

Abstract
The sterol storage disorder cerebrotendinous xanthomatosis (CTX) is characterized by abnormal deposition of cholesterol and cholestanol in multiple tissues. Deposition in the central nervous system leads to neurological dysfunction marked by dementia, spinal cord paresis, and cerebellar ataxia. Deposition in other tissues causes tendon xanthomas, premature atherosclerosis, and cataracts. In two unrelated patients with CTX, we have identified different point mutations in the gene (CYP27) encoding sterol 27-hydroxylase, a key enzyme in the bile acid biosynthesis pathway. Transfection of mutant cDNAs into cultured cells results in the synthesis of immunoreactive sterol 27-hydroxylase protein with greatly diminished enzyme activity. We have localized the CYP27 gene to the q33-qter interval of human chromosome 2, and to mouse chromosome 1, in agreement with the autosomal recessive inheritance pattern of CTX. These findings underscore the essential role played by sterols in the central nervous system and suggest that mutations in other sterol metabolizing enzymes may contribute to diseases with neurological manifestations.
Approximately one-fourth of the cholesterol present in an adult mammal is located in the nervous system (1), where it is a major constituent of the myelin sheath that both insulates and facilitates axonal conduction (2). The central role played by cholesterol in the nervous system is made readily apparent in certain genetic diseases in which alterations in genes involved in sterol metabolism have profound effects on neural tissues.
One such disease is cerebrotendinous xanthomatosis (CTX),1 a rare sterol storage disorder first described in 1937 in individuals with neurological dysfunction and tendon xanthomas (3, 4). Subsequent studies demonstrated that cholesterol and the 5α-reduced form of cholesterol, cholestanol, accumulate in neural and other tissues of CTX subjects (5), and that these individuals have a reduced capacity to synthesize bile acids, the normal end products of cholesterol catabolism (6). The metabolic defect underlying the failure to synthesize bile acids is controversial and has been postulated to involve either a sterol 27-hydroxylase (7) or a sterol 24-hydroxylase (8). The mechanism by which a defect in hepatic bile acid synthesis leads to the diverse symptoms observed in CTX is at present not known (4); however, the pleiotropic nature of the disease does not appear to be related to hypercholesterolemia as a majority of CTX patients have normal serum cholesterol levels (4).
To gain insight into this disease and to explore the link between bile acids, sterols, and the nervous system, we recently isolated cDNA clones for the rabbit (9) and human sterol 27-hydroxylase mRNAs (10). The enzyme encoded by these cDNAs is a mitochondrial cytochrome P-450 that together with two protein cofactors, adrenodoxin and adrenodoxin reductase, hydroxylates a variety of sterols at the C-27 position (9–11). In the bile acid synthesis pathway, sterol 27-hydroxylase catalyzes the first step in the oxidation of the side chain of sterol intermediates (12, 13). Hybridization studies using the cloned rabbit cDNA revealed that sterol 27-hydroxylase mRNA was present in many different tissues and that the mRNA was encoded by a single copy gene (9). In this paper, we describe the use of the human sterol 27-hydroxylase cDNA to determine a molecular basis for CTX and to map the chromosomal location of the gene.
EXPERIMENTAL PROCEDURES
Materials
Enzymes used in cloning, DNA sequencing, and polymerase chain reactions were obtained from New England Biolabs, GIBCO, U.S. Biochemicals, and Perkin-Elmer Cetus Instruments. Nylon filters (Biotrans) for blot hybridization experiments were obtained from ICN Pharmaceuticals. Oligonucleotides for DNA sequence analysis and polymerase chain reactions were synthesized on an Applied Biosystems Model 380A DNA synthesizer. Genomic DNA was extracted from human diploid fibroblasts with an Applied Biosystems Model 340A extractor.
Fibroblast Culture
The normal and CTX fibroblasts used in these studies were obtained from Drs. M. S. Brown and J. L. Goldstein, University of Texas Southwestern Medical Center, Dallas, TX. The clinical features of the CTX subjects from which the fibroblasts were biopsied have been described previously. Subject CTX1 was a Canadian female, designated patient SC in Pastershank et al. (14). Subject CTX2 was a black American, originally described as patient JC in Harlan and Still (15), and characterized extensively with respect to sterol and bile acid metabolism by Salen and colleagues (6). CTX and normal fibroblasts were cultured as described previously (16). Genomic DNA and total RNA were isolated from these cells and analyzed by blot hybridization as described by Lehrman et al. (17, 18).
