture the peculiar conditions of muscle and liver in human.82 Cells from these organs have high amounts of mtDNA and intense mtDNA turnover. However, muscle are characterized by lowest dTTP pool and liver by lowest dGTP pool, making their cells especially vulnerable to mutations in TK2 (mitochondrial thymidine kinase 2) and DGUOK (deoxyguanosine kinase), respectively. Experiments with perfused rat heart have not identified any de novo synthesis of deoxyribonucleosides in mitochondria,91 questioning whether mitochondrial salvage enzymes can be considered inadequate across all tissue types. Moreover, the model has been constructed by considering a constant nucleoside concentration of 0.5 μM,92 a level that is 50-fold lower than estimates found in the literature93 and one that leads to a conclusion that the salvage pathway plays a minimal role in mtDNA synthesis in all tissues. These examples illustrate the impact of tissue-specific differences in biological pathways that can limit the generalizability of the model to other species or cell and tissue types.
Example 3: HCY Metabolism
The HCY metabolism in the cytoplasm is among the most refined pathways within models of FOCM. The most updated model provides an explicit spatial description, accounting for the subcellular distribution of folate pools and the influence of other FOCM pathways localized in mitochondria.66 The FOCM model was extended to include the transsulfuration pathway and glutathione transport into the blood, providing an important link between cellular metabolism and blood biomarkers. Furthermore, the FOCM network in the cytoplasm, including the HCY remethylation pathway, has been shown to be connected through the activity of MTR, and also display ‘long-range’ interactions [e.g., MTHFR and GNMT (glycine N-methyltransferase) are inhibited by SAM and 5mTHF, respectively; CBS (cystathionine β-synthase) is activated by SAM] that contribute to the maintenance of stable SAM levels and therefore stable DNA methylation rates in the face of fluctuations in MET input.70,71 MTHFR catalyzes the NADPH-dependent conversion of 5,10-methyleneTHF to 5mTHF. MTHFR reaction velocity has been modeled with a second-order Michaelis–Menten equation (Vmax,MTHFR = 5300 μM h−1, Km,CH2F = 50 μM, and Km,NADPH = 16 μM; see Refs 74, 94, and 95) with NADPH concentration assumed to be constant (50 μM).66 The binding of SAM to MTHFR leads to an allosteric inhibition of the enzyme that has been derived by nonlinear regression of experimental data.96,97 The inhibition factor has no effect (i.e., it equals 1) when the MET concentration in the blood is 30 μM.70,71
The reaction catalyzed by GNMT generates sarcosine from glycine, using SAM as methyl donor. Deterministic models66,70,71 describe GNMT activity by means of a second-order Michaelis–Menten equation (Vmax = 245 μM h−1, Km,SAM = 32 μM and Km,Glycine = 130 μM; see Ref 98) including additional terms for product inhibition of SAH (Ki = 18 μM)63,71 and inhibition caused by the allosteric binding of 5mTHF.99
In case of a decline in the SAM concentration, the stabilization of the DNA methylation rate emerges from a complex mechanism of action, which involves MTHFR, GNMT, and DNMT. The decline of SAM releases the inhibition of MTHFR, thus determining an increase in the 5mTHF. The raise of 5mTHF inhibits GNMT activity. With the inhibition of GNMT, DNMT becomes responsible for the largest fraction of flows from SAM to SAH. Thus, even though the total flux from SAM to SAH is lower, the prevalence of DNMT activity buffers the consequences of the SAM decline on the DNA methylation reaction rate. This description provided by deterministic models of FOCM matches the hypothesis of Wagner et al.100
The FOCM was also shown to influence glutathione metabolism by modulating CBS activity, and a recent application described interconnections that link the MET cycle to glutathione metabolism, which can result in paradoxical responses of glutathione-related biomarkers in the blood.101 The model of Martinov et al. describes how elevated rates of HCY remethylation and high MET concentrations stimulate transsulfuration to remove excess MET through its conversion to cysteine.65 The model also describes how impairments in the MET cycle, resulting from deficiencies in key enzymes such as CBS and GNMT, and connections to transsulfuration pathway and polyamine biosynthesis can explain elevations in plasma HCY levels and other related biomarkers in cancer and other pathologies.67,102
The mathematical model of glutathione metabolism in hepatocytes66 represents the most advanced effort to simulate the dynamics of the folate-mediated FOCM; it has been developed extending specific models on folate62 and MET cycles,63,65,67 highlighting connections with the transsulfuration pathway and exploring the role of oxidative stress on enzyme regulation. It includes regulatory mechanisms involved in the transsulfuration pathway, and can predict the consequences of oxidative stress on the metabolic profiles associated with Down syndrome and autism. Oxidative stress has been modeled in terms of H2O2 concentration. Under more oxidative conditions, the inhibition of the enzymes MTR and betaine-homocysteine methyltransferases (BHMT) is predicted, whereas CBS and GCS (γ -glutamylcysteine synthetase) are activated.65,103 Oxidative stress and overexpression of the CBS gene characterize individuals with Down syndrome.104 The increase in CBS activity deprives the MTR reaction of HCY and promotes the accumulation of its other substrate, 5mTHF.105 Oxidative stress also impairs MTR activity, limiting the conversion of 5mTHF to THF. The concurrent effect of oxidative stress on CBS and MTR, combined with the irreversible kinetics of the 5mTHF synthesis, make 5mTHF accumulation particularly severe (methyl trap). The methyl trap compromises the generation of THF, a fundamental active form of folate that is required for purine and dTMP biosynthesis.106 This mechanism has been suggested by the outcomes of a deterministic model66 and might help to explain functional folate deficiency in spite of normal levels of folate and vitamin B12 in the blood. Using a similar approach, Reed et al.71 and Nijhout et al.107 modeled the complex interplay between nutrition and genetics (e.g., gene polymorphisms and vitamin deficiencies regulating enzymatic activities) on the dynamics within the FOCM network. The catalytic activity of SHMT can be affected by vitamin B6 deficiency108 (modeled as lower Vmax) and changes in gene expression (which alter both enzyme activity and binding affinity; these have been modeled as lower binding capacity with respect to 5mTHF).107 In the case of vitamin B6 deficiency, the inhibition of the SHMT1 activity resulted in almost no changes in serine and glycine levels, despite that SHMT interconverts both of these metabolites. This is consistent with the lack of substantial deleterious effects observed in case of SHMT knockout in mice.109 The most responsive target affecting serine and glycine concentrations, in case of vitamin B6 deficiency, is GDC in the mitochondria. This is because high reaction rates are required for sustaining the GDC-catalyzed reaction of glycine synthesis.110 SHMT overexpression and knockdown have consequences on the intensity of the tight inhibitory binding of 5mTHF to SHMT.21,25 When the SHMT concentration increases, 5mTHF is sequestered,21,27 thereby inhibiting MET synthesis in a glycine-independent manner. The decrease in the propensity of HCY remethylation to MET leads to lower levels of SAM.107 Consequences of SHMT overexpression on the fate of 5,10-methyleneTHF are still under debate. It is likely that pathways for HCY remethylation and dTMP biosynthesis escape any competition mechanism. This might be proven by the preferential enrichment of SHMT-derived 5,10-methyleneTHF into dTMP, an outcome resulting from the cell cycle-dependent nuclear localization of SHMT, TYMS, and DHFR.14 Also vitamin B12 deficiency and the common polymorphism in human MTHFR gene (677 C→T) reduce FOCM performance. Vitamin B12 deficiency impairs MTR reaction velocity. Because the MTHFR-catalyzed conversion of 5,10-methyleneTHF to 5mTHF is essentially irreversible, loss of MTR activity due to vitamin B12 deficiency (or genetic mutation) leads to a functional folate deficiency (i.e., a folate methyl trap). Deterministic models of FOCM described this effect by reducing the Vmax of the reaction catalyzed by MTR.62,63 As reported in the literature,111 a raise in the concentration of 5mTHF results in hypomethionemia and homocysteinuria. The genetic variant in the gene MTHFR (677 C→T) has been associated with a decrease in the activity of the enzyme MTHFR.54,56 Simulations tested the consequences of this genotypic change, predicting an increase of HCY and a decrease in the concentrations of SAM and 5mTHF.71 These results are comparable with patterns identified for MTHFR genotypes in human populations.112,113 Reed et al.71 suggested that the consequences of a loss of MTR activity (methyl trap) can be partly mitigated by the concurrent presence of a gene polymorphism which reduces MTHFR activity, a hypothesis that should be further validated by clinical data. These models provide a fairly simple but effective tool to estimate metabolic fluxes. The Michaelis–Menten equations and their parameters (Vmax and Km) capture the dynamics of folates and glutathione metabolites with a standard mathematical notation, independent of the underlying mechanisms which are now known to include an intricate set of phenomena (e.g., formation of complexes and translocation to different cellular compartments).
Example 4: Formation and Regulatory Properties of SAM and SAH
The conversion of MET to SAM is catalyzed by the activities of MET adenosyltransferase enzymes MAT I, MAT II, and MAT III, which function to generate the essential cofactor required for cellular transmethylation reactions. SAH is a product of the methylation reaction, and functions as an effective inhibitor of SAM-dependent methylases.53 Therefore, the SAM/SAH ratio is often referred to as the cellular methylation potential. SAM is the link to three metabolic pathways: polyamine synthesis, transmethylation, and transsulfuration. In all cells, SAM is the principal methyl donor for transmethylation reactions, but liver plays a crucial role in its homeostasis, as it is the major site for SAM synthesis and degradation.114 It is estimated that 85% of all methylation reactions occur in the liver and up to half of the MET dietary intake is converted to SAM by hepatocytes.115 As much as 48% of MET metabolism occurs in hepatocytes and MAT1A is specifically expressed in adult liver.116 Upon malignant transformation, hepatocytes cease to express MAT I and MAT III and induce the expression of the MAT II isoenzyme.67 The shift toward a MAT II enzyme contributes to reduced SAM content and stimulates DNA synthesis, offering a growth advantage to liver cells.117 The MAT2A gene encodes for MAT II, an isoenzyme expressed in extrahepatic tissues and in the fetal liver, where it is progressively replaced by MAT1A during development.118,119 Although MAT isoenzymes catalyze the same reaction, they differ in their kinetic parameters and are differentially regulated by SAM.114 Lowest Km Michaelis constant values are associated to MAT II, whereas the highest are exhibited by MAT III. SAM has a minimal inhibitory effects on MAT I, stimulates MAT III120 and elicits the strongest influence by its inhibition of MAT II.121 Upon malignant transformation, hepatocytes experience significant changes due to suppression of GNMT (see above, an enzyme that regulates the cellular methylation potential in the liver) and MAT I/III expression paralleled by induction of the MATII isoenzyme.122
Prudova et al.67 used the Hill equation to model the MAT II reaction velocity and to consider inhibition by SAM.
