

Microglia have been implicated in amyotrophic lateral sclerosis (ALS) progression, but the underlying mechanisms are unclear. Mesci et al. reveal that suppression of system (a cystine/glutamate transporter) reduces microglia-triggered glutamate excitotoxicity and switches microglia to a neuroprotective phenotype: this slows disease progression and increases neuronal survival in ALS mice.
Keywords: ALS, neuroinflammation, excitotoxicity, motoneuron, xCT
Abstract
Amyotrophic lateral sclerosis is the most common adult-onset motor neuron disease and evidence from mice expressing amyotrophic lateral sclerosis-causing SOD1 mutations suggest that neurodegeneration is a non-cell autonomous process where microglial cells influence disease progression. However, microglial-derived neurotoxic factors still remain largely unidentified in amyotrophic lateral sclerosis. With excitotoxicity being a major mechanism proposed to cause motor neuron death in amyotrophic lateral sclerosis, our hypothesis was that excessive glutamate release by activated microglia through their system (a cystine/glutamate antiporter with the specific subunit xCT/Slc7a11) could contribute to neurodegeneration. Here we show that xCT expression is enriched in microglia compared to total mouse spinal cord and absent from motor neurons. Activated microglia induced xCT expression and during disease, xCT levels were increased in both spinal cord and isolated microglia from mutant SOD1 amyotrophic lateral sclerosis mice. Expression of xCT was also detectable in spinal cord post-mortem tissues of patients with amyotrophic lateral sclerosis and correlated with increased inflammation. Genetic deletion of xCT in mice demonstrated that activated microglia released glutamate mainly through system . Interestingly, xCT deletion also led to decreased production of specific microglial pro-inflammatory/neurotoxic factors including nitric oxide, TNFa and IL6, whereas expression of anti-inflammatory/neuroprotective markers such as Ym1/Chil3 were increased, indicating that xCT regulates microglial functions. In amyotrophic lateral sclerosis mice, xCT deletion surprisingly led to earlier symptom onset but, importantly, this was followed by a significantly slowed progressive disease phase, which resulted in more surviving motor neurons. These results are consistent with a deleterious contribution of microglial-derived glutamate during symptomatic disease. Therefore, we show that system participates in microglial reactivity and modulates amyotrophic lateral sclerosis motor neuron degeneration, revealing system inactivation, as a potential approach to slow amyotrophic lateral sclerosis disease progression after onset of clinical symptoms.
Introduction
Amyotrophic lateral sclerosis (ALS) is characterized by loss of upper and lower motor neurons leading to progressive paralysis and death of patients. Although mainly sporadic, ∼10% of ALS cases are inherited and the second most frequent cause of familial ALS are dominant mutations in the gene encoding Cu/Zn superoxide dismutase (SOD1) (Rosen et al., 1993). As of today, the animal models recapitulating closest ALS symptoms are mice and rats expressing mutant human SOD1 (while expression of wild-type human SOD1 does not lead to disease) (Boillee et al., 2006a). In these models, ubiquitous expression of mutant SOD1 leads to progressive motor neuron death through an unknown gain-of-toxic function for which several hypotheses have been proposed including excitotoxicity and neuroinflammation (Boillee et al., 2006a). In addition, motor neuron death involves a non-cell autonomous mechanism, with neighbouring non-neuronal cells participating in the disease (Clement et al., 2003; Beers et al., 2006; Boillee et al., 2006b; Yamanaka et al., 2008; Lobsiger et al., 2009; Kang et al., 2013). In particular, reducing mutant SOD1 expression selectively in microglial cells/macrophages, strongly slowed down disease progression in ALS mice (Beers et al., 2006; Boillee et al., 2006b; Wang et al., 2009).
Microglial cells are the macrophages of the CNS. They are known to respond to any CNS insult including during neurodegenerative diseases and their responses can have both neuroprotective and neurotoxic effects (Czeh et al., 2011). By analogy to macrophages, microglial cells are able to modify their phenotypes from an anti-inflammatory/neuroprotective (M2) to a pro-inflammatory/neurotoxic (M1) state, with M1/M2 being the two extremes of a phenotype continuum. These phenotype changes are thought to occur during chronic neurodegenerative conditions, and have been detected in spinal cords of ALS mice (Beers et al., 2011; Liao et al., 2012). The consequences of these changes on the actual ALS disease course are still largely unknown. A hint comes from administration of minocycline in ALS mice, which impacted global M1/M2 microglial polarization (Kobayashi et al., 2013), however, minocycline is also known to have direct anti-apoptotic actions on motor neurons themselves (Zhu et al., 2002). In addition, genetic deletion of the superoxide-producing enzyme NOX2/cybb (strongly expressed in microglia), suggested that reducing a specific M1-phenotype related factor could benefit disease in ALS mice (Wu et al., 2006; Marden et al., 2007).
Therefore, as a strong candidate implicated in disease, targeting microglia to slow down ALS disease progression could best be achieved by both maintaining microglia in an anti-inflammatory/neuroprotective (M2) state, and blocking the disease-relevant pro-inflammatory/neurotoxic (M1) microglial-derived factors.
As part of preventing neurotoxicity, we focused on glutamate excitotoxicity, one of the longest standing proposed pathological mechanisms for ALS. Motor neurons are highly sensitive to glutamate excitotoxicity, especially in the context of ALS-linked mutations (Rothstein et al., 1995; Van Den Bosch et al., 2006), as most recently shown using neurons derived from human-induced pluripotent stem cells carrying the C9orf72 mutation (Donnelly et al., 2013). ALS-associated glutamate excitotoxicity could also be promoted by non-cell autonomous mechanisms, as motor neuron neighbouring astrocytes show decreased expression of reuptake transporters for synaptic glutamate, both in ALS mice/rats and human tissues (Rothstein et al., 1995; Howland et al., 2002). Importantly, microglia on their own could also directly contribute to ALS-associated excitotoxicity, as they can release glutamate (Qin et al., 2006; Maezawa and Jin, 2010; Takeuchi et al., 2011). We now propose that system could be a major contributor of microglial-derived glutamate. System is a cystine/glutamate antiporter capturing extracellular cystine, used for glutathione synthesis, in exchange for glutamate release. It is composed of two subunits, one common to several amino acid transporters, SLC3A2, and a specific one, xCT/Slc7a11, a 12 transmembrane domain protein that forms the channel (Sato et al., 1999; Gasol et al., 2004). System , although considered a more glial than neuronal transporter, is also known to be expressed by the macrophage lineage (Watanabe and Bannai, 1987; Piani and Fontana, 1994; Sato et al., 2001) and has been detected in reactive microglia of Alzheimer’s disease model mice (Qin et al., 2006) however, its microglial expression and regulation in ALS mice has so far not been assessed.
We hypothesized that modulation of system in ALS could be beneficial in two ways. First, activated microglia could release excessive glutamate. Thus, system deletion would reduce glutamate excitotoxicity. Second, both extracellular glutamate and intracellular glutathione can influence microglial activation, by acting through microglial expressed glutamate receptors (Kaindl et al., 2012; Lewerenz et al., 2013) or by acting (in the case of glutathione) on redox modulated intracellular microglial signalling cascades (Ogunrinu and Sontheimer, 2010; Lewerenz et al., 2013). Thus, system suppression could directly influence the microglial M1/M2 polarization state during ALS disease progression.
With the present study, we therefore used xCT (Slc7a11) gene deletion, to assess if modulation of system can influence overall microglial reactivity and therefore disease course and motor neuron degeneration in mutant SOD1 ALS mice.
Materials and methods
Additional information is supplied in the online Supplementary material.
Animals
Mice were hSOD1G85R, hSOD1G37R (Boillee et al., 2006b), control hSOD1WT [C57Bl/6-Tg(SOD1)3Cje/J, Jackson Laboratory], and mice deleted for the Slc7a11 gene (xCT−/− mice) (Sato et al., 2005), all on a C57BL/6J background (Janvier). All procedures were performed in accordance with the guidelines for care and use of experimental animals of the European Union and approved by the ethics committee for animal experimentation n°5 in Ile-de-France.
Survival analysis
A two-step mating strategy gave hSOD1G37R:xCT−/− (n = 21), hSOD1G37R:xCT+/− (n = 35) and hSOD1G37R:xCT+/+ (n = 24) mice, which were weighted weekly as an objective and unbiased measure of disease course (Boillee et al., 2006b; Lobsiger et al., 2009) and their grip strength measured (Bioseb, grip test). The three groups of mice started with comparable weight and grip-strength means.