Polymerase Chain Reactions
Amplification of sterol 27-hydroxylase mRNA from fibroblasts was accomplished as described by Saiki et al. (19). Total RNA (1 μg) was first converted into cDNA in a volume of 50 μl using a cDNA synthesis kit from GIBCO. Primers for the polymerase chain reaction (see below) were added directly to the reverse transcription reaction, and amplification was carried out after a 10-min incubation at 100 °C and Taq enzyme addition for 35 cycles of 1 min/95 °C and 5 min/68 °C. A final cycle of 1 min/95 °C and 10 min/68 °C was then carried out prior to purification of the amplified cDNA by Centricon 30 (Amicon Corp.) column filtration.
The coding region of the sterol 27-hydroxylase mRNA was amplified in three overlapping fragments. One pair of oligonucleotides representing nucleotides 170 to 194 (dTCCGGCGGCGGCAACGGAGCTTAGA) and nucleotides 1250 to 1274 (dAAGAGGAAGCCATCAACTTCAATTT) was used to amplify mRNA spanning nucleotides 195 to 1249. A second pair derived from nucleotides 1066 to 1090 (dTACCACCTCTCAAAGGACCCTGAGA) and nucleotides 1728 to 1752 (dGCAAGGAGTTCCTCCCACCTCTCGA) was used to amplify the 3′ region of the cDNA encompassing nucleotides 1091 to 1727.
We were unsuccessful in amplifying the extreme 5′ end of the mRNA using a variety of different primers and conditions. It did prove possible to amplify the 5′ region of the sterol 27-hydroxylase gene. To this end, a genomic clone harboring the gene was isolated, and DNA of the 5′-flanking region and intron 1 were subjected to DNA sequence analysis. Two primers were synthesized from this information, representing a portion of the 5′-flanking region (dACTCAGCACTCGACCCAAAGGTGCA) and a portion of intron 1 (dTCTCTGTTCCGGTGCCCACTCCCAT), and used for amplification as described above except that 1 μg of genomic DNA was the starting template.
DNA sequence analysis of the amplified fragments was accomplished by the methods of Sanger et al. (20), using bacteriophage T7 DNA polymerase (Sequenase, U.S. Biochemicals). For each CTX mutation, RNA and DNA from two separate aliquots of cells were amplified and sequenced.
Transfection Analysis of CTX Mutations
Two mutations detected in the CTX subjects were recreated in an expressible cDNA encoding human sterol 27-hydroxylase (10). Site-directed mutagenesis of the cDNA was accomplished in bacteriophage M13 vectors (21) with oligonucleotides 23 bases in length as described previously (22). The vector used to drive expression of both the normal and mutated cDNAs in simian COS-M6 cells was a pCMV derivative (9). COS cells were cultivated as described by Esser et al. (22) and transfected with the sterol 27-hydroxylase expression vectors as described by Andersson et al. (9) and Cali et al. (10), except that a bovine adrenodoxin cDNA was not employed. Sterol 27-hydroxylase enzyme activity in transfected cells was detected by thin layer chromatography assay (9). The substrate was 5β-[7β-3H]cholestane-3α,7α,12α-triol. In the experiments reported here (Fig. 4), “product” corresponds to the sum of both oxidized sterols generated by the enzyme, i.e. 5β-(7β-3H] cholestane-3α,7α,12α,27-tetrol and 3α,7α,12α-trihydroxy-5β-[7β-3H] cholestanoic acid (10). Cellular protein content was determined as described by Bradford (23).
Fig. 4. Expression of normal and CTX sterol 27-hydroxylase cDNAs.

COS cells were transfected with the indicated cDNA in the pCMV2 vector. Forty-eight hours after transfection, the medium was made 2.5 μM in substrate (5β-[7β-3H]cholestane-3α,7α,12α-triol) as outlined under “Experimental Procedures” and left on the cells for the indicated time periods. The conversion of substrate into products (5β-[7β-3H]cholestane-3α,7α,12α,27-tetrol plus 3α,7α,12α-trihydroxy-5β-[7β-3H]cholestanoic acid) was determined by thin layer chromatography analysis of media sterols. The results shown are representative of two separate transfection experiments.