The dimensionless value of 0.76 stands for the Hill coefficient, and Ki,SAM = 50 μM quantifies the inhibition effect of SAM on the reaction velocity; Vmax,MATH = 507 μmol h−1 L−1 (of cells) and Km,MET = 4μM.120,123
SAM is also known to regulate other enzymes involved in HCY metabolism that influence flux through the HCY remethylation pathway. Mathematical functions describing the inhibition and activation by SAM have been derived from experimental data and/or estimated from the literature. For example, CBS catalyzes the conversion of HCY and serine to cystathionine, a reaction that is regulated by SAM levels. The reaction catalyzed by CBS is modeled with a second-order Michaelis-Menten equation, and its velocity modified with a scaling factor that considers SAM and SAH activation. The first part of the equation refers to a second-order Michaelis–Menten function with Vmax,CBS =700,000 μM h−1, Km,HCY = 1000 μM124 and Km,Serine = 2000 μM.125 The second term relates to the allosteric activation by SAM and SAH. Allosteric activation by SAM and SAH has been derived from the literature.126,127 Among the assumptions used in the model, the function was scaled such that it equals 1 in case of steady-state conditions (i.e., when the model is at equilibrium), which occurs when the MET concentration in the blood is 30 μM (SAM = 81.1 μM and SAH = 19.1 μM).70,71
Deterministic models suggest that a decline in SAM reduces CBS activity, leading to a shift toward HCY remethylation.63,65,67,71 However, no effect of SAM deficiency was observed experimentally in rat liver128 questioning the relevance of the allosteric stimulation.
Models of FOCM also take into account the role of SAM as a substrate for polyamine biosynthesis. Cells preferentially use SAM to produce polyamines, at the expense of transmethylation reactions. When later stages of polyamine biosynthesis are inhibited, decarboxylated adeonosylmethionine (dcSAM) accumulates, and elevated levels of dcSAM maintain polyamine synthesis by inhibiting methylation, and therefore alternative uses of SAM.129–131 As in the case of FOCM, mathematical models on polyamine biosynthesis have been developed132,133; the authors emphasized the importance of the bottom-up approach allowed by mathematical models for discriminating the role of the different components of polyamine metabolism on their homeostasis. The complex regulation at transcriptional, translational, and metabolic levels, consequences of genetic and environmental perturbations, and relationships with other metabolic pathways, preclude crude analysis of reaction pathways of polyamine metabolism and reliance on in vivo experiments. Mathematical models permit regulatory patterns that are specific of the polyamine metabolism to be distinguished from contributions of other processes with which it closely interacts.
These examples illustrate the complex set of interactions that characterize FOCM, and the necessity to include all regulatory features in the model. Michaelis–Menten equations represent the backbone for describing most of the reaction velocities, but are not sufficient to capture the dynamics of the system including feedback and feedforward signaling by metabolites. Such interactions are common and, besides the activation of CBS by SAM and SAH, additional terms are required for considering long-range effects and allosteric controls on other enzymes63,66,70: BHMT is inhibited by SAM and SAH; GNMT is inhibited by SAH and 5mTHF; MAT I is inhibited by SAM and glutathione disulfide; MAT III is activated by SAM and inhibited by glutathione disulfide; and MTHFR is inhibited by SAM, while SAH competes with SAM preventing its inhibitory effect. Knowledge of the intracellular concentrations of these regulatory metabolites can be limiting, requiring assumptions that may or may not reflect the in vivo state.
LIMITATIONS OF DATA DERIVED IN VITRO AND GAPS IN UNDERSTANDING OF THE BIOLOGICAL MECHANISMS THAT REGULATE FOCM
The availability of reliable and consistent data is one of several major limitations to modeling FOCM. Specifically, the availability of single source kinetic and metabolic data and gaps in understanding of the biological mechanisms that regulate FOCM need to be addressed.
Availability of Single Source Kinetic and Metabolic Data
The construction of FOCM models requires existing literature to provide a coherent set of kinetic parameters and metabolite concentrations. However, there can be wide ranges of variation in the Michaelis–Menten parameters published for a given enzyme, and there is always uncertainty regarding the accuracy of kinetic parameters that are obtained in vitro to describe activity in vivo. Many of the kinetic parameters reported in the literature are obtained from recombinant enzymes and can fail to capture in vivo dynamics (e.g., the in vitro conditions may not reflect the cellular environment adequately, affecting enzyme conformation, modification, and activity). This is important because in addition to equation structure, kinetic parameters can also impact model outcomes. Existing FOCM models mainly rely on literature data, which raises concerns of consistency. Rarely are kinetic constants obtained from the same organism, organ, tissue, and cell culture. Mathematical models of FOCM are focused primarily on enzymes that are mainly expressed in the mammalian liver cells because of the availability of data.65–67,71 Nijhout et al.62,70 defined the structure of the folate cycle in cytoplasm based on the scheme described by Wagner,134 whereas the MET cycle modeled by Reed et al.63,71 referred to the set of reactions sketched by Martinov et al.65 Although the choice of data from several mammalian species (human, rat, and mouse) confers a degree of robustness, it simultaneously poses serious concerns regarding generalizability of the findings to other organs, including blood. There are numerous other examples of tissue-specific differences on FOCM. The conversion of 10-FormylTHF to THF and carbon dioxide is catalyzed by a liver-specific 10-formylTHF dehydrogenase,135 and the folate-dependent catabolism of histidine occurs in only a few tissues such as liver and kidney.136 Pathologies associated with polymorphisms in folate metabolism and impaired B vitamin intake tend to exhibit tissue specificity, including colorectal carcinogenesis137–139 and neural tube defects,140 and models based on liver architecture may not accurately describe FOCM in these systems.
Compartmentation and Channeling in FOCM
The recent advances in understanding the cell biology and biochemistry of FOCM present challenges for existing deterministic models, which are based on the assumption that the de novo synthesis of purines, dTMP, and HCY remethylation all compete for a common pool of folate cofactors in the cytoplasm. As reviewed in Section Pathways and Their Subcellular Localization, ongoing research is indicating that these pathways are isolated both temporally through cell cycle, and spatially through the formation of multienzyme complexes. These findings require new parameters to be included into models of FOCM, and/or can fundamentally challenge the assumptions upon which deterministic models are based. For example, Michaelis–Menten constants often do not apply to enzymes present in complexes where the spatial localization of all the protein subunits can affect the kinetics of their activity (e.g., sigmoidal dependence of reaction velocity on substrate concentration displayed by allosteric interactions, see Ref 141). The colocalization of multiple enzymes can shape the active sites, allow allosteric cooperativity, provide an additional level of signaling or regulation, and permit channeling of intermediates during an enzymatic turnover.142,143 The formation of complexes can increase the stability and improve the functionality of monomeric enzymes144 thus requiring novel mathematical formulations for representing the mechanisms of action. Furthermore, estimations of cellular metabolite concentrations, including their enrichment in subcellular compartments (e.g., cytosol, mitochondria, nucleus) can be challenging or impossible,145 and their mode of transport (active or passive) and the associated kinetics may not be well characterized. Knowledge of the details of a specific transport mechanism can permit accurate modeling of the interaction between compartments, but increases the complexity of the system in terms of number of reactions or differential equations (depending on the modeling approach used), which may turn into a higher computational complexity when integrating the system. More commonly, one is able to fit experimental data assuming either an abstract limited capacity communication channel standing between two separated volumes, and therefore approximating its kinetics (vt = actual transport velocity) by setting a maximum transport velocity (Vmax), e.g., using a (firstorder) Michaelis–Menten expression (Km = Michaelis constant value; [A] = substrate concentration),
or the unbounded exchange of material, using a simple linear rate of transport (Kt).
These are the approaches used by Reed et al.66 for their model. However, it is often hard to correctly predict the trends of metabolite concentrations over time without taking into account their involvement in reactions occurring in physically separated cellular compartments that continuously exchange material and consequently have a certain degree of inter-dependence.145 Different transport mechanisms require that the appropriate kinetics be applied, and therefore knowledge of the details of a specific transport mechanism are needed to precisely model the interaction between compartments. This comes at the expense of (1) details of the process, which are not always available and (2) the complexity of the system, which may turn into a higher computational complexity when attempting to simulate the dynamics.