Primary microglial cell culture
Cells were derived from postnatal Day 0 or 1 mouse pup cerebral cortex according to Thery et al. (1991). Highly pure microglia (>99%, quantified after immunostaining with microglial specific antibodies CD11b/Itgam, F4/80/Emr1 and Hoechst 33342 nuclear staining dye) were plated for the different assays and their survival measured. For immunostaining anti-xCT (Novus Bio; 1:5000), anti-CD11b (Serotec, 1:400) and F4/80 (Serotec, 1:100) antibodies were used. Fluorescence signal (integrated density) was measured per cell and compared to the control condition (xCT+/+ microglia without lipopolysaccharide treatment), using ImageJ software (n = 3 experiments per genotype).
Glutamate assay
Glutamate released (for 30 h after adding lipopolysaccharide, 20 000 cells/well, n = 3–4 experiments per genotype and treatment) was measured with the glutamate dehydrogenase-based colorimetric assay of Beutler (1985).
Nitric oxide assay
Nitric oxide production was assessed by measuring nitrite levels (a stable by-product of nitric oxide) with the colorimetric Griess method for 50 000 cells/well (n = 3 experiments per genotype and treatment).
Luminex assay
Microglia were plated at a density of 5 × 104 cells/96-well-plates (5–6 wells/condition, n = 3–4 experiments per genotype and treatment). All samples (25 µl of medium) were run in duplicates with Milliplex Map kits and analysed with the Magpix system (Life Technologies).
Glutathione assay
Total glutathione levels were measured in the spinal cord of 1-year-old mice using the QuantiChrom Glutathione Assay Kit (BioAssay Systems).
RNA extraction and real-time PCR
RNA extraction for tissues and cells was performed with Qiagen RNeasy® Kits (Qiagen). Reverse transcription was performed with SuperScript® III (Life Technologies) using 500 ng of RNA for spinal cord tissues, 250 ng for cultures and human spinal cord tissues, and 20 ng or 2 ng for laser-microdissected motor neurons and adult mouse spinal cord microglia, respectively. Quantitative PCRs were performed with SYBR® Green Master Mix (Applied Biosystems).
Laser microdissection of motor neurons
Motor neuron laser microdissection was performed as previously described (Lobsiger et al., 2007). From one lumbar (L4–L6) spinal cord, 350 motor neurons were collected yielding ∼20 ng of RNA.
Isolation of microglia from adult mouse spinal cord
Microglial cells were isolated as described by De Groot et al. (2000), followed by purification with anti-CD11b microbead-coupled antibodies and Miltenyi MS columns. Approximately 7–10 × 104 CD11b+ cells were recovered from one control spinal cord.
Immunohistochemistry on spinal cord tissues
Thirty-micrometre paraformaldehyde fixed cryosections were stained with: anti-Iba1 (Wako Chemicals, 1:500), anti-Mac-2 (Cederlane, 1:500), anti-GFAP (Dako, 1:4000) and anti-ChAT (Millipore, 1:200) for n = 3–4 animals per genotype and per time point.
Motor neuron counts
Motor neuron numbers were determined from 30-μm serial sections across the entire lumbar spinal cord and counted in every 12th ChAT stained section corresponding to a total of 23–26 sections per animal for n = 3 animals per genotype and time point.
Human samples
Patients with ALS were enrolled in a brain donation programme declared to the Ministry of Research and Universities, as requested by French Law. An explicit consent was signed by the patient, or by the next of kin, in the name of the patient, in accordance with the French Bioethical Laws.
Immunoblots
Proteins (16 µg) were separated on NuPAGE™ 4–12% Bis–Tris Gel (Invitrogen) and electrophoretically transferred to nitrocellulose membranes stained with anti-xCT (Abcam; 1:1500) and anti-beta-actin (Sigma; 1:6000) antibodies.
Statistical analysis
Two-way ANOVA tests followed by a Bonferroni multiple comparison test were used to compare groups with different genotypes and different treatments (with or without lipopolysaccharide). One-way ANOVA tests followed by a Bonferroni multiple comparison test were used to compare multiple groups, Student’s t-test to compare means of two groups and Kaplan-Meier Log Rank test for survival curves.
Results
Primary microglial cells express system and xCT levels are increased on microglial activation
To study the implication of system and microglial glutamate release in ALS, we first studied the expression of system in primary mouse microglial cells. The mRNA for xCT, the specific subunit of system , was expressed by control (xCT+/+) microglia and strongly increased (18-fold) when cells were activated with lipopolysaccharide, the most commonly used factor to activate microglia (Fig. 1A). At a protein level, xCT was present in the cytoplasm and on the plasma membrane of xCT+/+ microglia (with the signal absent in xCT−/− cells) (Fig. 1B) and likewise strongly increased (3-fold) upon activation with lipopolysaccharide (Fig. 1B and C). Next, we confirmed that primary microglial cells from both mutant (hSOD1G37R, hSOD1G85R) and wild-type (hSOD1WT) human SOD1 expressing mice were also able to induce xCT levels after lipopolysaccharide activation (Fig. 1A and C).
Figure 1.

xCT, the specific subunit of system is expressed by primary mouse microglial cells and its level increases upon microglial activation. xCT levels were measured in primary microglial cells from mice expressing xCT (xCT+/+), or not (xCT−/−), heterozygous for xCT (xCT+/−) or expressing different forms of mutant (hSOD1G37R, hSOD1G85R) or wild-type (hSOD1WT) hSOD1, after lipopolysaccharide (LPS, 100 ng/ml) treatment or in (non-treated) control conditions. (A) Microglial xCT mRNA levels measured by reverse-transcription quantitative PCR and normalized to the housekeeping gamma-actin (Actg1) gene. Results are shown relative to non-treated control microglia (xCT+/+). (B) Representative pictures of cultured microglial cells stained with anti-xCT antibodies (green) and fluorescent nuclear Hoechst stain (blue). (C) xCT immunofluorescent intensities (integrated densities per cell) measured for each cell. Bars represent means of fold changes, ± SEM; *P < 0.05, **P < 0.01 and ***P < 0.001, two-way ANOVA with Bonferroni multiple comparison test, relative to non-treated cells of the same genotype; n = 3 experiments per genotype; n.s. = non-significant. Scale bar in B = 50 µm.
Importantly, as no differences between wild-type hSOD1, mutant hSOD1 or non-transgenic microglia were detected, these results suggest that induction of system was solely dependent on the microglial activation state and not the presence of a specific ALS-linked gene mutation.
Deletion of xCT reduces the pro-inflammatory phenotype of activated primary microglia
To study system involvement in glutamate release by primary microglial cells (and linked to it, its potential contribution in excitotoxicity), we measured glutamate concentration in culture medium. Unstimulated microglia released glutamate at 10–50 μM after 30 h in culture and this increased by 70% upon lipopolysaccharide activation (Fig. 2A). Importantly, xCT deletion in microglial cells from xCT−/− mice, led to a 70% decrease in glutamate release compared to control (xCT+/+) microglia (Fig. 2A). In addition, treating xCT−/− microglial cells with lipopolysaccharide did not raise glutamate production (Fig. 2A). Therefore, our findings show that microglial cells release glutamate mainly through their system . Of note, mutant or wild-type hSOD1 expressing microglia released similar amounts of glutamate as control microglia, both with and without lipopolysaccharide activation (Fig. 2B). These data show (similar to the xCT expression, above) that glutamate release did not depend on the presence of mutant or wild-type hSOD1 but solely on the microglial activation state.
Figure 2.