Immunochemical Analysis
An anti-peptide antibody was raised against an amino acid sequence (Cys-Pro-Gly-Val-Arg-Arg-Arg-Gln-Arg-Ser-Leu-Glu-Glu-Ile-Pro) corresponding to residues 15 to 28 of the mature sterol 27-hydroxylase protein (10) and containing an extra cysteine at the amino terminus for coupling purposes. The peptide was synthesized by Multipeptide Systems (San Diego, CA). Antibodies were raised in New Zealand White rabbits following a regimen described previously (24). Immunoblotting was as described (24), except that an alkaline phosphatase detection system (Bio-Rad) was used to visualize immune complexes.
Chromosomal Localization Studies
A panel of 11 Chinese hamster × human hybrid cell clones from five independent fusion experiments between Chinese hamster and human cell lines (25) was used for primary chromosomal assignment of the human sterol 27-hydroxylase gene (CYP27, Ref. 26). Regional mapping of the human CYP27 gene was carried out with four hybrids from series X,XVIII, and XIX containing different regions of human chromosome 2. For localization of the murine CYP27 gene, a panel of 13 rodent × mouse hybrid clones or subclones derived from four fusion experiments were used. The origin and characterization of hybrids in both human and mouse mapping panels have been described (27).
Southern blotting experiments were carried out as detailed previously (28) using radiolabeled probes derived from the 5′ or 3′ regions of the human sterol 27-hydroxylase cDNA (10). Radiolabeling with 32P was done by the random priming method (29).
RESULTS
The sterol 27-hydroxylase gene in CTX subjects was first examined in Southern blotting experiments with genomic DNA isolated from the fibroblasts of two unrelated affected individuals. As shown in Fig. 1, these studies did not reveal any gross abnormalities in the gene structure: digestion of the CTX DNAs with three different restriction enzymes yielded a simple hybridization pattern identical with that obtained with the DNA of a normal individual. The relative intensities of the normal and CTX hybridizing fragments in the autoradiograms were similar, suggesting that both copies of the sterol 27-hydroxylase gene were present in the CTX subjects (Fig. 1).
Fig. 1. Southern blot hybridization of genomic DNA isolated from subjects CTX1 and CTX2.

Genomic DNA (10 μg) from normal and CTX fibroblasts was digested with the indicated restriction enzyme and subjected to Southern blot analysis as described by Lehrman et al. (17). Radioactive single-stranded DNA probes (45) corresponding to approximately nucleotides 500 to 710, 711 to 914, and 1400 to 1594 of the human sterol 27-hydroxylase cDNA (10) were used in the hybridization reactions. The positions to which HindIII fragments of bacteriophage λ-DNA migrated in the agarose gel and their sizes in kilobases (kb) are indicated on the left side of the figure. Filters were exposed to Kodak XRP x-ray film for 120 h in the presence of an intensifying screen.
Total RNA was isolated from the fibroblasts of subjects CTX1 and CTX2 and analyzed by blot hybridization. As shown in Fig. 2, a single 2.0-kilobase species of sterol 27-hydroxylase mRNA was detected that was present in near equal amounts in both normal and CTX fibroblasts. These studies suggested that if mutations in sterol 27-hydroxylase were present in these CTX subjects, they would most likely be either missense or nonsense mutations.
Fig. 2. Blot hybridization analysis of normal and CTX sterol 27-hydroxylase mRNAs.

Different amounts of total cellular RNA isolated from normal and CTX fibroblasts were analyzed by blot hybridization after electrophoresis through 1.5% (w/v) agarose gels. Hybridization probes labeled with 32P were derived from nucleotides 510 to 710, 711 to 914, and 1400 to 1594 of the sterol 27-hydroxylase cDNA. After washing, the filters were exposed to Kodak XAR-5 x-ray film for 96 h in the presence of an intensifying screen. The positions to which RNAs of known size (kilobases, kb) migrated to in the gel are shown on the left of the figure.
The presence of sterol 27-hydroxylase mRNA in the CTX fibroblasts allowed the use of the polymerase chain reaction to amplify and then sequence this mRNA. The data from these experiments are summarized in Fig. 3 and illustrate the locations of two different mutations that were present in subjects CTX1 and CTX2. Both mutations involved single base pair substitutions that convert arginine codons (CGPy) to cysteine codons (TGPy). No other nucleotide changes were detected in the two CTX mRNAs, which together with the Southern blotting data of Fig. 1, suggests that both individuals were homozygous at this locus. In addition, the CTX2 mutation was not present in the sterol 27-hydroxylase mRNAs of five unrelated individuals of the same ethnic background analyzed in a similar manner (data not shown).