Local metabolite concentration gradients can be generated in the cell, and thereby create additional challenges to mathematical modeling. Mitochondria are crucial for the functioning of FOCM in the cytoplasm and nucleus, as they exchange serine and glycine with the cytoplasm and are an essential source of formate for cytoplasmic and nuclear FOCM. In some cases, mitochondria may be enriched near the nucleus.146 Serine and glycine are exchanged between cytoplasm and mitochondria; their ratio can not only regulate the production of formate in the mitochondria but also affect the reaction catalyzed by SHMT in the nucleus. Better understanding of formate, serine, and glycine fluxes and their transport into subcellular compartments is needed, especially for nuclear FOCM where the current dogma assumes they are freely exchanged based on nuclear pore sizes.15
Compartmentation also requires consideration of protein modifications and the role of essential but nonenzymatic functions in the model. Although the enzymes that constitute the de novo dTMP synthesis pathway are present in the cytoplasm, the pathway is not active until it translocates into the nucleus at S-phase.9,15,25 Modification by SUMO enables the localization of three enzymes in the nucleus (TYMS, SHMT, and DHFR), and the efficiency of this process can determine total de novo dTMP synthesis capacity.25 Existing deterministic models do not capture contributions of folate-dependent enzymes to FOCM outside their kinetic role within metabolic pathways. For example, SHMT has been shown recently to be essential for nuclear de novo dTMP synthesis, yet does not make major contributions as an enzyme catalyst to the biosynthetic pathway. Rather, SHMT plays an important role as scaffold protein that is essential for the assembly and stability of the metabolic pathway at the nuclear lamina9 while MTHFD1 provides most of the 5,10-methyleneTHF required for TYMS activity.9,21 Furthermore, the histone demethylase KDM1 interacts with SHMT and may also provide 5,10-methyleneTHF for de novo dTMP synthesis. Modeling the interplay among the three enzymes capable of generating 5,10-methyleneTHF is key for understanding the regulation of FOCM in the nucleus and its consequences on dTMP synthesis.9,10,14
The model developed by Reed et al.,66 while not currently describing the role of the nucleus in de novo dTMP biosynthesis, does consider the pathway with the assumption that it functions in the cytoplasm and mitochondria. In their model, except for a small initial transient phase, almost all of the 5,10-methyleneTHF are generated by MTHFD1, whereas SHMT removes 5,10-methyleneTHF (and glycine) to produce THF (and serine). Although this model does not accurately account for compartmentation and metabolic complex formation, it does nonetheless correctly identify the importance of MTHFD1 in generating 5,10-methyleneTHF (through a three-step reaction which starts from THF and mitochondria-produced formate) for de novo dTMP synthesis, but fails to account for the essentiality of SHMT in determining de novo dTMP biosynthesis capacity in its scaffold role in complex formation and the directionality of the reaction. This model was tested in light of current knowledge of nuclear compartmentation of the de novo dTMP biosynthesis, including its ability to illustrate the scaffold function of SHMT in the nucleus, and its ability to supply one-carbons to TYMS in case of MTHFD1 knockout (the reduced activity of the trifunctional enzyme has been simulated by decreasing the Vmax of the reactions catalyzed by MTHFD1). Under these circumstances, the direction of the reaction catalyzed by SHMT is reversed (i.e., we observed a contribution of SHMT in producing 5,10-methyleneTHF). Although conceived for the cytoplasm and crudely based on Michaelis–Menten kinetics, the equations used by Reed et al.66 to model de novo dTMP biosynthesis are robust enough for capturing a realistic dynamics in the nucleus. However, the model does not account for the presence of folate-cofactors in the nucleus, the impact this has on rates of de novo dTMP biosynthesis in the nucleus, or the impact of folate cofactors localizing to the nucleus in HCY and glutathione biosynthesis.66
Similarly, there is increasing evidence that de novo purine nucleotide biosynthesis is spatially and temporally isolated from both dTMP biosynthesis and HCY remethylation due to the formation of a complex referred to as purinosome147 in G1.14,16 This complex is composed of six enzymes, and the pathway may not be functional until the purinosome is established. This raises questions regarding the mechanism of ‘delivery’ of folate cofactors to the metabolic complex, as current deterministic models assume the THF cofactors are delivered to enzymes by diffusion rather than by trafficking folate to the complex. Interestingly, MTHFD1, the enzyme responsible for generating 10-formylTHF, is not present within the purinosome.17 However, methenylTHF synthetase (MTHFS), an enzyme that binds 10-formylTHF tightly16,148 colocalizes with the purinosome, and has been proposed to deliver the chemically labile 10-formylTHF cofactors to the activities of GAR and AICAR.16 Posttranslational modification by SUMO is required for localization to the purinosome, and reductions in MTHFS expression exhibit nearly proportional decreases in de novo purine nucleotide synthesis.16 Neither folate channeling nor protein-mediated trafficking of folate cofactors are considered in existing deterministic models of FOCM.66,71 Rather, they adopt second-order Michaelis–Menten reactions to describe both the activities of GAR and AICART, which has consequences on the structure of the model.
OPPORTUNITIES FOR STOCHASTIC MODELING OF FOCM
The two most common approaches to simulating the behavior of a biological system are deterministic and stochastic modeling. The former relies on the formulation of the model through coupled ODEs (reaction rate equations, RRE) that describe each step in the pathway and the associated changes in metabolite concentrations (continuously) over time; the solution provides a unique, single trajectory. This approach accounts for the number of reactions occurring per unit of time and supplies solutions, which can be reliable and accurate numerical values of the system outputs and the concentration of intermediates in the system that is being modeled. Clearly, when considering small numbers of molecules, it becomes more and more evident that the deterministic approach is a simplification, as it is known that reactions involve a discrete number of reactants (molecules), and they occur randomly at discrete time points, so the system is expected to exhibit some variability in its behavior from one experiment to the other. Furthermore, the combinatorial complexity of the system might rapidly become a limiting factor for the deterministic modeling process.149 From the dynamics point of view, it is quite clear that small alterations of some key parameters of the reactions may alter the steady-state concentrations of some metabolites, or even lead to unexpected behavior. This is both because of the high nonlinearities in the terms defining the velocities of each single reaction (see the examples in the previous section) and because of the dense network of interactions that could hide unpredictable long-term consequences. Consequently, accuracy in measuring or estimating kinetic parameters of the enzymes and metabolites involved in the pathway are often key requirements for obtaining a model with which perform in silico experiments and predictions.
On the other hand, one can assume that the system is well-stirred, ignore the exact position and velocity of each molecule, so that the propensity function of a reaction is proportional to the probability for that reaction to occur in the next infinitesimal time step. On the basis of these assumptions, Gillespie150 derived a simple method, often referred to as the ‘Stochastic Simulation Algorithm’ (SSA), which samples a trajectory from the set of all the possible evolutions of the system by selecting and executing one reaction at a time. This model captures the variability of the system behavior, which is especially relevant when low reactant populations are involved and the system architecture contains uncertainties and is assembled with incomplete knowledge, including variables such as regulation, substrate channeling, and compartmentation. The stochastic regime also allows for different modeling strategies to be included other than the simple fixed-entities and fixed-reactions representations that are characteristic of (reaction-based) deterministic models. Stochastic modeling can provide a more useful approach to describing complex systems for which there is no prior knowledge of all the reactants and their precise concentrations, and reactions and their regulation. Molecules are permitted to assume different configurations and/or assemble in more or less complex structures following general ‘rules’ (as opposed to more specific reactions) that allow for the evolution of groups of species and partial complexes151 so to obtain a compact description of the dynamics. When dealing with a two-substrate enzyme, for example, only three interactions need to be modeled: the binding/unbinding of the two substrates S1 and S2 to the enzyme E, and the formation of the product P once both substrates are bound. In terms of differential equations, instead, there are seven quantities to be represented (namely E, S1, S2, E:S1, E:S2, E:S1:S2, P; consider that E:S1 and E:S2 represent configurations where the substrates are bound with the enzyme, while E:S1:S2 consists of the enzyme bound with both of the substrates prior their conversion into the product P). In the case of FOCM, modeling the formation of multienzyme complexes, or even the activity of multisubstrate enzymes, could take advantage of this paradigm. If we take, for example, the case of the multifunctional enzyme MTHFD1, its action can be modeled through two folate binding sites5: the first one performing the dehydrogenase and cyclohydrolase activities; the second being the synthetase domain. Given that each enzyme has the necessary binding affinities for all the substrates, one direction of the folate transformation (the one we are interested in the most in FOCM, THF → 10-formylTHF → 5,10-methenylTHF → 5,10-methyleneTHF) is represented by the following 21 reactions involving 10 reactants:
By using a rule-based description of the different interactions, instead, only the binding/unbinding/synthesis events (for each of the three substrates) need to be represented for a total of nine rules (Figure 4). This is not just a twofold reduction in the model complexity (indeed, more complex examples exist), but it allows the model to be written, read, edited, and updated more easily. Different implementations have been given for such rule-based modeling strategies (see for example, Refs 151–153) but it is clear that the simulation environment is best fit with a discrete-stochastic approach described above.
FIGURE 4.
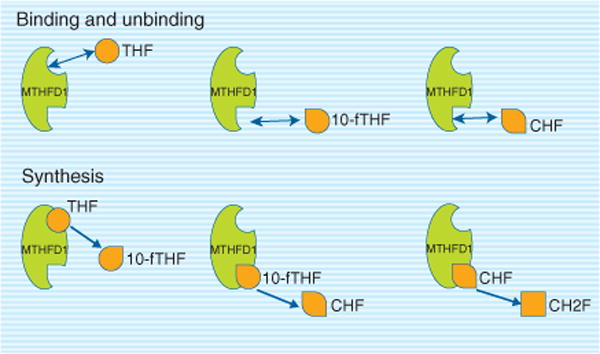
Methylenetetrahydrofolate dehydrogenase (MTHFD1) is a trifunctional enzyme with three enzymatic activities methenyltetrahydrofolate cyclohydrolase, 10-formyltetrahydrofolate synthetase, and methylenetetrahydrofolate dehydrogenase. The MTHFD1L isozyme only exhibits 10-formyltetrahydrofolate synthetase, whereas the MTHFD2 and MTHFD2L isozymes only exhibit methenyltetrahydrofolate cyclohydrolase and methylenetetrahydrofolate dehydrogenase activities. THF, tetrahydrofolate; 10-fTHF, 10-formyltetrahydrofolate; CHF, 5,10-methenyltetrahydrofolate.
The performance of SSA approaches can be limited by the computational expense that increases as the number of molecules and reactions grows. However, efficient (although inexact) implementations of the SSA have been derived: τ-leaping methods154,155 perform multiple reactions at each step, allowing for much faster simulations. Furthermore, the stochastic approach may not be required for large numbers of molecules, because for such quantities the variability displayed by the system behavior, due to stochastic noise, is usually negligible compared to the observed quantities, which tend to match to the average (i.e., their time course is, practically speaking, deterministic). The inefficiency of any numerical method for the simulation of dynamic systems is often due to the presence of different time scales, with the fastest reactions requiring the simulation to proceed with small time steps. In biochemistry and molecular biology, such stiff behavior appears when species with very different concentrations are present in the system. More recently, hybrid stochastic-deterministic simulation techniques have been developed156 that correctly exhibit the stochastic behavior of the slower (less populated) time scales but rapidly move forward along the fastest (more populated) time scales.