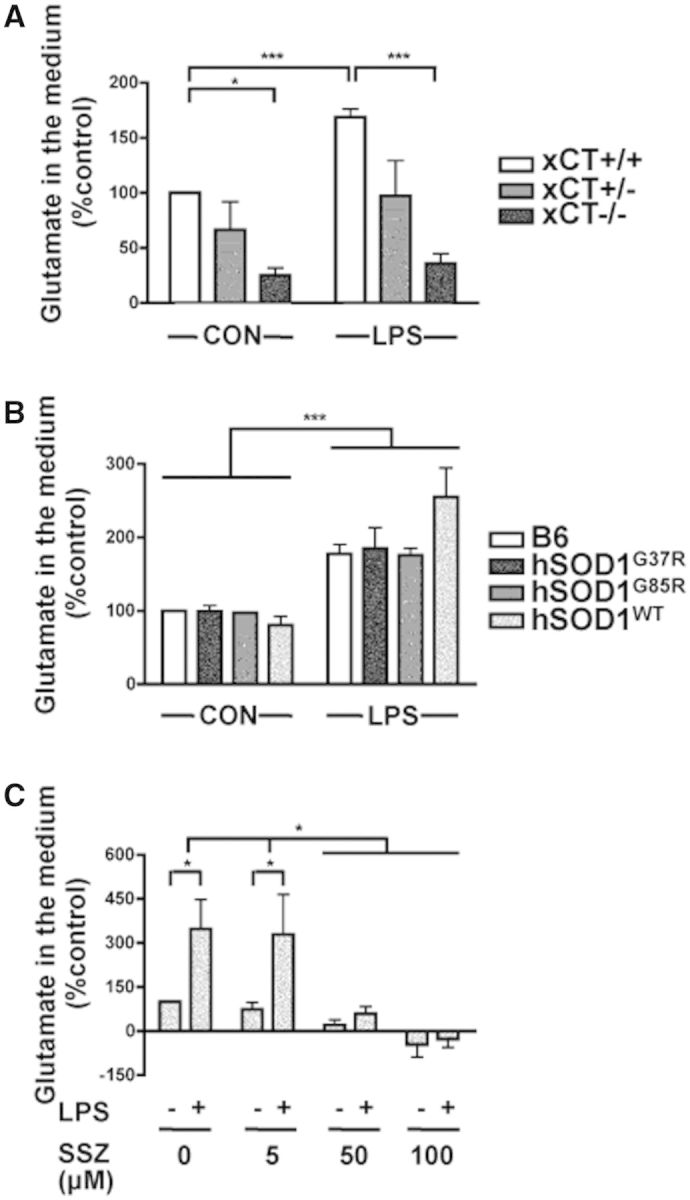
System deletion or inhibition strongly reduces glutamate release from primary mouse microglial cells. Glutamate release was measured in the culture medium of primary microglial cells from mice expressing (xCT+/+) or not (xCT−/−) xCT, heterozygous for xCT (xCT+/−) or expressing different forms of mutant (hSOD1G37R, hSOD1G85R) or wild-type (hSOD1WT) hSOD1, after lipopolysaccharide (LPS, 100 ng/ml) treatment or in (non-treated) control (CON) conditions. (A and B) Glutamate levels are shown relative to non-treated control microglia (xCT+/+) (A), or non-transgenic littermate control C57BL/6J microglia (B6) (B). Note that because of variability, the apparent trend of increased glutamate release in lipopolysaccharide-treated hSOD1WT microglia was not statistically significant. (C) Glutamate levels in media of control microglia (B6), treated or not with lipopolysaccharide and increasing concentrations of sulfasalazine (SSZ). Bars represent means of percentages, ± SEM; *P < 0.05 and ***P < 0.001, two-way ANOVA with Bonferroni multiple comparison test; n = 3 experiments per genotype in A, and B and per dose of sulfasalazine in C.
Although system blockers are known to lack specificity (Lewerenz et al., 2013), we tested one, sulfasalazine, and found that it blocked microglial glutamate release both in control conditions and after lipopolysaccharide treatment (Fig. 2C), consistent with system being responsible for the measured glutamate release.
To test our hypothesis that xCT deletion would not just reduce excessive glutamate release, but could also directly influence the global M1/M2 activation state, we assessed microglial production of known pro-inflammatory/neurotoxic (M1) and anti-inflammatory/neurotrophic (M2) factors. First, the classic microglial neurotoxic (M1) factor nitric oxide was assessed. Lipopolysaccharide strongly induced (by 10-fold) microglial nitric oxide production, whereas xCT deletion led to 65% less nitric oxide production upon lipopolysaccharide activation (Fig. 3A). Next, using a multiplex enzyme-linked immunosorbent assay (Luminex), different growth factors (G-CSF/Csf3, GM-CSF/Csf2, VEGFa), cytokines (TNFa, IFNg, IL1a, IL1b, IL3, IL4, IL6, IL10, IL12b, IL13, IL17) and chemokines (CCL5, CCL2, CCL3, CCL4) were measured in media conditioned by control (xCT+/+) or xCT−/− microglia, activated or not by lipopolysaccharide (Fig. 3B–E and Supplementary Fig. 1). Except for IL13 and IFNg, which were not detectable, all the factors assessed were released by microglia. Interestingly, compared to controls, in xCT-deleted microglia, we found a significant downregulation of the lipopolysaccharide-induced production of the growth factor G-CSF/Csf3 (−80%, Fig. 3D), but also of the pro-inflammatory factors TNFa (−60%, Fig. 3B), IL6 (−70%, Fig. 3C), and the chemokine CCL3 (−25%, Fig. 3E). Of note, not all measured factors were dependent on xCT expression; CCL2 production was stable, underlining the specificity of the changes found (Supplementary Fig. 1).
Figure 3.

xCT deletion reduces production of the pro-inflammatory/neurotoxic factors nitric oxide, TNFa and IL6 by lipopolysaccharide activated primary mouse microglial cells. Different factors were measured in the culture medium of primary microglial cells from mice expressing xCT (xCT+/+) or not (xCT−/−), after lipopolysaccharide (LPS, 100 ng/ml) treatment or in (non-treated) control conditions. (A) Levels of nitric oxide (NO) production (measured in the culture medium through its stable by-product, nitrite) by microglial cells are shown relative to non-treated control microglia (xCT+/+). Note that xCT−/− microglial cells produce less nitric oxide than xCT+/+ microglial cells when activated by lipopolysaccharide. (B–E) Results from microglial cytokines: TNFa (B), IL6 (C); growth factors: G-CSF/Csf3 (D) and chemokines: MIP-1α/CCL3 (E). Results represent amounts of factors released in the culture medium as measured by a multiplex Luminex assay. Here are shown the factors which were different between xCT−/− and xCT+/+ microglial cells. For additional protein factors measured, see Supplementary Fig. 1. Bars represent means of amounts ± SEM; all lipopolysaccharide treated xCT+/+ groups are significantly different (P < 0.01) compared with non-treated; *P < 0.05, **P < 0.01, two-way ANOVA with Bonferroni multiple comparison test, n = 3–4 experiments for each genotype.
Thus, as upon activation, xCT-deleted microglial cells released not just less glutamate, but also less nitric oxide and other pro-inflammatory/neurotoxic (M1) factors, system can therefore be considered a regulator of the microglial activation state.
xCT expression is increased in microglia of ALS mice and correlates with inflammation in human ALS spinal cord
Although system is considered a rather glial transporter, its lack of expression by adult motor neurons was not known, but was an essential requirement for our non-cell autonomous hypothesis. Thus, we established, by comparing xCT mRNA expression from isolated, laser-microdissected, adult motor neurons to whole lumbar spinal cord, that motor neurons did not express significant levels of system in control and mutant SOD1 ALS mice (Fig. 4A and Supplementary Fig. 2B), indicating that potential changes of xCT expression and effects of xCT deletion during disease, cannot be linked to a contribution of motor neurons.
Figure 4.

During disease, xCT mRNA expression is increased in spinal cord microglia of mutant SOD1 ALS mice and its expression level correlates with increased inflammation in post-mortem human ALS spinal cord tissues. (A–C) xCT mRNA expression was analysed by reverse-transcription quantitative PCR normalized to gamma-actin (Actg1). (A) xCT mRNA expression results are shown from fresh frozen adult non-transgenic control mouse whole lumbar spinal cord tissue [SC (whole)], laser-microdissected motor neurons from adult non-transgenic control (LMD MN) or hSOD1G37R [LMD MN (G37R)] mouse spinal cords, remains of the spinal cord sections devoid of laser microdissected motor neurons [SC (w/o MN)] and microglial cells isolated from spinal cords of adult non-transgenic control mice (adult microglia). Note that xCT mRNA was not detected in motor neurons and was enriched in microglial cells compared to the spinal cord. (B) xCT mRNA expression results are shown from non-transgenic control C57BL/6J (B6) or hSOD1G37R mouse lumbar spinal cord tissues during disease progression. (C) xCT mRNA expression results are shown from lumbar spinal cord tissues of presymptomatic hSOD1G37R mice or adult microglial cells isolated from hSOD1G37R mouse spinal cords at different disease stages. Bars represent means of fold-changes, ± SEM; results are shown relative to non-transgenic control whole lumbar spinal cord in A, non-transgenic hSOD1G37R littermates at 2–4 months of age in B, and to presymptomatic whole spinal cords (2–4 months of age) in C; *P < 0.05, ***P < 0.001, one-way ANOVA with Bonferroni multiple comparison test; n = 3 mice per genotype and time point. presympt = presymptomatic (2–4 months old); onset = age when reaching weight peak, considered as the beginning of the disease (in average, at 8.5 months of age); sympt = the beginning of the symptomatic phase, defined by 10% of weight loss and when first motor symptoms become apparent (in average, at 12 months of age); late sympt = late symptomatic stage, defined by 15% of weight loss (in average, at 13 months of age) and end stage of disease with complete hindlimb paralysis (in average, at 13.5 months of age). (D) Expression of xCT protein (top) and mRNA (bottom) in human ALS post-mortem spinal cord tissues. xCT protein (37 kDa) expression was measured by immunoblotting from spinal cord extracts of five human ALS cases (1–5) and beta-actin (42 kDa) used as a normalizer (top). For clinical data see Supplementary Table 1. Levels of xCT mRNA were measured by quantitative PCR and correlated to levels of the microglial inflammation marker CD68. Samples were normalized with the housekeeping gene GAPDH (bottom). Linear regression P = 0.0086, β = 1.472, R2 = 0.9270. Note that the samples that contain the highest levels of mRNA for xCT are also the ones that have the highest content of CD68 in agreement with increased expression of xCT under inflammatory conditions.