Fig. 3. CTX mutations in sterol 27-hydroxylase.
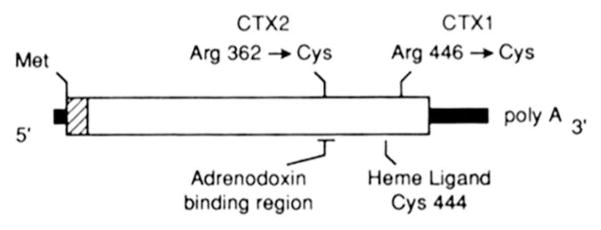
The 532-amino-acid coding region of the sterol 27-hydroxylase mRNA specifies a 33-residue mitochondrial signal sequence (hatched region) and a 499-residue mature protein (white block). Noncoding sequences (thick black lines) are present at the 5′ and 3′ ends of the mRNA. The locations of the amino acid changes detected in subjects CTX1 and CTX2 after amplification and DNA sequencing are indicated above the line. Two protein domains of this mitochondrial cytochrome P-450 that may be affected by these changes are indicated below the schematic.
In order to determine the effects of the observed amino acid substitutions on sterol 27-hydroxylase enzyme activity, each of the mutations shown in Fig. 3 was recreated by site-directed mutagenesis in an expressible cDNA and analyzed by transfection into simian COS cells. As substrate, we used 5β-[7β-3H]cholestane-3α,7α,12α-triol, an intermediate in the bile acid synthesis pathway that is converted to 5β-[7β-3H]cholestane-3α,7α,12α,27-tetrol and further oxidized to 3α,7α,12α-trihydroxy-5β-[7β-3H]cholestanoic acid by the action of sterol 27-hydroxylase (10). As shown in Fig. 4 (upper left panel), mock transfection of COS cells with the expression vector alone did not give rise to sterol 27-hydroxylase activity. However, transfection of cells with a vector containing the normal sterol 27-hydroxylase cDNA (upper right panel), led to a 50% conversion of substrate into product over an 18-h time period. In contrast, transfection with a cDNA containing the CTX1 mutation led to the detection of a low, but demonstrable level of activity (lower left panel), whereas transfection with a CTX2 cDNA did not result in detectable enzyme activity (lower right panel). Reversion of the CTX2 mutation by site-directed mutagenesis restored the capacity to synthesize a fully active enzyme (data not shown).
To confirm that the transfected cells did indeed express a sterol 27-hydroxylase protein, cell extracts were subjected to immunoblot analysis using an antiserum against a peptide from the predicted protein sequence. In this analysis (Fig. 5), mock transfected cells did not produce any immunoreactive protein (lane 1), whereas cells transfected with the normal cDNA (lanes 2 and 6) or the CTX2-revertant cDNA (lane 4) expressed two immunoreactive proteins corresponding to the precursor (upper band) and mature (lower band) forms of the mitochondrial sterol 27-hydroxylase protein. Both CTX2 (lane 3) and CTX1 (lane 5) cDNAs expressed comparable levels of these two proteins; however, the amount of the mature form appeared somewhat lower in both cases.
Fig. 5. Immunoblotting of proteins synthesized from transfected sterol 27-hydroxylase cDNAs.