SSA approaches can provide new and useful insights into FOCM, especially for all cases in which the rule-based approach might facilitate the phase of model construction and promote the more realistic representation of biological interactions (i.e., modeling the propensity of elementary binding/unbinding events involving enzymes and reactants, rather than abstracting the dynamics in terms of Michaelis—Menten equations). Stochastic approaches are useful for modeling reactions where assumptions of equilibrium, and metabolite and cofactor equilibration with the bulk solvent may not apply. Published FOCM (deterministic) models tend to address phenomena such as formation of complexes, inhibition and activation by using a simple mathematical description (i.e., Michaelis–Menten functions), and completing the model with additional terms, rather than describing the molecular behavior of multienzyme metabolic complexes, including their assembly, regulation, and unique metabolic behaviors. Although Michaelis—Menten approximations are an efficient tool for describing the intricacy emerging from complex formation and posttranslational modifications, they can fail to capture the correct dynamics because they rely on the assumptions that either (1) the enzyme has a high (≫1) dissociation constant or (2) substrates are present in a much higher concentration than enzymes. Future efforts could maintain the simple representation of Michaelis–Menten reactions but should focus on spatial (i.e., nucleus vs cytoplasm) and functional properties that cannot be elucidated from in vitro reconstitution experiments.
New FOCM models should also study the translocation between compartments. Hypothesis of diffusion and active transport in the communication between nucleus and cytoplasm should be tested. The involvement of enzymes in the formation of complexes, essential also for transferring metabolites through different compartments, calls for the need of considering the enzymes as model variables. Reed et al.66 based their description of the cytosolic and mitochondrial dynamics on Vmax; this means that enzymatic concentrations are included—‘masked’—into Vmax values (Vmax is computed as the product of enzyme concentration times Kcat—turnover number). The formation of a complex involving the SHMT enzyme and 5mTHF has been modeled by Nijhout et al.,107 under a deterministic context. They have added SHMT concentration as a model variable and studied the effects of its binding with 5mTHF; this example represents an almost unique attempt of modeling complex formation. We assert future FOCM models should express some enzyme activities as a function of their concentration, making evident the contribution of this term to Vmax. Similarly, Michaelis–Menten functions can account for the consequences of single nucleotide polymorphisms and food intake (e.g., the role of vitamins as cofactors), with these changes in enzymatic binding affinities reflected in Km values.157
Rule-based modeling enables the explicit description of key processes in the de novo dTMP biosynthesis in the nucleus: (1) SUMOylation and nuclear import of SHMT, TYMS, and DHFR; (2) preferential inclusion of one-carbons from SHMT into the dTMP; (3) formate and 5,10-methyleneTHF transportation into the nucleus (i.e., freely exchange through pores vs active transportation mediated by a complex). This mechanistic description could help to (1) shedding light on the relative contribution of MTHFD1, SHMT, and KDM1 in the production of 5,10-methyleneTHF; (2) studying how different forms of folate can regulate the scaffold reactions; (3) identifying the more realistic mechanism of transport for the formate that is generated into mitochondria; (4) clarifying how formate levels can regulate maximal rates of de novo dTMP and MET synthesis in nucleus and cytoplasm (this should be analyzed together with the role of serine/glycine ratio in regulating the production of formate in the mitochondria). Furthermore, de novo purine biosynthesis exhibits controversial aspects that could be analyzed with a stochastic rule-based modeling approach. They involve the regulatory inhibition of the salvage pathway, the presence of a multienzyme complex called purinosome and the central role of MTHFS in its formation through the delivery of 10-formylTHF.14,16 Curto et al.158 constructed a deterministic model on purine metabolism, highlighting the need of including SAM for a complete description; however, their focus on MET cycle, and connections to FOCM remained marginal. The understanding of the cell-cycle dependence of de novo purine biosynthesis calls for a rule-based strategy, in order to better integrate purinosome formation and 10-step reactions of de novo purine biosynthesis.
EXTENDING MATHEMATICAL MODELS TO IDENTIFY AND PREDICT BIOMARKERS OF IMPAIRED FOCM
Modeling of metabolic networks is a powerful approach to identify gaps in pathway components and their regulation, and allows a better understanding of cellular behavior in both normal and pathogenic states. In general, epidemiological studies refer to metabolites in terms of their blood concentrations, while mathematical models often describe concentrations in the cytoplasm. Extending these models to identify intermediates, including metabolites or genomic markers, whose levels change as a result of disease initiation or progression is critical for the discovery and validation of biomarkers that inform preventative, management, or therapeutic treatments,66,107 and to set dietary reference intakes for essential nutrients.159 Modeling the connection between cellular and blood concentrations is of central importance for transferring knowledge from simulations to clinical applications.101 The inherent flexibility of stochastic models permits the mechanistic explanation of unexpected patterns (e.g., changes in the biomarker concentration in the blood), an otherwise impossible perspective if multiple effects were lumped into the abstraction of Michaelis–Menten equations.
CONCLUSIONS
Deterministic models have been widely applied to represent the dynamics of metabolic networks. They make use of ODEs, listing all chemical species that could exist in a system and often relying on Michaelis–Menten equations to approximate reaction velocity. Deterministic models describe macroscopic behavior of biological systems and call for an explicit enumeration of molecular states and interactions. However, many cellular constituents (e.g., enzymes and metabolites) are present in small numbers, and significant fluctuations characterize processes as complex formation, posttranslational modification of enzymes, gene expression, and competitive phosphorylation of multiple sites in signaling proteins. Combining the stochastic approach with a rule-based representation can help (1) to model the intrinsic noise displayed by biological systems, (2) to address the limited flexibility of standard simulation methods for coarse-graining biochemical processes, and (3) to manage combinatorial complexity emerging from complex reactions that would otherwise be intractable.149,160 Trafficking of intracellular folates among compartmentalized metabolic pathways requires mechanisms to mobilize folate cofactors. Formation of complexes (e.g., purinosome to supply the labile form of 10-formylTHF to GAR and AICAR, in de novo purine biosynthesis), posttranslational modifications (e.g., SUMOylation of the enzymes involved in nuclear dTMP biosynthesis), inhibition of enzyme activity (e.g., tight-binding between 5mTHF and SHMT), and transportation of precursors through different cellular compartments (e.g., mitochondria-derived formate carried into the nucleus to support de novo dTMP biosynthesis) are some of the processes responsible for spatial and temporal compartmentation in FOCM. We argue that applying the rule-based approach would better address the combinatorial challenge posed by their modeling. Moreover, stochastic simulations would enable to elucidate the functioning of FOCM and its relationship to human health and disease.
Acknowledgments
Bianca Baldacci is kindly acknowledged for the graphic design contribution. We are also grateful to Alessandro Romanel, Gianluca Fantaccini and Martha S. Field for stimulating discussions.
References
- 1.Kitano H. Computational systems biology. Nature. 2002;420:206–210. doi: 10.1038/nature01254. [DOI] [PubMed] [Google Scholar]
- 2.Kitano H. Looking beyond the details: a rise in system-oriented approaches in genetics and molecular biology. Curr Genet. 2002;41:1–10. doi: 10.1007/s00294-002-0285-z. [DOI] [PubMed] [Google Scholar]
- 3.Kitano H. Systems biology: a brief overview. Science. 2002;295:1662–1664. doi: 10.1126/science.1069492. [DOI] [PubMed] [Google Scholar]
- 4.Pahle J. Biochemical simulations: stochastic, approximate stochastic and hybrid approaches. Brief Bioinform. 2009;10:53–64. doi: 10.1093/bib/bbn050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fox JT, Stover PJ. Folate-mediated one-carbon metabolism. Vitam Horm. 2008;79:1–44. doi: 10.1016/S0083-6729(08)00401-9. [DOI] [PubMed] [Google Scholar]
- 6.Miranda TB, Jones PA. DNA methylation: the nuts and bolts of repression. J Cell Physiol. 2007;213:384–390. doi: 10.1002/jcp.21224. [DOI] [PubMed] [Google Scholar]