Because primary microglial cells, when activated, increased xCT expression and as microglia are activated during ALS, we analysed lumbar spinal cord xCT mRNA expression at four different disease stages in slowly progressing mutant hSOD1G37R ALS mice, which reach end stage on average at 13.5 months (Fig. 4B). In non-transgenic control mice, xCT levels were not modified during ageing, and wild-type hSOD1WT did not influence xCT expression (Supplementary Fig. 2A). Importantly, however, in ALS mice, xCT expression, while unchanged presymptomatically, was strongly increased at both disease onset and symptomatic stages, and then decreased at end stage (Fig. 4B).
To analyse whether microglial cells were a major source of disease-associated increased xCT expression, we isolated adult microglia from spinal cords of ALS mice during disease. First, compared to whole lumbar spinal cord, xCT mRNA was clearly enriched in microglia (Fig. 4A). Isolated microglia expressed mRNA for the microglial/macrophage marker CD68 and to higher levels when isolated from symptomatic compared to presymptomatic ALS mouse spinal cords (Supplementary Fig. 2C). Consistent with a contribution from activated microglia to disease-associated xCT induction, xCT mRNA levels were strongly increased in isolated microglia during disease progression (by 45-fold between symptomatic and presymptomatic stages) (Fig. 4C).
Importantly, xCT mRNA and protein were also expressed in the spinal cord of human ALS cases and xCT mRNA levels correlated with levels of CD68 (Fig. 4D and Supplementary Table 1) in agreement with our results in mice where xCT expression was increased when microglial cells were activated.
Deletion of xCT in ALS mice leads to increased expression of M2 markers, suggesting a switch towards a more neuroprotective microglial phenotype
As deleting xCT reduced neurotoxic factor production of cultured microglia, and microglial xCT expression was increased during disease in ALS mice, we next investigated the impact of xCT deletion on microglial phenotype, in vivo, over the course of the disease. Therefore, we crossed hSOD1G37R mice with xCT−/− mice.
First, we analysed general microgliosis in lumbar spinal cords of ALS mice, deleted or not for xCT, over the course of the disease, by using the classic microglial markers Iba1/Aif1 and Mac-2/Lgals3 (Fig. 5 and Supplementary Fig. 3). Although microglial activation increased during the course of the disease, overall microgliosis (as assessed by Iba1/Aif1 and Mac-2/Lgals3 immunostainings) was comparable between hSOD1G37R ALS mice with or without xCT, at every tested disease stage (Fig. 5 and Supplementary Fig. 3).
Figure 5.

xCT deletion does not affect general microgliosis revealed by Iba1/Aif1 immunoreactivity in mutant SOD1 ALS mice. Representative pictures of immunostainings against the classic microglial marker Iba1/Aif1 in the lumbar spinal cord of hSOD1G37R:xCT+/+, hSOD1G37R:xCT+/− and hSOD1G37R:xCT−/− ALS mice, shown at five different stages during the disease course (for definitions of disease stages, see Fig. 4 legend). For an additional commonly used microglial marker of general microgliosis (Mac-2/Lgals3), see Supplementary Fig. 3. Scale bar in O = 100 µm.
As classic microglial activation markers (Iba1/Aif1, Mac-2/Lgals3) do not necessarily predict more specific M1/M2 microglial activation states, we compared mRNA expression levels of specific microglial-associated M1 (iNOS/Nos2, NOX2/Cybb, Il1b, IL12p40/Il12b) and M2 (Ym1/Chil3, Arg1, Igf1) markers, during disease progression in the lumbar spinal cord of hSOD1G37R:xCT+/+ and hSOD1G37R:xCT−/− ALS mice (Fig. 6 and Supplementary Fig. 4).
Figure 6.

xCT deletion changes the profile of microglial associated M1/M2 markers in spinal cords of mutant SOD1 ALS mice during disease progression. (A) mRNA expression levels [analysed by reverse-transcription quantitative PCR, normalized to gamma-actin, (Actg1)] of the M1 markers iNOS/Nos2, NOX2/Cybb, Il1b, IL12p40/Il12b and the M2 markers Ym1/Chil3, Arg1 and Igf1, were measured in whole lumbar spinal cords of xCT−/− and xCT+/+ mice. The same M1/M2 markers were also measured in whole lumbar spinal cords of control (xCT+/+) as well as hSOD1G37R:xCT+/+ and hSOD1G37R:xCT−/− ALS mice over the disease course (B–F); for the M1 markers Il1b (B), Nox2 (C) and the M2 markers Ym1/Chil3 (D), Arg1 (E) and Igf1 (F). Bars represent means ± SEM; *P < 0.05, **P < 0.01, *** P < 0.001, Student’s t-test, compared to control (xCT+/+) mice; n = 3 mice per genotype and time point. For additional markers, see Supplementary Fig. 4. For definitions of disease stages, see Fig. 4 legend.
In presymptomatic hSOD1G37R ALS mice, the expression of both M1 and M2 markers was comparable to control (xCT+/+) mice (Supplementary Fig. 4A). For the M1 markers, Il1b and NOX2/Cybb were increased over the course of the disease (Fig. 6B and C). For the M2 markers, Igf1 was increased over the course of the disease (Fig. 6F), whereas the patterns of expression of Arg1 and Ym1/Chil3 were more complex. Indeed, Arg1 expression was stable during the disease and only increased at end stage (Fig. 6E). Ym1/Chil3 levels were increased at the symptomatic stage, but transiently decreased to almost control levels at the late symptomatic stage, to strongly increase again at disease end stage (Fig. 6D).
Upon deletion of xCT, surprisingly, already at baseline in non-ALS mice as well as in presymptomatic ALS mice, we detected a striking difference between xCT−/− and xCT+/+ mice for the M2 marker Ym1/Chil3, with mRNA levels more than 10-fold higher in xCT−/− mice (Fig. 6A and D). This difference (increase in Ym1/Chil3 expression) was even more striking when directly comparing adult spinal cord microglial cells isolated from xCT+/+ and xCT−/− mice (Supplementary Fig. 5).
Over the disease course, comparing ALS mice with and without xCT, we could distinguish two different disease phases, with an early disease phase (onset and symptomatic stage), where xCT deletion led to an increased expression of Il1b (M1) and a suppression of Ym1/Chil3 (M2) (Fig. 6B and D) and a late disease phase (late symptomatic stage), where xCT deletion led to an increased expression of the neuroprotective M2 markers Ym1/Chil3 and Arg1 (Fig. 6D and E).
Consistent with what we already detected in vitro, deletion of system in ALS mice, conferred a more anti-inflammatory/neuroprotective microglial-associated phenotype and this was especially evident in the late disease phase. This suggested a switch towards a more neuroprotective microglial phenotype.
A dual action from xCT deletion leads to an initial speed-up followed by a beneficial prolongation of the symptomatic disease phase
As xCT deletion led to a more anti-inflammatory/neuroprotective environment in ALS mouse spinal cords, we followed hSOD1G37R mice, deleted or not for xCT to analyse the consequence of removing xCT on the ALS disease course.
First, baseline xCT-deleted mice showed no phenotype and had a normal lifespan (958 ± 27.7 days, n = 20 and 978 ± 25.3 days, n = 24; for xCT+/+ and xCT−/− mice, respectively; equally gender-matched). Nevertheless, to ensure absence of subtle pathological abnormalities related to ALS, we confirmed lack of motor neuron loss and general glial activation in 1-year-old xCT−/− mice (Supplementary Fig. 6A and C). Likewise, although system can contribute to glutathione synthesis, previous studies already showed that global brain glutathione content (measured in the striatum and hippocampus) was not reduced in xCT-deleted mice (De Bundel et al., 2011; Massie et al., 2011), which we now also confirmed for spinal cord (Supplementary Fig. 6B).