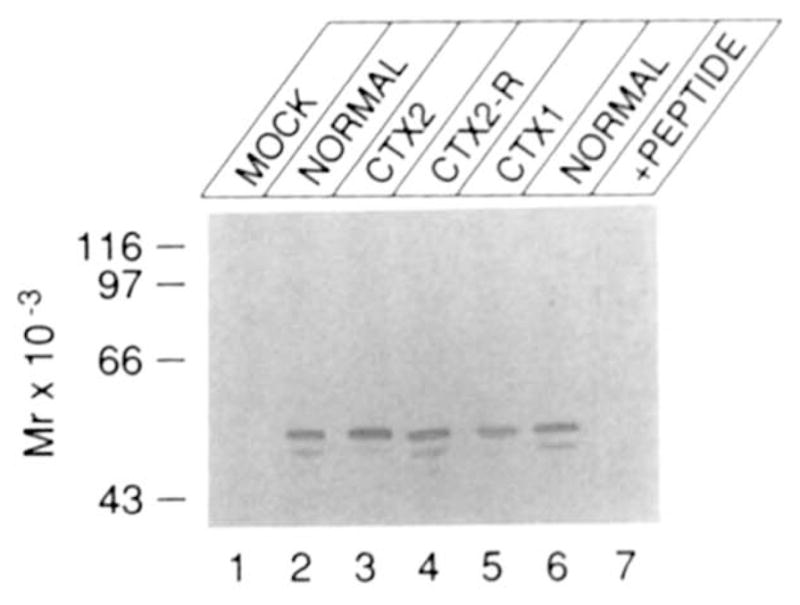
COS cells were transfected with the indicated cDNA. Forty-eight hours after transfection, the cells were lysed with detergent-containing buffers and fractionated by centrifugation into a nuclear, mitochondrial, and microsomal pellet (46). Twenty micrograms of the mitochondrial pellet was electrophoresed and subjected to immunoblot analysis as described under “Experimental Procedures.” In lane 7, 20 μg of mitochondrial pellet from cells transfected with the normal cDNA was electrophoresed and subjected to immunoblot analysis as described above, except that the antisera was preincubated with 10 μg/ml peptide prior to inclusion in the blotting reactions.
The assignment of precursor and mature protein labels to the upper and lower bands, respectively, is based on the observation that a radiolabeled preparation of sterol 27-hydroxylase translated in vitro from synthetic RNA in a rabbit reticulocyte lysate co-migrates with the upper of the two proteins visualized by immunoblotting (data not shown), and on the predicted size of the mitochondrial signal sequence (33 residues) that is removed to generate the mature human enzyme (10). Although not tested formally, we believe that the excess of the precursor form of sterol 27-hydroxylase (Fig. 5) is a consequence of overexpression of the cDNA in the transfected COS-M6 cells. Regardless, the data of Fig. 5 indicate that the low to absent enzyme activity in COS cells transfected with the CTX cDNAs cannot be attributed to lack of expression.
The inheritance pattern of CTX suggests that the defective gene should map to one of the autosomal chromosomes (4). Thus, we next determined the chromosomal location of the sterol 27-hydroxylase gene (CYP27) by Southern blotting analysis of a panel of Chinese hamster × human somatic cell hybrids. The data in Table I, derived with a near full length cDNA probe, indicate that the presence or absence of human chromosome 2 in hybrid cell lines was in perfect concordance with human HindIII (14.5, 13.0, and 6.5 kilobases) and BglII (10.5 and 5.0 kilobases) fragments hybridizing with this probe. All other human chromosomes were excluded by 2 to 7 discordant hybrids.
Table I. Correlation of human CYP27 sequences with human chromosomes in rodent× human somatic cell hybrids.
The numbers of hybrids that are concordant (+/+ or −/−) and discordant (+/− or −/+) with the human CYP27 sequence are given for each chromosome. Hybrids in which a particular chromosome was structurally rearranged or present in fewer than 10% of cells were excluded.
| Hybridization/chromosome | Human chromosomes
|
||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | 18 | 19 | 20 | 21 | 22 | X | |
| +/+ | 1 | 2 | 1 | 1 | 0 | 2 | 0 | 1 | 1 | 0 | 1 | 2 | 1 | 2 | 1 | 1 | 0 | 1 | 1 | 0 | 2 | 1 | 1 |
| −/− | 7 | 9 | 5 | 5 | 6 | 5 | 5 | 6 | 7 | 7 | 4 | 7 | 5 | 3 | 3 | 6 | 7 | 4 | 5 | 5 | 4 | 2 | 1 |
| +/− | 1 | 0 | 0 | 1 | 2 | 0 | 2 | 1 | 1 | 2 | 1 | 0 | 1 | 0 | 1 | 1 | 2 | 0 | 1 | 2 | 0 | 1 | 0 |
| −/+ | 2 | 0 | 4 | 1 | 2 | 4 | 2 | 3 | 2 | 2 | 3 | 2 | 4 | 4 | 6 | 2 | 2 | 5 | 4 | 4 | 4 | 6 | 3 |
| Discordant hybrids | 3 | 0 | 4 | 2 | 4 | 4 | 4 | 4 | 3 | 4 | 4 | 2 | 5 | 4 | 7 | 3 | 4 | 5 | 5 | 6 | 4 | 7 | 3 |
| Informative hybrids | 11 | 11 | 10 | 8 | 10 | 11 | 9 | 11 | 11 | 11 | 9 | 11 | 11 | 9 | 11 | 10 | 11 | 10 | 11 | 11 | 10 | 10 | 5 |
Regional localization of the CYP27 gene was accomplished by Southern blot analysis of four hybrid cell lines containing defined portions of human chromosome 2 in a Chinese hamster or rat background. The results are summarized in Fig. 6 and indicate that the CYP27 gene maps to the distal portion (q33-qter) of the long arm of chromosome 2. Thus CPY27 is asyntenic with genes for 12 other subfamilies of cytochrome P-450s and with the adrenodoxin and adrenodoxin reductase genes (26, 30, 31). In experiments not shown, the CYP27 gene was similarly mapped to chromosome 1 of the mouse within a large syntenic group of genes that have homologs on human chromosome 2 (32).