- 7.Winter-Vann AM, Kamen BA, Bergo MO, Young SG, Melnyk S, James SJ, Casey PJ. Targeting Ras signaling through inhibition of carboxyl methylation: an unexpected property of methotrexate. Proc Natl Acad Sci U S A. 2003;100:6529–6534. doi: 10.1073/pnas.1135239100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stead LM, Jacobs RL, Brosnan ME, Brosnan JT. Methylation demand and homocysteine metabolism. Adv Enzyme Regul. 2004;44:321–333. doi: 10.1016/j.advenzreg.2003.11.012. [DOI] [PubMed] [Google Scholar]
- 9.Anderson DD, Woeller CF, Chiang EP, Shane B, Stover PJ. Serine hydroxymethyltransferase anchors de novo thymidylate synthesis pathway to nuclear lamina for DNA synthesis. J Biol Chem. 2012;287:7051–7062. doi: 10.1074/jbc.M111.333120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luka Z, Moss F, Loukachevitch LV, Bornhop DJ, Wagner C. Histone demethylase LSD1 is a folate-binding protein. Biochemistry. 2011;50:4750–4756. doi: 10.1021/bi200247b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tibbetts AS, Appling DR. Compartmentalization of mammalian folate-mediated one-carbon metabolism. Annu Rev Nutr. 2010;30:57–81. doi: 10.1146/annurev.nutr.012809.104810. [DOI] [PubMed] [Google Scholar]
- 12.Christensen KE, MacKenzie RE. Mitochondrial one-carbon metabolism is adapted to the specific needs of yeast, plants and mammals. Bioessays. 2006;28:595–605. doi: 10.1002/bies.20420. [DOI] [PubMed] [Google Scholar]
- 13.Suh JR, Herbig AK, Stover PJ. New perspectives on folate catabolism. Annu Rev Nutr. 2001;21:255–282. doi: 10.1146/annurev.nutr.21.1.255. [DOI] [PubMed] [Google Scholar]
- 14.Stover PJ, Field MS. Trafficking of intracellular folates. Adv Nutr. 2011;2:325–331. doi: 10.3945/an.111.000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woeller CF, Anderson DD, Szebenyi DM, Stover PJ. Evidence for small ubiquitin-like modifier-dependent nuclear import of the thymidylate biosynthesis pathway. J Biol Chem. 2007;282:17623–17631. doi: 10.1074/jbc.M702526200. [DOI] [PubMed] [Google Scholar]
- 16.Field MS, Anderson DD, Stover PJ. Mthfs is an essential gene in mice and a component of the purinosome. Front Genet. 2011;2:36. doi: 10.3389/fgene.2011.00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.An S, Kumar R, Sheets ED, Benkovic SJ. Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science. 2008;320:103–106. doi: 10.1126/science.1152241. [DOI] [PubMed] [Google Scholar]
- 18.An S, Kyoung M, Allen JJ, Shokat KM, Benkovic SJ. Dynamic regulation of a metabolic multi-enzyme complex by protein kinase CK2. J Biol Chem. 2010;285:11093–11099. doi: 10.1074/jbc.M110.101139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamaoka T, Yano M, Kondo M, Sasaki H, Hino S, Katashima R, Moritani M, Itakura M. Feedback inhibition of amidophosphoribosyltransferase regulates the rate of cell growth via purine nucleotide, DNA, and protein syntheses. J Biol Chem. 2001;276:21285–21291. doi: 10.1074/jbc.M011103200. [DOI] [PubMed] [Google Scholar]
- 20.Anderson DD, Stover PJ. SHMT1 and SHMT2 are functionally redundant in nuclear de novo thymidylate biosynthesis. PLoS One. 2009;4:e5839. doi: 10.1371/journal.pone.0005839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herbig K, Chiang EP, Lee LR, Hills J, Shane B, Stover PJ. Cytoplasmic serine hydroxymethyltransferase mediates competition between folate-dependent deoxyribonucleotide and S-adenosylmethionine biosyntheses. J Biol Chem. 2002;277:38381–38389. doi: 10.1074/jbc.M205000200. [DOI] [PubMed] [Google Scholar]
- 22.Prem veer Reddy G. Catalytic function of thymidylate synthase is confined to S phase due to its association with replitase. Biochem Biophys Res Commun. 1982;109:908–915. doi: 10.1016/0006-291x(82)92026-5. [DOI] [PubMed] [Google Scholar]
- 23.Anderson DD, Woeller CF, Stover PJ. Small ubiquitin-like modifier-1 (SUMO-1) modification of thymidylate synthase and dihydrofolate reductase. Clin Chem Lab Med. 2007;45:1760–1763. doi: 10.1515/CCLM.2007.355. [DOI] [PubMed] [Google Scholar]
- 24.Fox JT, Shin WK, Caudill MA, Stover PJ. A UV-responsive internal ribosome entry site enhances serine hydroxymethyltransferase 1 expression for DNA damage repair. J Biol Chem. 2009;284:31097–31108. doi: 10.1074/jbc.M109.015800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MacFarlane AJ, Anderson DD, Flodby P, Perry CA, Allen RH, Stabler SP, Stover PJ. Nuclear localization of de novo thymidylate biosynthesis pathway is required to prevent uracil accumulation in DNA. J Biol Chem. 2011;286:44015–44022. doi: 10.1074/jbc.M111.307629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shane B, Stokstad EL. Vitamin B12-folate interrelationships. Annu Rev Nutr. 1985;5:115–141. doi: 10.1146/annurev.nu.05.070185.000555. [DOI] [PubMed] [Google Scholar]
- 27.Stover P, Schirch V. 5-Formyltetrahydrofolate polyglutamates are slow tight binding inhibitors of serine hydroxymethyltransferase. J Biol Chem. 1991;266:1543–1550. [PubMed] [Google Scholar]
- 28.Stover P, Schirch V. The metabolic role of leucovorin. Trends Biochem Sci. 1993;18:102–106. doi: 10.1016/0968-0004(93)90162-g. [DOI] [PubMed] [Google Scholar]
- 29.Anderson DD, Quintero CM, Stover PJ. Identification of a de novo thymidylate biosynthesis pathway in mammalian mitochondria. Proc Natl Acad Sci U S A. 2011;108:15163–15168. doi: 10.1073/pnas.1103623108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bogenhagen D, Clayton DA. Mouse L cell mitochondrial DNA molecules are selected randomly for replication throughout the cell cycle. Cell. 1977;11:719–727. doi: 10.1016/0092-8674(77)90286-0. [DOI] [PubMed] [Google Scholar]
- 31.Di Pietro E, Sirois J, Tremblay ML, MacKenzie RE. Mitochondrial NAD-dependent methylenetetrahydrofolate dehydrogenase-methenyltetrahydrofolate cyclohydrolase is essential for embryonic development. Mol Cell Biol. 2002;22:4158–4166. doi: 10.1128/MCB.22.12.4158-4166.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Di Pietro E, Wang XL, MacKenzie RE. The expression of mitochondrial methylenetetrahydrofolate dehydrogenase-cyclohydrolase supports a role in rapid cell growth. Biochim Biophys Acta. 2004;1674:78–84. doi: 10.1016/j.bbagen.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 33.Bolusani S, Young BA, Cole NA, Tibbetts AS, Momb J, Bryant JD, Solmonson A, Appling DR. Mammalian MTHFD2L encodes a mitochondrial methyl enetetrahydrofolate dehydrogenase isozyme expressed in adult tissues. J Biol Chem. 286:5166–5174. doi: 10.1074/jbc.M110.196840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pike ST, Rajendra R, Artzt K, Appling DR. Mitochondrial C1-tetrahydrofolate synthase (MTHFD1L) supports the flow of mitochondrial one-carbon units into the methyl cycle in embryos. J Biol Chem. 285:4612–4620. doi: 10.1074/jbc.M109.079855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shin YS, Chan C, Vidal AJ, Brody T, Stokstad EL. Subcellular localization of γ -glutamyl carboxypeptidase and of folates. Biochim Biophys Acta. 1976;444:794–801. doi: 10.1016/0304-4165(76)90326-3. [DOI] [PubMed] [Google Scholar]
- 36.Lin BF, Huang RF, Shane B. Regulation of folate and one-carbon metabolism in mammalian cells. III. Role of mitochondrial folylpoly-γ-glutamate synthetase. J Biol Chem. 1993;268:21674–21679. [PubMed] [Google Scholar]
- 37.McCarthy EA, Titus SA, Taylor SM, Jackson-Cook C, Moran RG. A mutation inactivating the mitochondrial inner membrane folate transporter creates a glycine requirement for survival of chinese hamster cells. J Biol Chem. 2004;279:33829–33836. doi: 10.1074/jbc.M403677200. [DOI] [PubMed] [Google Scholar]
- 38.Schirch V, Strong WB. Interaction of folylpolyglutamates with enzymes in one-carbon metabolism. Arch Biochem Biophys. 1989;269:371–380. doi: 10.1016/0003-9861(89)90120-3. [DOI] [PubMed] [Google Scholar]
- 39.Spivey HO, Ovadi J. Substrate channeling. Methods. 1999;19:306–321. doi: 10.1006/meth.1999.0858. [DOI] [PubMed] [Google Scholar]
- 40.Pawelek PD, Allaire M, Cygler M, MacKenzie RE. Channeling efficiency in the bifunctional methylenetetrahydrofolate dehydrogenase/cyclohydrolase domain: the effects of site-directed mutagenesis of NADP binding residues. Biochim Biophys Acta. 2000;1479:59–68. doi: 10.1016/s0167-4838(00)00058-3. [DOI] [PubMed] [Google Scholar]
- 41.Bulock KG, Beardsley GP, Anderson KS. The kinetic mechanism of the human bifunctional enzyme ATIC (5-amino-4-imidazolecarboxamide ribonucleotide transformylase/inosine 5′-monophosphate cyclohydrolase). A surprising lack of substrate channeling. J Biol Chem. 2002;277:22168–22174. doi: 10.1074/jbc.M111964200. [DOI] [PubMed] [Google Scholar]
- 42.Kim DW, Huang T, Schirch D, Schirch V. Properties of tetrahydropteroylpentaglutamate bound to 10-formyltetrahydrofolate dehydrogenase. Biochemistry. 1996;35:15772–15783. doi: 10.1021/bi9619684. [DOI] [PubMed] [Google Scholar]
- 43.Strong WB, Schirch V. In vitro conversion of formate to serine: effect of tetrahydropteroylpolyglutamates and serine hydroxymethyltransferase on the rate of 10-formyltetrahydrofolate synthetase. Biochemistry. 1989;28:9430–9439. doi: 10.1021/bi00450a028. [DOI] [PubMed] [Google Scholar]