Next, analysis of disease course showed that xCT deletion in mutant SOD1 ALS mice had no impact on disease onset, defined by two methods (Fig. 7A and A’), (i) the age when reaching weight peak (hSOD1G37R:xCT+/+: 247 ± 6.5 days, n = 28; hSOD1G37R:xCT+/−: 254 ± 8.8 days, n = 40; hSOD1G37R:xCT−/−: 242.8 ± 7.8 days, n = 24); and (ii) the age when reaching grip strength peak (hSOD1G37R:xCT+/+: 197.6 ± 1.9 days, n = 28; hSOD1G37R:xCT+/−: 191.3 ± 1.2 days, n = 40; hSOD1G37R:xCT−/−: 183.9 ± 1.9 days, n = 24), consistent with the idea that effects linked to modulation of microglia should rather contribute to the symptomatic phase of the disease. Surprisingly, hSOD1G37R :xCT−/− mice reached the specific early disease stage (the beginning of the symptomatic phase, defined by appearance of motoric symptoms and 10% weight loss) 1 month earlier than hSOD1G37R :xCT+/+ mice (−32.5 days; P = 0.0233) (328.4 ± 9.2 days, n = 24 and 360.5 ± 7.7 days, n = 26, respectively) (Fig. 7B), with a similar speed-up, when defining early disease by 35% grip strength loss (−30.8 days; P = 0.0329) (Fig. 7B’).
Figure 7.

xCT deletion in mutant SOD1 ALS mice initially speeds up early disease but then promotes longer survival after reaching the symptomatic disease stage. (A–C) Kaplan-Meier plots of ages when onset [at weight peak (A) or at grip strength peak (A’)], early disease [at 10% of weight loss (B) or at 35% of grip strength loss (B’)] and end stage (at complete hindlimb paralysis) (C) were reached in hSOD1G37R:xCT+/+ (blue), hSOD1G37R:xCT+/− (green) and hSOD1G37R:xCT−/− (orange) mice. Animal numbers (equally gender-mixed) are indicated in brackets. (B and B’) statistical significant differences (P < 0.05, log-rank) when reaching early disease stage, are indicated between the hSOD1G37R:xCT−/− and the hSOD1G37R:xCT+/+ groups. (D, left), early disease duration corresponding to means of differences between ages when individual animals reached onset and 10% of weight loss. (D, right), late disease duration corresponding to means of differences between ages when individual animals reached 10% of weight loss and end stage. (E) Survival duration (in days) after reaching a defined advanced symptomatic disease stage [20% of weight loss (WL)]. Bars represent the proportion of mice (relative to the total number of mice per genotype) still alive within the indicated time frames. Note that hSOD1G37R:xCT−/− mice survive longer after reaching symptoms than hSOD1G37R:xCT+/+ mice. (F) Percentage of the remaining grip strength score at the late symptomatic stage (15% of weight loss) compared to the maximum grip strength score (at onset). Bars represent means, ± SEM, *P < 0.05, **P < 0.01, Student’s t-test; with n = 24 animals for hSOD1G37R:xCT+/+ and n = 21 for hSOD1G37R:xCT−/− mice. Additional analyses of disease progression are provided in Supplementary Fig. 7.
However, this earlier reaching of the symptomatic stage (10% weight loss) did not decrease survival, with hSOD1G37R:xCT−/− mice reaching end stage (complete hind limb paralysis) at the same time as hSOD1G37R:xCT+/+ mice (Fig. 7C) (410 ± 6 days, n = 21 and 405 ± 7.5 days, n = 24, respectively). This dual action of xCT deletion led to: (i) a speed-up to reach early disease (Fig. 7B, B’ and D) but in contrast, and importantly; (ii) an increase of the duration of the late disease phase after appearance of symptoms (the time between 10% weight loss and end-stage or 35% grip strength loss and end-stage; +28 days, P = 0.0278) in hSOD1G37R:xCT−/− mice (85.5 ± 8.2 days, n = 21) as compared to hSOD1G37R:xCT+/+ mice (57.4 ± 5.9 days, n = 24) (Fig. 7D and Supplementary Fig. 7).
Our tracking of the mouse cohort consistently indicated that, after appearance of symptoms, hSOD1G37R :xCT−/− mice seemed to live longer and behave better than hSOD1G37R :xCT+/+ mice during the symptomatic phase. To quantify, we measured the survival time to reach a defined advanced symptomatic disease stage (20% of weight loss). The time elapsed, after appearance of symptoms, to reach this advanced symptomatic stage was indeed increased (Supplementary Fig. 7A). Interestingly, and consistent with our observation, while 40% of hSOD1G37R :xCT+/+ mice had already died at this disease stage, this was the case for only 20% of the hSOD1G37R :xCT−/− mice (Fig. 7E). Even more so, compared to hSOD1G37R :xCT+/+ mice, hSOD1G37R :xCT−/− mice lived longer after reaching this disease stage (Fig. 7E and Supplementary Fig. 7B, P = 0.01, which is underestimated as n = 17/21 for the hSOD1G37R:xCT−/− group and only n = 15/24 for the hSOD1G37R :xCT+/+ group). In line with this finding, during the late symptomatic disease phase, hSOD1G37R:xCT−/− mice had ∼20% (P < 0.05) more remaining grip strength than hSOD1G37R:xCT+/+ mice (57 ± 1.6% and 39 ± 1.7%, respectively) (Fig. 7F).
Deletion of xCT in ALS mice results in more surviving motor neurons at disease end stage
The three groups of ALS mice, hSOD1G37R:xCT+/+, hSOD1G37R:xCT+/− and hSOD1G37R:xCT−/−, started out with the same number of motor neurons (30.9 ± 0.4, 35.5 ± 4.6 and 33.7 ± 1.0 per spinal cord section, respectively) (Fig. 8) and, as expected, during disease, motor neurons degenerated in hSOD1G37R:xCT+/+ mice with only 35% remaining at end stage (Fig. 8). Interestingly, however, hSOD1G37R:xCT−/− mice had, with 45% remaining, a significant increase in survival of motor neurons (Fig. 8). This suggested an overall less neurotoxic environment and slowing of motor neuron degeneration due to xCT deletion, consistent with our results, in vitro, showing decreased secretion of pro-inflammatory/neurotoxic (M1) factors from primary microglial cells (Fig. 3) and, in vivo, with the switch to a spinal cord environment associated with a more anti-inflammatory/neuroprotective (M2) microglial phenotype in late symptomatic xCT-deleted ALS mice (Fig. 6).
Figure 8.

xCT deletion in mutant SOD1 ALS mice leads to an increased number of surviving motor neurons at disease end stage. (A) Representative pictures of lumbar spinal cords of hSOD1G37R:xCT+/+ (left column), hSOD1G37R:xCT+/− (middle column) and hSOD1G37R:xCT−/− (right column) mice at presymptomatic (top) and at end stage (bottom), stained with the ChAT antibody to reveal motor neurons. (B) Motor neuron numbers counted in the lumbar spinal cords at different time points. For definitions of disease stages, see Fig. 4 legend. Bars represent means, ± SEM; * P < 0.05, Student’s t-test, n = 3 mice per genotype and time point. Scale bar in A = 100 µm.
Discussion
In the present study, we found xCT to be expressed in the spinal cord, consistent with its presence in CNS tissues (Sato et al., 2002; Burdo et al., 2006; Massie et al., 2008), but not in motor neurons, in accordance with system considered a glial transporter (Pow, 2001). The main hurdle to fully characterize cell-specific expression is a lack of good xCT antibodies, which so far only work in culture, while for tissues, we also detected signals in xCT-deleted mice. In culture, both astrocytes and microglia express xCT (Gochenauer and Robinson, 2001; Allen et al., 2002; Qin et al., 2006; Markowitz et al., 2007; Lewerenz et al., 2009). Our findings of xCT enrichment in isolated microglia compared to whole spinal cord, now show xCT expression by microglia and give support to a previous report suggesting (using in situ hybridization) xCT expression in mouse hippocampal microglia (Qin et al., 2006). We showed that xCT mRNA levels were increased in ALS mouse spinal cords, lipopolysaccharide-activated primary microglia and in isolated microglia from ALS mouse spinal cords during the disease, suggesting that both microglial activation and proliferation participated in the xCT increase in the spinal cord (without excluding other glial cells, such as astrocytes, to contribute).
Only one other study has focused on system in ALS mice, and found a transient increase in system activity in acute spinal cord slices from fast progressing hSOD1G93A mice (Albano et al., 2013). We did not detect differences compared to control xCT expression levels in slow progressing hSOD1G37R mice, and our results rather suggested that xCT expression was dependent on microglial activation than presence of mutant SOD1. As microglial activation is a pathological sign of both familial and sporadic ALS (Henkel et al., 2004; Turner et al., 2004; Corcia et al., 2012) and as we found xCT to be expressed in spinal cords of sporadic ALS cases and to parallel CD68 levels, increased xCT expression and potential deleterious effects following from it, could be part of a more general disease mechanism not restricted to defined ALS gene mutations.