- 44.Sidi Y, Mitchell BS. Z-nucleotide accumulation in erythrocytes from Lesch-Nyhan patients. J Clin Investig. 1985;76:2416–2419. doi: 10.1172/JCI112255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fenech M. Folate (vitamin B9) and vitamin B12 and their function in the maintenance of nuclear and mitochondrial genome integrity. Mutat Res. 2012;733:21–33. doi: 10.1016/j.mrfmmm.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 46.Mashiyama ST, Courtemanche C, Elson-Schwab I, Crott J, Lee BL, Ong CN, Fenech M, Ames BN. Uracil in DNA, determined by an improved assay, is increased when deoxynucleosides are added to folate-deficient cultured human lymphocytes. Anal Biochem. 2004;330:58–69. doi: 10.1016/j.ab.2004.03.065. [DOI] [PubMed] [Google Scholar]
- 47.Basten GP, Duthie SJ, Pirie L, Vaughan N, Hill MH, Powers HJ. Sensitivity of markers of DNA stability and DNA repair activity to folate supplementation in healthy volunteers. Br J Cancer. 2006;94:1942–1947. doi: 10.1038/sj.bjc.6603197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blount BC, Mack MM, Wehr CM, MacGregor JT, Hiatt RA, Wang G, Wickramasinghe SN, Everson RB, Ames BN. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: implications for cancer and neuronal damage. Proc Natl Acad Sci USA. 1997;94:3290–3295. doi: 10.1073/pnas.94.7.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Melnyk S, Pogribna M, Miller BJ, Basnakian AG, Pogribny IP, James SJ. Uracil misincorporation, DNA strand breaks, and gene amplification are associated with tumorigenic cell transformation in folate deficient/repleted Chinese hamster ovary cells. Cancer Lett. 1999;146:35–44. doi: 10.1016/s0304-3835(99)00213-x. [DOI] [PubMed] [Google Scholar]
- 50.Kapiszewska M, Kalemba M, Wojciech U, Milewicz T. Uracil misincorporation into DNA of leukocytes of young women with positive folate balance depends on plasma vitamin B12 concentrations and methylenetetrahydrofolate reductase polymorphisms. A pilot study. J Nutr Biochem. 2005;16:467–478. doi: 10.1016/j.jnutbio.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 51.van den Donk M, Pellis L, Crott JW, van Engeland M, Friederich P, Nagengast FM, van Bergeijk JD, de Boer SY, Mason JB, Kok FJ, et al. Folic acid and vitamin B-12 supplementation does not favorably influence uracil incorporation and promoter methylation in rectal mucosa DNA of subjects with previous colorectal adenomas. J Nutr. 2007;137:2114–2120. doi: 10.1093/jn/137.9.2114. [DOI] [PubMed] [Google Scholar]
- 52.Nilsen H, Stamp G, Andersen S, Hrivnak G, Krokan HE, Lindahl T, Barnes DE. Gene-targeted mice lacking the Ung uracil-DNA glycosylase develop B-cell lymphomas. Oncogene. 2003;22:5381–5386. doi: 10.1038/sj.onc.1206860. [DOI] [PubMed] [Google Scholar]
- 53.Clarke S, Banfield K. S-adenosylmethionine-depen dent methyltransferases. In: Carmel R, Jacobson DW, editors. Homocysteine in Health and Disease. Cambridge: Cambridge Press; 2001. [Google Scholar]
- 54.Kang SS, Zhou J, Wong PW, Kowalisyn J, Strokosch G. Intermediate homocysteinemia: a thermolabile variant of methylenetetrahydrofolate reductase. Am J Hum Genet. 1988;43:414–421. [PMC free article] [PubMed] [Google Scholar]
- 55.Friso S, Choi SW, Girelli D, Mason JB, Dolnikowski GG, Bagley PJ, Olivieri O, Jacques PF, Rosenberg IH, Corrocher R, et al. A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proc Natl Acad Sci U S A. 2002;99:5606–5611. doi: 10.1073/pnas.062066299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Clarke R, Halsey J, Bennett D, Lewington S. Homocysteine and vascular disease: review of published results of the homocysteine-lowering trials. J Inherit Metab Dis. 2011;34:83–91. doi: 10.1007/s10545-010-9235-y. [DOI] [PubMed] [Google Scholar]
- 57.Friso S, Choi SW, Dolnikowski GG, Selhub J. A method to assess genomic DNA methylation using high-performance liquid chromatography/electrospray ionization mass spectrometry. Anal Chem. 2002;74:4526–4531. doi: 10.1021/ac020050h. [DOI] [PubMed] [Google Scholar]
- 58.Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33(suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- 59.Huang C, Sloan EA, Boerkoel CF. Chromatin remodeling and human disease. Curr Opin Genet Dev. 2003;13:246–252. doi: 10.1016/s0959-437x(03)00054-6. [DOI] [PubMed] [Google Scholar]
- 60.Lamarre SG, Molloy AM, Reinke SN, Sykes BD, Brosnan ME, Brosnan JT. Formate can differentiate between hyperhomocysteinemia due to impaired remethylation and impaired transsulfuration. Am J Physiol Endocrinol Metab. 2012;302:E61–E67. doi: 10.1152/ajpendo.00345.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zeisel SH. Dietary choline deficiency causes DNA strand breaks and alters epigenetic marks on DNA and histones. Mutat Res. 2012;733:34–38. doi: 10.1016/j.mrfmmm.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nijhout HF, Reed MC, Budu P, Ulrich CM. A mathematical model of the folate cycle: new insights into folate homeostasis. J Biol Chem. 2004;279:55008–55016. doi: 10.1074/jbc.M410818200. [DOI] [PubMed] [Google Scholar]
- 63.Reed MC, Nijhout HF, Sparks R, Ulrich CM. Amathematical model of the methionine cycle. J Theor Biol. 2004;226:33–43. doi: 10.1016/j.jtbi.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 64.Ullah M, Schmidt H, Cho KH, Wolkenhauer O. Deterministic modelling and stochastic simulation of biochemical pathways using MATLAB. Syst Biol. 2006;153:53–60. doi: 10.1049/ip-syb:20050064. [DOI] [PubMed] [Google Scholar]
- 65.Martinov MV, Vitvitsky VM, Mosharov EV, Banerjee R, Ataullakhanov FI. A substrate switch: a new mode of regulation in the methionine metabolic pathway. J Theor Biol. 2000;204:521–532. doi: 10.1006/jtbi.2000.2035. [DOI] [PubMed] [Google Scholar]
- 66.Reed MC, Thomas RL, Pavisic J, James SJ, Ulrich CM, Nijhout HF. A mathematical model of glutathione metabolism. Theor Biol Med Model. 2008;5:8. doi: 10.1186/1742-4682-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Prudova A, Martinov MV, Vitvitsky VM, Ataullakhanov FI, Banerjee R. Analysis of pathological defects in methionine metabolism using a simple mathematical model. Biochim Biophys Acta. 2005;1741:331–338. doi: 10.1016/j.bbadis.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 68.Goel G, Chou IC, Voit EO. System estimation from metabolic time-series data. Bioinformatics. 2008;24:2505–2511. doi: 10.1093/bioinformatics/btn470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vilela M, Vinga S, Maia MA, Voit EO, Almeida JS. Identification of neutral biochemical network models from time series data. BMC Syst Biol. 2009;3:47. doi: 10.1186/1752-0509-3-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nijhout HF, Reed MC, Lam SL, Shane B, Gregory JF, 3rd, Ulrich CM. In silico experimentation with a model of hepatic mitochondrial folate metabolism. Theor Biol Med Model. 2006;3:40. doi: 10.1186/1742-4682-3-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reed MC, Nijhout HF, Neuhouser ML, Gregory JF, 3rd, Shane B, James SJ, Boynton A, Ulrich CM. A mathematical model gives insights into nutritional and genetic aspects of folate-mediated one-carbon metabolism. J Nutr. 2006;136:2653–2661. doi: 10.1093/jn/136.10.2653. [DOI] [PubMed] [Google Scholar]
- 72.Martinov MV, Plotnikov AG, Vitvitsky VM, Ataullakhanov FI. Deficiencies of glycolytic enzymes as a possible cause of hemolytic anemia. Biochim Biophys Acta. 2000;1474:75–87. doi: 10.1016/s0304-4165(99)00218-4. [DOI] [PubMed] [Google Scholar]
- 73.Strong WB, Tendler SJ, Seither RL, Goldman ID, Schirch V. Purification and properties of serine hydroxymethyltransferase and C1-tetrahydrofolate synthase from L1210 cells. J Biol Chem. 1990;265:12149–12155. [PubMed] [Google Scholar]
- 74.Daubner SC, Matthews RG. Purification and properties of methylenetetrahydrofolate reductase from pig liver. J Biol Chem. 1982;257:140–145. [PubMed] [Google Scholar]
- 75.Jackson RC, Harrap KR. Studies with a mathematical model of folate metabolism. Arch Biochem Biophys. 1973;158:827–841. doi: 10.1016/0003-9861(73)90579-1. [DOI] [PubMed] [Google Scholar]
- 76.Schirch V, Hopkins S, Villar E, Angelaccio S. Serine hydroxymethyltransferase from Escherichia coli: purification and properties. J Bacteriol. 1985;163:1–7. doi: 10.1128/jb.163.1.1-7.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schirch V. Purification of folate-dependent enzymes from rabbit liver. Methods Enzymol. 1997;281:146–161. doi: 10.1016/s0076-6879(97)81021-x. [DOI] [PubMed] [Google Scholar]
- 78.Schirch L, Peterson D. Purification and properties of mitochondrial serine hydroxymethyltransferase. J Biol Chem. 1980;255:7801–7806. [PubMed] [Google Scholar]
- 79.Seither RL, Trent DF, Mikulecky DC, Rape TJ, Goldman ID. Folate-pool interconversions and inhibition of biosynthetic processes after exposure of L1210 leukemia cells to antifolates. Experimental and network thermodynamic analyses of the role of dihydrofolate polyglutamylates in antifolate action in cells. J Biol Chem. 1989;264:17016–17023. [PubMed] [Google Scholar]
- 80.Vorontzov IN, Greshilov MM, Belousova AK, Gerasimova GK. Mathematical description and investigation of the principles of functioning of the folic acid cycle. Biokhimiya. 1980;45:83–97. [PubMed] [Google Scholar]
- 81.Morrison PF, Allegra CJ. Folate cycle kinetics in human breast cancer cells. J Biol Chem. 1989;264:10552–10566. [PubMed] [Google Scholar]
- 82.Gandhi VV, Samuels DC. Enzyme kinetics of the mitochondrial deoxyribonucleoside salvage pathway are not sufficient to support rapid mtDNA replication. PLoS Comput Biol. 2011;7:e1002078. doi: 10.1371/journal.pcbi.1002078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bradshaw PC, Li J, Samuels DC. A computational model of mitochondrial AZT metabolism. Biochem J. 2005;392:363–373. doi: 10.1042/BJ20050749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bradshaw PC, Samuels DC. A computational model of mitochondrial deoxynucleotide metabolism and DNA replication. Am J Physiol Cell Physiol. 2005;288:C989–C1002. doi: 10.1152/ajpcell.00530.2004. [DOI] [PubMed] [Google Scholar]
- 85.Poovathingal SK, Gruber J, Lakshmanan L, Halliwell B, Gunawan R. Is mitochondrial DNA turnover slower than commonly assumed? Biogerontology. 2012;13:557–564. doi: 10.1007/s10522-012-9390-7. [DOI] [PubMed] [Google Scholar]
- 86.Favre C, Zhdanov A, Leahy M, Papkovsky D, O’Connor R. Mitochondrial pyrimidine nucleotide carrier (PNC1) regulates mitochondrial biogenesis and the invasive phenotype of cancer cells. Oncogene. 2010;29:3964–3976. doi: 10.1038/onc.2010.146. [DOI] [PubMed] [Google Scholar]
- 87.Floyd S, Favre C, Lasorsa FM, Leahy M, Trigiante G, Stroebel P, Marx A, Loughran G, O’Callaghan K, Marobbio CM, et al. The insulin-like growth factor-I-mTOR signaling pathway induces the mitochondrial pyrimidine nucleotide carrier to promote cell growth. Mol Biol Cell. 2007;18:3545–3555. doi: 10.1091/mbc.E06-12-1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ferraro P, Nicolosi L, Bernardi P, Reichard P, Bianchi V. Mitochondrial deoxynucleotide pool sizes in mouse liver and evidence for a transport mechanism for thymidine monophosphate. Proc Natl Acad Sci U S A. 2006;103:18586–18591. doi: 10.1073/pnas.0609020103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bridges EG, Jiang Z, Cheng YC. Characterization of a dCTP transport activity reconstituted from human mitochondria. J Biol Chem. 1999;274:4620–4625. doi: 10.1074/jbc.274.8.4620. [DOI] [PubMed] [Google Scholar]
- 90.Wang L. Deoxynucleoside salvage enzymes and tissue specific mitochondrial DNA depletion. Nucleosides Nucleotides Nucleic Acids. 2010;29:370–381. doi: 10.1080/15257771003729732. [DOI] [PubMed] [Google Scholar]
- 91.Morris GW, Iams TA, Slepchenko KG, McKee EE. Origin of pyrimidine deoxyribonucleotide pools in perfused rat heart: implications for 3′-azido-3′-deoxythymidine-dependent cardiotoxicity. Biochem J. 2009;422:513–520. doi: 10.1042/BJ20082427. [DOI] [PubMed] [Google Scholar]
- 92.Traut TW. Physiological concentrations of purines and pyrimidines. Mol Cell Biochem. 1994;140:1–22. doi: 10.1007/BF00928361. [DOI] [PubMed] [Google Scholar]
- 93.Li KM, Rivory LP, Hoskins J, Sharma R, Clarke SJ. Altered deoxyuridine and thymidine in plasma following capecitabine treatment in colorectal cancer patients. Br J Clin Pharmacol. 2007;63:67–74. doi: 10.1111/j.1365-2125.2006.02710.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Matthews RG. Methylenetetrahydrofolate reductase from pig liver. Methods Enzymol. 1986;122:372–381. doi: 10.1016/0076-6879(86)22196-5. [DOI] [PubMed] [Google Scholar]
- 95.Green JM, MacKenzie RE, Matthews RG. Substrate flux through methylenetetrahydrofolate dehydrogenase: predicted effects of the concentration of methylenetetrahydrofolate on its partitioning into pathways leading to nucleotide biosynthesis or methionine regeneration. Biochemistry. 1988;27:8014–8022. doi: 10.1021/bi00421a007. [DOI] [PubMed] [Google Scholar]
- 96.Jencks DA, Mathews RG. Allosteric inhibition of methylenetetrahydrofolate reductase by adenosylmethionine. Effects of adenosylmethionine and NADPH on the equilibrium between active and inactive forms of the enzyme and on the kinetics of approach to equilibrium. J Biol Chem. 1987;262:2485–2493. [PubMed] [Google Scholar]
- 97.Yamada K, Chen Z, Rozen R, Matthews RG. Effects of common polymorphisms on the properties of recombinant human methylenetetrahydrofolate reductase. Proc Natl Acad Sci USA. 2001;98:14853–14858. doi: 10.1073/pnas.261469998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ogawa H, Fujioka M. Induction of rat liver glycine methyltransferase by high methionine diet. Biochem Biophys Res Commun. 1982;108:227–232. doi: 10.1016/0006-291x(82)91855-1. [DOI] [PubMed] [Google Scholar]
- 99.Yeo EJ, Wagner C. Purification and properties of pancreatic glycine N-methyltransferase. J Biol Chem. 1992;267:24669–24674. [PubMed] [Google Scholar]
- 100.Wagner C, Briggs WT, Cook RJ. Inhibition of glycine N-methyltransferase activity by folate derivatives: implications for regulation of methyl group metabolism. Biochem Biophys Res Commun. 1985;127:746–752. doi: 10.1016/s0006-291x(85)80006-1. [DOI] [PubMed] [Google Scholar]
- 101.Geenen S, du Preez FB, Reed M, Nijhout HF, Kenna JG, Wilson ID, Westerhoff HV, Snoep JL. A mathematical modelling approach to assessing the reliability of biomarkers of glutathione metabolism. Eur J Pharm Sci. 2012;46:233–243. doi: 10.1016/j.ejps.2011.08.017. [DOI] [PubMed] [Google Scholar]
- 102.Neuhouser ML, Nijhout HF, Gregory JF, 3rd, Reed MC, James SJ, Liu A, Shane B, Ulrich CM. Mathematical modeling predicts the effect of folate deficiency and excess on cancer-related biomarkers. Cancer Epidemiol Biomarkers Prev. 2011;20:1912–1917. doi: 10.1158/1055-9965.EPI-10-1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Deplancke B, Gaskins HR. Redox control of the transsulfuration and glutathione biosynthesis pathways. Curr Opin Clin Nutr Metab Care. 2002;5:85–92. doi: 10.1097/00075197-200201000-00015. [DOI] [PubMed] [Google Scholar]
- 104.Pogribna M, Melnyk S, Pogribny I, Chango A, Yi P, James SJ. Homocysteine metabolism in children with Down syndrome: in vitro modulation. Am J Hum Genet. 2001;69:88–95. doi: 10.1086/321262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ueland PM, Refsum H, Christensen B. Methotrexate sensitivity in Down’s syndrome: a hypothesis. Cancer Chemother Pharmacol. 1990;25:384–386. doi: 10.1007/BF00686245. [DOI] [PubMed] [Google Scholar]
- 106.Shane B, Stokstad EL. The interrelationships among folate, vitamin B12, and methionine metabolism. Adv Nutr Res. 1983;5:133–170. doi: 10.1007/978-1-4613-9937-7_7. [DOI] [PubMed] [Google Scholar]
- 107.Nijhout HF, Gregory JF, Fitzpatrick C, Cho E, Lamers KY, Ulrich CM, Reed MC. A mathematical model gives insights into the effects of vitamin B-6 deficiency on 1-carbon and glutathione metabolism. J Nutr. 2009;139:784–791. doi: 10.3945/jn.109.104265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Perry C, Yu S, Chen J, Matharu KS, Stover PJ. Effect of vitamin B6 availability on serine hydroxymethyl-transferase in MCF-7 cells. Arch Biochem Biophys. 2007;462:21–27. doi: 10.1016/j.abb.2007.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.MacFarlane AJ, Liu X, Perry CA, Flodby P, Allen RH, Stabler SP, Stover PJ. Cytoplasmic serine hydroxymethyltransferase regulates the metabolic partitioning of methylenetetrahydrofolate but is not essential in mice. J Biol Chem. 2008;283:25846–25853. doi: 10.1074/jbc.M802671200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Runyan TJ, Gershoff SN. Glycine metabolism in vitamin B6-deficient and deoxypyridoxine-treated rats. J Nutr. 1969;98:113–118. doi: 10.1093/jn/98.1.113. [DOI] [PubMed] [Google Scholar]
- 111.Rosenblatt D. Inborn errors of folate and cobalamin metabolism. In: Carmel R, Jacobsen D, editors. Homocysteine in Health and Disease. Cambridge: Cambridge University Press; 2001. pp. 244–258. [Google Scholar]
- 112.Kluijtmans LA, Young IS, Boreham CA, Murray L, McMaster D, McNulty H, Strain JJ, McPartlin J, Scott JM, Whitehead AS. Genetic and nutritional factors contributing to hyperhomocysteinemia in young adults. Blood. 2003;101:2483–2488. doi: 10.1182/blood.V101.7.2483. [DOI] [PubMed] [Google Scholar]
- 113.McNulty H, McKinley MC, Wilson B, McPartlin J, Strain JJ, Weir DG, Scott JM. Impaired functioning of thermolabile methylenetetrahydrofolate reductase is dependent on riboflavin status: implications for riboflavin requirements. Am J Clin Nutr. 2002;76:436–441. doi: 10.1093/ajcn/76.2.436. [DOI] [PubMed] [Google Scholar]
- 114.Mato JM, Lu SC. Role of S-adenosyl-L-methionine in liver health and injury. Hepatology. 2007;45:1306–1312. doi: 10.1002/hep.21650. [DOI] [PubMed] [Google Scholar]
- 115.Mudd SH, Poole JR. Labile methyl balances for normal humans on various dietary regimens. Metabolism. 1975;24:721–735. doi: 10.1016/0026-0495(75)90040-2. [DOI] [PubMed] [Google Scholar]
- 116.Mato JM, Corrales FJ, Lu SC, Avila MA. S-Adenosyl methionine: a control switch that regulates liver function. FASEB J. 2002;16:15–26. doi: 10.1096/fj.01-0401rev. [DOI] [PubMed] [Google Scholar]
- 117.Cai J, Mao Z, Hwang JJ, Lu SC. Differential expression of methionine adenosyltransferase genes influences the rate of growth of human hepatocellular carcinoma cells. Cancer Res. 1998;58:1444–1450. [PubMed] [Google Scholar]
- 118.Kotb M, Mudd SH, Mato JM, Geller AM, Kredich NM, Chou JY, Cantoni GL. Consensus nomenclature for the mammalian methionine adenosyltransferase genes and gene products. Trends Genet. 1997;13:51–52. doi: 10.1016/s0168-9525(97)01013-5. [DOI] [PubMed] [Google Scholar]
- 119.Gil B, Casado M, Pajares MA, Bosca L, Mato JM, Martin-Sanz P, Alvarez L. Differential expression pattern of S-adenosylmethionine synthetase isoenzymes during rat liver development. Hepatology. 1996;24:876–881. doi: 10.1002/hep.510240420. [DOI] [PubMed] [Google Scholar]