Deletion of xCT in mutant SOD1 ALS mice surprisingly accelerated onset of symptoms for which we propose two hypotheses: (i) as system is involved in glutathione synthesis (Sato et al., 1999), early reduced antioxidant glutathione levels could be responsible. However, total spinal cord glutathione content was unchanged in xCT−/− mice, consistent with previous studies in striatum and hippocampus (De Bundel et al., 2011; Massie et al., 2011). Nevertheless, subtle, cell-specific effects could occur, particularly via astrocytes, which provide neurons with glutathione (Sagara et al., 1993; Dringen and Hirrlinger, 2003); and (ii) a transient decrease in neuroprotective/anti-inflammatory factors could be implicated. Indeed, we detected decreased production of G-CSF/Csf3 by activated xCT-deleted primary microglia, which in the CNS, is considered neuroprotective (Schneider et al., 2005; Pitzer et al., 2008). Likewise, in xCT-deleted ALS mice, we detected transient increased expression of pro-inflammatory IL1b (at onset) and transient decreased expression of anti-inflammatory Ym1/Chil3 markers (at the early symptomatic stage).
In numerous previous studies, in which a specific disease-modifying treatment of mutant SOD1 ALS mice resulted in an accelerated onset of symptoms, a similar reduced survival was usually found and mice died earlier (Lambrechts et al., 2003; Van Damme et al., 2005; Zhang et al., 2011; Lerman et al., 2012). These examples are consistent with treatments affecting mainly onset of symptoms. Once symptoms appeared, the disease progressed at the same rate and the final length of the symptomatic disease phase (from onset of symptoms until end stage) was unchanged.
In our case, xCT deletion accelerated onset of symptoms. However, and in contrast to the mentioned examples, xCT deletion in ALS mice did not lead to a reduced survival and mice did not die earlier, suggesting that xCT deletion, once symptoms appeared, slowed the rate of disease progression. Indeed, we found that in xCT-deleted ALS mice (i) time elapsed after onset of symptoms to reach a defined late symptomatic stage was increased; (ii) levels of spinal cord M2 markers were increased; (iii) grip strength during the late symptomatic phase was better (although, actual motor neuron numbers were still similar, suggesting that the remaining ones were less affected); and finally (iv) more surviving motor neurons remained at end stage. While it is possible that the dual actions of xCT deletion cannot be separated, our results strongly suggest a neuroprotective consequence if system activity would be reduced during the progressive disease phase that follows onset of symptoms.
Our hypothesis for the slowed disease progression is a beneficial consequence of xCT-deletion to lower glutamate secretion. Thus, xCT deletion would help reduce ALS-associated glutamate excitotoxicity. Indeed, microdialysis in the striatum of xCT-deleted mice showed a 70% decrease of extracellular glutamate (Massie et al., 2011). Excitotoxicity remains one of the major mechanistic hypotheses in ALS and riluzole, the only FDA-approved drug for ALS, has anti-excitotoxic properties (Bensimon et al., 1994; Foran and Trotti, 2009). Motor neurons are highly sensitive to glutamate excitotoxicity (Takuma et al., 1999; Kawahara et al., 2004; Van Den Bosch et al., 2006; Hideyama et al., 2012), and astrocytic glutamate transporters are reduced in rodent models and patients with ALS (Rothstein et al., 1995; Howland et al., 2002). Additionally, elevated neuronal glutamate release was described in ALS mice (Milanese et al., 2011). Now, with our microglia data a new player is added to this excitotoxicity hypothesis for ALS. Indeed, although microglia can release glutamate through connexins (Takeuchi et al., 2006; Maezawa and Jin, 2010), we show that system was the main source of glutamate release by microglia, and that their activation led to increased xCT expression and glutamate release.
Finally, we show a direct effect of system deletion on the phenotype of microglia. Targeting of system modified release of glutamate but also changed the profile of microglial-associated pro-inflammatory/neurotoxic (M1) and anti-inflammatory/neuroprotective (M2) markers. Of note, glutamate was shown to directly act on microglia through their NMDA receptors, triggering their activation and leading to secretion of pro-inflammatory/neurotoxic (M1) factors (Kaindl et al., 2012). Our data show that deletion of system resulted in less nitric oxide production by activated microglial cells and decreased release of pro-inflammatory cytokines TNFa and IL6 and the chemokine MIP-1α/CCL3. Likewise, expression analysis of microglial associated M1/M2 markers in the spinal cord of xCT-deleted mice showed levels of the anti-inflammatory/neuroprotective (M2) marker Ym1/Chil3 to be strongly increased, both in spinal cord and in isolated adult microglial cells.
Importantly, we also measured the influence of xCT-deletion on the profile of microglial associated M1/M2 markers, during disease progression in spinal cords of ALS mice. Indeed, M1/M2 profiles had previously been suggested to reflect disease progression in hSOD1G93A ALS mice, with a more pro-inflammatory (M1) profile rather linked to fast disease progression and an anti-inflammatory (M2) profile rather linked to slow disease progression (Beers et al., 2011; Liao et al., 2012). Comparing our results from slow progressing hSOD1G37R ALS mice with the data from fast progressing hSOD1G93A mice, we found that for the M1 markers, NOX2 presented the same pattern of increased expression until end stage; IL1b remained increased until end stage in hSOD1G93A mice, whereas in slow progressing hSOD1G37R mice, levels slightly decreased at the late symptomatic disease stage. Concerning the M2 marker Ym1/Chil3, although its increase during the early symptomatic phase was similar in both models, decreased expression at the late disease stage remained low until end stage in hSOD1G93A mice, but surprisingly, increased again in slow progressing hSOD1G37R mice. We believe that these apparent discrepancies highlight that M1/M2 modulation strongly depends on the inherent differences during the symptomatic phase of fast and slow progressing ALS mouse models.
With respect to the effect of xCT-deletion, two M2 markers, Ym1/Chil3 and Arg1, showed a striking effect. When xCT-deleted ALS mice reached symptom onset earlier, Ym1/Chil3 expression was decreased in the spinal cord compared to xCT-containing ALS mice, whereas during the late symptomatic stages, Ym1/Chil3 and Arg1 levels were increased. These data suggested that a switch from a M1 to a M2-like environment occurred and that this could account for the observed slowing of ALS disease progression after onset of symptoms, in xCT-deleted ALS mice.
To date, it is not clear whether a switch from M1 to M2 could confer direct neuroprotection. In addition, classic M1 and M2 patterns are extremes of a spectrum of different cell states (Xue et al., 2014) and in different injury and disease models including ALS, microglia adopt a dual phenotype, expressing factors that are hypothesized to be both neurotoxic and neuroprotective (Chiu et al., 2013). Futhermore, although M2 cells should be beneficial, peripheral injection of bone marrow cells differentiated to a M2-phenotype worsened symptoms in hSOD1G93A mice (Vaknin et al., 2011) (although actual CNS infiltration was not detected). Additionally, decreasing the expression of microglial M1 markers does not necessarily increase the production of neurotrophic M2 factors, as seen after minocycline injection (Kobayashi et al., 2013). However, deleting NOX2 (known to increase ALS mouse survival) (Wu et al., 2006; Marden et al., 2007) has been shown, both in culture and acute CNS lesions, to decrease M1 marker expression (Choi et al., 2012).
Our finding of a beneficial effect of xCT deletion after symptom onset in ALS mice is consistent with findings from other models of neurodegenerative diseases. Suppression of system in mouse or in in vitro models of Alzheimer’s disease, Parkinson’s disease, multiple sclerosis and gliomas demonstrated protection from neurodegeneration (Chung et al., 2005; Qin et al., 2006; Domercq et al., 2007; Massie et al., 2011). So far, contribution of system to neurodegeneration was hinted at, but only using rather acute models of neurodegenerative diseases. Our findings with ALS mice now reveal disease contribution of system in an actual progressive and chronic model of a neurodegenerative disease. These studies, combined with our finding, strongly support the idea of a disease contribution of system in neurodegenerative disorders in general.
In summary, while xCT−/− microglial cells release less M1-related factors (nitric oxide, TNFa, IL6, CCL3) and xCT−/− mice show increased expression of M2-related Ym1/Chil3 in spinal cord and isolated microglial cells, system seems to be a new regulator of microglial function. In addition, with its capacity to release glutamate and the potential to contribute to excitotoxicity, we show evidence that system could be implicated in motor neuron degeneration in ALS mice. Thus, pharmacological blockage of system , during the symptomatic phase (but not before, to avoid negative early effects) could increase survival in ALS mice. However, for this, first better and more specific system antagonists are needed (as commercially available antagonists are not specific enough and their bioavailability for the CNS not optimal). As ALS is a mainly sporadic disease, targeting the symptomatic phase of the disease would be relevant and could include focusing on the M1/M2 microglial cell phenotype, possibly via system .