- 120.Sullivan DM, Hoffman JL. Fractionation and kinetic properties of rat liver and kidney methionine adenosyltransferase isozymes. Biochemistry. 1983;22:1636–1641. doi: 10.1021/bi00276a017. [DOI] [PubMed] [Google Scholar]
- 121.Finkelstein JD. Methionine metabolism in mammals. J Nutr Biochem. 1990;1:228–237. doi: 10.1016/0955-2863(90)90070-2. [DOI] [PubMed] [Google Scholar]
- 122.Avila MA, Berasain C, Torres L, Martin-Duce A, Corrales FJ, Yang H, Prieto J, Lu SC, Caballeria J, Rodes J, et al. Reduced mRNA abundance of the main enzymes involved in methionine metabolism in human liver cirrhosis and hepatocellular carcinoma. J Hepatol. 2000;33:907–914. doi: 10.1016/s0168-8278(00)80122-1. [DOI] [PubMed] [Google Scholar]
- 123.Kotb M, Kredich NM. S-Adenosylmethionine synthetase from human lymphocytes. Purification and characterization. J Biol Chem. 1985;260:3923–3930. [PubMed] [Google Scholar]
- 124.J F. Regulation of homocysteine metabolism in homocysteine. In: Carmel R, Jacobson DW, editors. Homocysteine in Health and Disease. Cambridge: Cambridge University Press; 2001. pp. 92–99. [Google Scholar]
- 125.Taoka S, Ohja S, Shan X, Kruger WD, Banerjee R. Evidence for heme-mediated redox regulation of human cystathionine β-synthase activity. J Biol Chem. 1998;273:25179–25184. doi: 10.1074/jbc.273.39.25179. [DOI] [PubMed] [Google Scholar]
- 126.Janosik M, Kery V, Gaustadnes M, Maclean KN, Kraus JP. Regulation of human cystathionine β-synthase by S-adenosyl-L-methionine: evidence for two catalytically active conformations involving an autoinhibitory domain in the C-terminal region. Biochemistry. 2001;40:10625–10633. doi: 10.1021/bi010711p. [DOI] [PubMed] [Google Scholar]
- 127.Kluijtmans LA, Boers GH, Stevens EM, Renier WO, Kraus JP, Trijbels FJ, van den Heuvel LP, Blom HJ. Defective cystathionine β-synthase regulation by S-adenosylmethionine in a partially pyridoxine responsive homocystinuria patient. J Clin Invest. 1996;98:285–289. doi: 10.1172/JCI118791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Martinez M, Cuskelly GJ, Williamson J, Toth JP, Gregory JF., 3rd Vitamin B-6 deficiency in rats reduces hepatic serine hydroxymethyltransferase and cystathionine β-synthase activities and rates of in vivo protein turnover, homocysteine remethylation and transsulfuration. J Nutr. 2000;130:1115–1123. doi: 10.1093/jn/130.5.1115. [DOI] [PubMed] [Google Scholar]
- 129.Heby O. DNA methylation and polyamines in embryonic development and cancer. Int J Dev Biol. 1995;39:737–757. [PubMed] [Google Scholar]
- 130.Heby O, Persson L, Smith SS. Polyamines, DNA methylation and cell differentiation. Adv Exp Med Biol. 1988;250:291–299. doi: 10.1007/978-1-4684-5637-0_26. [DOI] [PubMed] [Google Scholar]
- 131.Frostesjo L, Holm I, Grahn B, Page AW, Bestor TH, Heby O. Interference with DNA methyltransferase activity and genome methylation during F9 teratocarcinoma stem cell differentiation induced by polyamine depletion. J Biol Chem. 1997;272:4359–4366. doi: 10.1074/jbc.272.7.4359. [DOI] [PubMed] [Google Scholar]
- 132.Montanez R, Rodriguez-Caso C, Sanchez-Jimenez F, Medina MA. In silico analysis of arginine catabolism as a source of nitric oxide or polyamines in endothelial cells. Amino Acids. 2008;34:223–229. doi: 10.1007/s00726-007-0502-7. [DOI] [PubMed] [Google Scholar]
- 133.Rodriguez-Caso C, Montanez R, Cascante M, Sanchez-Jimenez F, Medina MA. Mathematical modeling of polyamine metabolism in mammals. J Biol Chem. 2006;281:21799–21812. doi: 10.1074/jbc.M602756200. [DOI] [PubMed] [Google Scholar]
- 134.Wagner C. Biochemical role of folate in cellular metabolism. In: Bailey LB, editor. Folate in Health and Disease. New York: Marcel Dekker; 1995. pp. 23–42. [Google Scholar]
- 135.Min H, Shane B, Stokstad EL. Identification of 10-formyltetrahydrofolate dehydrogenase-hydrolase as a major folate binding protein in liver cytosol. Biochim Biophys Acta. 1988;967:348–353. doi: 10.1016/0304-4165(88)90097-9. [DOI] [PubMed] [Google Scholar]
- 136.Fowler B. The folate cycle and disease in humans. Kidney Int Suppl. 2001;78:S221–S229. doi: 10.1046/j.1523-1755.2001.59780221.x. [DOI] [PubMed] [Google Scholar]
- 137.Giovannucci E. Epidemiologic studies of folate and colorectal neoplasia: a review. J Nutr. 2002;132:2350S–2355S. doi: 10.1093/jn/132.8.2350S. [DOI] [PubMed] [Google Scholar]
- 138.MacFarlane AJ, Stover PJ. Convergence of genetic, nutritional and inflammatory factors in gastrointestinal cancers. Nutr Rev. 2007;65:S157–S166. doi: 10.1111/j.1753-4887.2007.tb00355.x. [DOI] [PubMed] [Google Scholar]
- 139.Choi SW, Mason JB. Folate and carcinogenesis: an integrated scheme. J Nutr. 2000;130:129–132. doi: 10.1093/jn/130.2.129. [DOI] [PubMed] [Google Scholar]
- 140.Beaudin AE, Stover PJ. Insights into metabolic mechanisms underlying folate-responsive neural tube defects: a minireview. Birth Defects Res A Clin Mol Teratol. 2009;85:274–284. doi: 10.1002/bdra.20553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fatmi MQ, Chang CE. The role of oligomerization and cooperative regulation in protein function: the case of tryptophan synthase. PLoS Comput Biol. 2010;6:e1000994. doi: 10.1371/journal.pcbi.1000994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Marianayagam NJ, Sunde M, Matthews JM. The power of two: protein dimerization in biology. Trends Biochem Sci. 2004;29:618–625. doi: 10.1016/j.tibs.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 143.Bahar I, Chennubhotla C, Tobi D. Intrinsic dynamics of enzymes in the unbound state and relation to allosteric regulation. Curr Opin Struct Biol. 2007;17:633–640. doi: 10.1016/j.sbi.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Woolf PJ, Linderman JJ. Self organization of membrane proteins via dimerization. Biophys Chem. 2003;104:217–227. doi: 10.1016/s0301-4622(02)00369-1. [DOI] [PubMed] [Google Scholar]
- 145.Klitgord N, Segre D. The importance of compartmentalization in metabolic flux models: yeast as an ecosystem of organelles. Genome Inform. 2010;22:41–55. [PubMed] [Google Scholar]
- 146.Garofalo T, Tinari A, Matarrese P, Giammarioli AM, Manganelli V, Ciarlo L, Misasi R, Sorice M, Malorni W. Do mitochondria act as “cargo boats” in the journey of GD3 to the nucleus during apoptosis? FEBS Lett. 2007;581:3899–3903. doi: 10.1016/j.febslet.2007.07.020. [DOI] [PubMed] [Google Scholar]
- 147.An S, Deng Y, Tomsho JW, Kyoung M, Benkovic SJ. Microtubule-assisted mechanism for functional metabolic macromolecular complex formation. Proc Natl Acad Sci U S A. 2010;107:12872–12876. doi: 10.1073/pnas.1008451107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Field MS, Szebenyi DM, Perry CA, Stover PJ. Inhibition of 5,10-methenyltetrahydrofolate synthetase. Arch Biochem Biophys. 2007;458:194–201. doi: 10.1016/j.abb.2006.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bachman JA, Sorger P. New approaches to modeling complex biochemistry. Nat Methods. 2011;8:130–131. doi: 10.1038/nmeth0211-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.E W, Liu D, Vanden-Eijnden E. Nested stochastic simulation algorithm for chemical kinetic systems with disparate rates. J Chem Phys. 2005;123:194107. doi: 10.1063/1.2109987. [DOI] [PubMed] [Google Scholar]
- 151.Danos V, Feret J, Fontana W, Harmer R, Krivine J. Rule-based modelling, symmetries, refinements. (Lecture Notes in Bioinformatics).Formal Methods in Systems Biology. 2008;5054:103–122. [Google Scholar]
- 152.Andrews SS. Spatial and stochastic cellular modeling with the Smoldyn simulator. Methods Mol Biol. 2012;804:519–542. doi: 10.1007/978-1-61779-361-5_26. [DOI] [PubMed] [Google Scholar]
- 153.Dematte L, Priami C, Romanel A. The Beta Workbench: a computational tool to study the dynamics of biological systems. Brief Bioinform. 2008;9:437–449. doi: 10.1093/bib/bbn023. [DOI] [PubMed] [Google Scholar]
- 154.Turner TE, Schnell S, Burrage K. Stochastic approaches for modelling in vivo reactions. Comput Biol Chem. 2004;28:165–178. doi: 10.1016/j.compbiolchem.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 155.Gillespie DT. Stochastic simulation of chemical kinetics. Annu Rev Phys Chem. 2007;58:35–55. doi: 10.1146/annurev.physchem.58.032806.104637. [DOI] [PubMed] [Google Scholar]
- 156.Alfonsi A, Cancés E, Turinici G, Di Ventura B, Huisinga W. Adaptive simulation of hybrid stochastic and deterministic models for biochemical systems. ESAIM: Proc. 2005;14:1–13. [Google Scholar]
- 157.Ames BN, Elson-Schwab I, Silver EA. High-dose vitamin therapy stimulates variant enzymes with decreased coenzyme binding affinity (increased K(m)): relevance to genetic disease and polymorphisms. Am J Clin Nutr. 2002;75:616–658. doi: 10.1093/ajcn/75.4.616. [DOI] [PubMed] [Google Scholar]
- 158.Curto R, Voit EO, Sorribas A, Cascante M. Mathematical models of purine metabolism in man. Math Biosci. 1998;151:1–49. doi: 10.1016/s0025-5564(98)10001-9. [DOI] [PubMed] [Google Scholar]
- 159.Stover PJ. Nutritional genomics. Physiol Genom. 2004;16:161–165. doi: 10.1152/physiolgenomics.00204.2003. [DOI] [PubMed] [Google Scholar]
- 160.Sneddon MW, Faeder JR, Emonet T. Efficient modeling, simulation and coarse-graining of biological complexity with NFsim. Nat Methods. 2011;8:177–183. doi: 10.1038/nmeth.1546. [DOI] [PubMed] [Google Scholar]