Acknowledgements
We would like to thank Annie Gervais, Catherine Colin, Jan Baijer, Nathalie Dechamps, Fabien Aubry, Aurélien Dauphin and the PICPS core facility, for their technical advice, as well as the technical staff from our animal housing facility CEF, the P3S core facility and the IHU-A-ICM.
Glossary
Abbreviations
- ALS
amyotrophic lateral sclerosis
- G-CSF/Csf3
granulocyte colony-stimulating factor 3
- Iba1/Aif
allograft inflammatory factor 1
- iNOS/Nos2
nitric oxide synthase 2, inducible
- NOX2/Cybb
cytochrome b-245, beta polypeptide (NADPH oxidase 2)
- TNFa/Tnf
tumor necrosis factor
- xCT/Slc7a11
solute carrier family 7 (cationic amino acid transporter, y+ system), member 11
- Ym1/Chil3
chitinase-like 3
Funding
This work was supported by the Thierry Latran Foundation, the Association pour la Recherche sur la Sclérose Latérale Amyotrophique et autres Maladies du Motoneurone, NRJ-Institut de France and European FP7 International Reintegration Marie Curie Grant. P.M. was supported by fellowships from the French ministry of research and the Fondation pour la Recherche Médicale.
Supplementary material
Supplementary material is available at Brain online.
References
- Albano R, Liu X, Lobner D. Regulation of system x(c)- in the SOD1-G93A mouse model of ALS. Exp Neurol. 2013;250:69–73. doi: 10.1016/j.expneurol.2013.09.008. [DOI] [PubMed] [Google Scholar]
- Allen JW, Shanker G, Tan KH, Aschner M. The consequences of methylmercury exposure on interactive functions between astrocytes and neurons. Neurotoxicology. 2002;23:755–9. doi: 10.1016/S0161-813X(01)00076-6. [DOI] [PubMed] [Google Scholar]
- Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA, et al. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci USA. 2006;103:16021–6. doi: 10.1073/pnas.0607423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers DR, Henkel JS, Zhao W, Wang J, Huang A, Wen S, et al. Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain. 2011;134:1293–314. doi: 10.1093/brain/awr074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N Engl J Med. 1994;330:585–91. doi: 10.1056/NEJM199403033300901. [DOI] [PubMed] [Google Scholar]
- Beutler HO. In: Methods of enzymatic analysis. 3rd edn. Bergmeyer HU, editor. Basel: Verlag Chemie, Weinheim, Deerfield Beach/Florida; 1985. pp. 369–76. [Google Scholar]
- Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006a;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Boillee S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006b;312:1389–92. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Burdo J, Dargusch R, Schubert D. Distribution of the cystine/glutamate antiporter system xc- in the brain, kidney, and duodenum. J Histochem Cytochem. 2006;54:549–57. doi: 10.1369/jhc.5A6840.2006. [DOI] [PubMed] [Google Scholar]
- Chiu IM, Morimoto ET, Goodarzi H, Liao JT, O'Keeffe S, Phatnani HP, et al. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 2013;4:385–401. doi: 10.1016/j.celrep.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SH, Aid S, Kim HW, Jackson SH, Bosetti F. Inhibition of NADPH oxidase promotes alternative and anti-inflammatory microglial activation during neuroinflammation. J Neurochem. 2012;120:292–301. doi: 10.1111/j.1471-4159.2011.07572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WJ, Lyons SA, Nelson GM, Hamza H, Gladson CL, Gillespie GY, et al. Inhibition of cystine uptake disrupts the growth of primary brain tumors. J Neurosci. 2005;25:7101–10. doi: 10.1523/JNEUROSCI.5258-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillee S, Rule M, et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302:113–7. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- Corcia P, Tauber C, Vercoullie J, Arlicot N, Prunier C, Praline J, et al. Molecular imaging of microglial activation in amyotrophic lateral sclerosis. PLoS One. 2012;7:e52941. doi: 10.1371/journal.pone.0052941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czeh M, Gressens P, Kaindl AM. The yin and yang of microglia. Dev Neurosci. 2011;33:199–209. doi: 10.1159/000328989. [DOI] [PubMed] [Google Scholar]
- De Bundel D, Schallier A, Loyens E, Fernando R, Miyashita H, Van Liefferinge J, et al. Loss of system x(c)- does not induce oxidative stress but decreases extracellular glutamate in hippocampus and influences spatial working memory and limbic seizure susceptibility. J Neurosci. 2011;31:5792–803. doi: 10.1523/JNEUROSCI.5465-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Groot CJ, Montagne L, Janssen I, Ravid R, Van Der Valk P, Veerhuis R. Isolation and characterization of adult microglial cells and oligodendrocytes derived from postmortem human brain tissue. Brain Res Brain Res Protoc. 2000;5:85–94. doi: 10.1016/s1385-299x(99)00059-8. [DOI] [PubMed] [Google Scholar]
- Domercq M, Sanchez-Gomez MV, Sherwin C, Etxebarria E, Fern R, Matute C. System xc- and glutamate transporter inhibition mediates microglial toxicity to oligodendrocytes. J Immunol. 2007;178:6549–56. doi: 10.4049/jimmunol.178.10.6549. [DOI] [PubMed] [Google Scholar]
- Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Mistry NA, Vidensky S, et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80:415–28. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dringen R, Hirrlinger J. Glutathione pathways in the brain. Biol Chem. 2003;384:505–16. doi: 10.1515/BC.2003.059. [DOI] [PubMed] [Google Scholar]
- Foran E, Trotti D. Glutamate transporters and the excitotoxic path to motor neuron degeneration in amyotrophic lateral sclerosis. Antioxid Redox Signal. 2009;11:1587–602. doi: 10.1089/ars.2009.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasol E, Jimenez-Vidal M, Chillaron J, Zorzano A, Palacin M. Membrane topology of system xc- light subunit reveals a re-entrant loop with substrate-restricted accessibility. J Biol Chem. 2004;279:31228–36. doi: 10.1074/jbc.M402428200. [DOI] [PubMed] [Google Scholar]
- Gochenauer GE, Robinson MB. Dibutyryl-cAMP (dbcAMP) up-regulates astrocytic chloride-dependent L-[3H]glutamate transport and expression of both system xc(-) subunits. J Neuroch. 2001;78:276–86. doi: 10.1046/j.1471-4159.2001.00385.x. [DOI] [PubMed] [Google Scholar]
- Henkel JS, Engelhardt JI, Siklos L, Simpson EP, Kim SH, Pan T, et al. Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann Neurol. 2004;55:221–35. doi: 10.1002/ana.10805. [DOI] [PubMed] [Google Scholar]
- Hideyama T, Yamashita T, Aizawa H, Tsuji S, Kakita A, Takahashi H, et al. Profound downregulation of the RNA editing enzyme ADAR2 in ALS spinal motor neurons. Neurobiol Dis. 2012;45:1121–8. doi: 10.1016/j.nbd.2011.12.033. [DOI] [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, et al. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci USA. 2002;99:1604–9. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaindl AM, Degos V, Peineau S, Gouadon E, Chhor V, Loron G, et al. Activation of microglial N-methyl-D-aspartate receptors triggers inflammation and neuronal cell death in the developing and mature brain. Ann Neurol. 2012;72:536–49. doi: 10.1002/ana.23626. [DOI] [PubMed] [Google Scholar]
- Kang SH, Li Y, Fukaya M, Lorenzini I, Cleveland DW, Ostrow LW, et al. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci. 2013;16:571–9. doi: 10.1038/nn.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Ito K, Sun H, Aizawa H, Kanazawa I, Kwak S. Glutamate receptors: RNA editing and death of motor neurons. Nature. 2004;427:801. doi: 10.1038/427801a. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Imagama S, Ohgomori T, Hirano K, Uchimura K, Sakamoto K, et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. 2013;4:e525. doi: 10.1038/cddis.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrechts D, Storkebaum E, Morimoto M, Del-Favero J, Desmet F, Marklund SL, et al. VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat Genet. 2003;34:383–94. doi: 10.1038/ng1211. [DOI] [PubMed] [Google Scholar]
- Lerman BJ, Hoffman EP, Sutherland ML, Bouri K, Hsu DK, Liu FT, et al. Deletion of galectin-3 exacerbates microglial activation and accelerates disease progression and demise in a SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Brain Behav. 2012;2:563–75. doi: 10.1002/brb3.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewerenz J, Albrecht P, Tien ML, Henke N, Karumbayaram S, Kornblum HI, et al. Induction of Nrf2 and xCT are involved in the action of the neuroprotective antibiotic ceftriaxone in vitro. J Neuroch. 2009;111:332–43. doi: 10.1111/j.1471-4159.2009.06347.x. [DOI] [PubMed] [Google Scholar]
- Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, et al. The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal. 2013;18:522–55. doi: 10.1089/ars.2011.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao B, Zhao W, Beers DR, Henkel JS, Appel SH. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp Neurol. 2012;237:147–52. doi: 10.1016/j.expneurol.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobsiger CS, Boillee S, Cleveland DW. Toxicity from different SOD1 mutants dysregulates the complement system and the neuronal regenerative response in ALS motor neurons. Proc Natl Acad Sci USA. 2007;104:7319–26. doi: 10.1073/pnas.0702230104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobsiger CS, Boillee S, McAlonis-Downes M, Khan AM, Feltri ML, Yamanaka K, et al. Schwann cells expressing dismutase active mutant SOD1 unexpectedly slow disease progression in ALS mice. Proc Natl Acad Sci USA. 2009;106:4465–70. doi: 10.1073/pnas.0813339106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa I, Jin LW. Rett syndrome microglia damage dendrites and synapses by the elevated release of glutamate. J Neurosci. 2010;30:5346–56. doi: 10.1523/JNEUROSCI.5966-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marden JJ, Harraz MM, Williams AJ, Nelson K, Luo M, Paulson H, et al. Redox modifier genes in amyotrophic lateral sclerosis in mice. J Clin Invest. 2007;117:2913–9. doi: 10.1172/JCI31265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz AJ, White MG, Kolson DL, Jordan-Sciutto KL. Cellular interplay between neurons and glia: toward a comprehensive mechanism for excitotoxic neuronal loss in neurodegeneration. Cellscience. 2007;4:111–46. [PMC free article] [PubMed] [Google Scholar]
- Massie A, Schallier A, Kim SW, Fernando R, Kobayashi S, Beck H, et al. Dopaminergic neurons of system x(c)(-)-deficient mice are highly protected against 6-hydroxydopamine-induced toxicity. FASEB J. 2011;25:1359–69. doi: 10.1096/fj.10-177212. [DOI] [PubMed] [Google Scholar]
- Massie A, Schallier A, Mertens B, Vermoesen K, Bannai S, Sato H, et al. Time-dependent changes in striatal xCT protein expression in hemi-Parkinson rats. Neuroreport. 2008;19:1589–92. doi: 10.1097/WNR.0b013e328312181c. [DOI] [PubMed] [Google Scholar]
- Milanese M, Zappettini S, Onofri F, Musazzi L, Tardito D, Bonifacino T, et al. Abnormal exocytotic release of glutamate in a mouse model of amyotrophic lateral sclerosis. J Neurochem. 2011;116:1028–42. doi: 10.1111/j.1471-4159.2010.07155.x. [DOI] [PubMed] [Google Scholar]
- Ogunrinu TA, Sontheimer H. Hypoxia increases the dependence of glioma cells on glutathione. J Biol Chem. 2010;285:37716–24. doi: 10.1074/jbc.M110.161190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piani D, Fontana A. Involvement of the cystine transport system xc- in the macrophage-induced glutamate-dependent cytotoxicity to neurons. J Immunol. 1994;152:3578–85. [PubMed] [Google Scholar]
- Pitzer C, Kruger C, Plaas C, Kirsch F, Dittgen T, Muller R, et al. Granulocyte-colony stimulating factor improves outcome in a mouse model of amyotrophic lateral sclerosis. Brain. 2008;131:3335–47. doi: 10.1093/brain/awn243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pow DV. Visualising the activity of the cystine-glutamate antiporter in glial cells using antibodies to aminoadipic acid, a selectively transported substrate. Glia. 2001;34:27–38. doi: 10.1002/glia.1037. [DOI] [PubMed] [Google Scholar]
- Qin S, Colin C, Hinners I, Gervais A, Cheret C, Mallat M. System Xc- and apolipoprotein E expressed by microglia have opposite effects on the neurotoxicity of amyloid-beta peptide 1-40. J Neurosci. 2006;26:3345–56. doi: 10.1523/JNEUROSCI.5186-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- Sagara JI, Miura K, Bannai S. Maintenance of neuronal glutathione by glial cells. J Neurochem. 1993;61:1672–6. doi: 10.1111/j.1471-4159.1993.tb09802.x. [DOI] [PubMed] [Google Scholar]
- Sato H, Kuriyama-Matsumura K, Hashimoto T, Sasaki H, Wang H, Ishii T, et al. Effect of oxygen on induction of the cystine transporter by bacterial lipopolysaccharide in mouse peritoneal macrophages. J Biol Chem. 2001;276:10407–12. doi: 10.1074/jbc.M007216200. [DOI] [PubMed] [Google Scholar]
- Sato H, Shiiya A, Kimata M, Maebara K, Tamba M, Sakakura Y, et al. Redox imbalance in cystine/glutamate transporter-deficient mice. J Biol Chem. 2005;280:37423–9. doi: 10.1074/jbc.M506439200. [DOI] [PubMed] [Google Scholar]
- Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999;274:11455–8. doi: 10.1074/jbc.274.17.11455. [DOI] [PubMed] [Google Scholar]
- Sato H, Tamba M, Okuno S, Sato K, Keino-Masu K, Masu M, et al. Distribution of cystine/glutamate exchange transporter, system x(c)-, in the mouse brain. J Neurosci. 2002;22:8028–33. doi: 10.1523/JNEUROSCI.22-18-08028.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider A, Kruger C, Steigleder T, Weber D, Pitzer C, Laage R, et al. The hematopoietic factor G-CSF is a neuronal ligand that counteracts programmed cell death and drives neurogenesis. J Clin Invest. 2005;115:2083–98. doi: 10.1172/JCI23559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, et al. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem. 2006;281:21362–8. doi: 10.1074/jbc.M600504200. [DOI] [PubMed] [Google Scholar]
- Takeuchi H, Mizoguchi H, Doi Y, Jin S, Noda M, Liang J, et al. Blockade of gap junction hemichannel suppresses disease progression in mouse models of amyotrophic lateral sclerosis and Alzheimer's disease. PLoS One. 2011;6:e21108. doi: 10.1371/journal.pone.0021108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuma H, Kwak S, Yoshizawa T, Kanazawa I. Reduction of GluR2 RNA editing, a molecular change that increases calcium influx through AMPA receptors, selective in the spinal ventral gray of patients with amyotrophic lateral sclerosis. Ann Neurol. 1999;46:806–15. doi: 10.1002/1531-8249(199912)46:6<806::aid-ana2>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Thery C, Chamak B, Mallat M. Cytotoxic effect of brain macrophages on developing. Eur J Neurosci. 1991;3:1155–64. doi: 10.1111/j.1460-9568.1991.tb00050.x. [DOI] [PubMed] [Google Scholar]
- Turner MR, Cagnin A, Turkheimer FE, Miller CC, Shaw CE, Brooks DJ, et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiol Dis. 2004;15:601–9. doi: 10.1016/j.nbd.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Vaknin I, Kunis G, Miller O, Butovsky O, Bukshpan S, Beers DR, et al. Excess circulating alternatively activated myeloid (M2) cells accelerate ALS progression while inhibiting experimental autoimmune encephalomyelitis. PLoS One. 2011;6:e26921. doi: 10.1371/journal.pone.0026921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P, Braeken D, Callewaert G, Robberecht W, Van Den Bosch L. GluR2 deficiency accelerates motor neuron degeneration in a mouse model of amyotrophic lateral sclerosis. J Neuropathol Exp Neurol. 2005;64:605–12. doi: 10.1097/01.jnen.0000171647.09589.07. [DOI] [PubMed] [Google Scholar]
- Van Den Bosch L, Van Damme P, Bogaert E, Robberecht W. The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim Biophys Acta. 2006;1762:1068–82. doi: 10.1016/j.bbadis.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Wang L, Deng HX, Grisotti G, Zhai H, Siddique T, Roos RP. Wild-type SOD1 overexpression accelerates disease onset of a G85R SOD1 mouse. Hum Mol Genet. 2009;18:1642–51. doi: 10.1093/hmg/ddp085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Bannai S. Induction of cystine transport activity in mouse peritoneal macrophages. J Exp Med. 1987;165:628–40. doi: 10.1084/jem.165.3.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Re DB, Nagai M, Ischiropoulos H, Przedborski S. The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc Natl Acad Sci USA. 2006;103:12132–7. doi: 10.1073/pnas.0603670103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40:274–88. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11:251–3. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Li L, Chen S, Yang D, Wang Y, Zhang X, et al. Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy. 2011;7:412–25. doi: 10.4161/auto.7.4.14541. [DOI] [PubMed] [Google Scholar]
- Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–8. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]