Fig. 6. Regional mapping of sterol 27-hydroxylase gene (CYP27) on human chromosome 2.

The regions present in four analyzed hybrid cell lines are indicated by vertical bars next to an idiogram of chromosome 2. Southern blot analysis of hybrid cell line DNA using a sterol 27-hydroxylase cDNA probe revealed the presence (+) or absence (−) of the human CYP27 fragments as indicated. The bracket demarks region q33-qter that contains the CYP27 locus.
DISCUSSION
We report different point mutations in the gene for sterol 27-hydroxylase in two unrelated subjects with CTX and the autosomal localization of this gene to chromosome 2 in the human and chromosome 1 in the mouse.
In patient CTX1, a mutation encoding a cysteine in place of an arginine was found (Fig. 3). The novel cysteine is located at position 446 and is two amino acids away from a ubiquitously conserved cysteine of cytochrome P-450 proteins that serves as a ligand for the heme cofactor (33). In patient CTX2, another arginine to cysteine encoding mutation was found (Fig. 3). In this case, the novel cysteine occurs at position 362 in a sequence that is highly conserved among mitochondrial cytochrome P-450s and that is thought to bind the adrenodoxin cofactor (34). Thus, in both cases, the observed mutations are present in functional domains of the sterol 27-hydroxylase and would be expected to disrupt enzyme activity. This expectation was met in expression studies involving cDNAs harboring the CTX mutations (Fig. 4). Interestingly, both CTX mutations occurred in CG dinucleotides, which are thought to be hypermutable in the human genome (35).
Although it is not yet certain that all forms of CTX are caused by mutations in the CYP27 gene, the results presented here, together with previous physiological (36), biochemical (7, 37), and immunochemical results (38) obtained with other CTX subjects, suggest that this conclusion is warranted. On the basis of our data, molecular assays can now be developed for early diagnosis and carrier detection of CTX in populations like Sephardic Jews of Moroccan descent in whom the frequency of this disease is very high (39, 40). Such assays should prove especially valuable in CTX, since early treatment with bile acids can retard or even reverse the course of the disease (41).
The presence of sterol 27-hydroxylase mRNA in a variety of tissues (9) suggests that this enzyme plays a multifaceted role in sterol metabolism that extends beyond the synthesis of bile acids in the liver. The deposition of sterols in the neural tissues of CTX patients together with the occurrence of a progressive neurological dysfunction (4) suggest that this enzyme performs a vital function in the nervous system. Evidence for such a role is bolstered by the observation that other sterol storage disorders that do not involve disruption of the sterol 27-hydroxylase gene, such as sistosterolemia (4), are not characterized by central nervous system defects (42).
Sterol 27-hydroxylase may facilitate reverse sterol transport (43) in neural tissues or participate in the redistribution of sterols among cells of the central nervous system (44). Alternatively, the enzyme may play a key regulatory role in maintaining sterol homeostasis, disruption of which may lead to sterol accumulation (10). Data to support these hypotheses may come from the future analysis of sterol 27-hydroxylase expression in extrahepatic tissues and in other disease states with neurological manifestations.
Acknowledgments
We thank Daphne Davis, Lisa Beatty, and Edith Womack for technical assistance, Helena Dahlbäck, Henry Danielsson, and Kjell Wikvall for radiolabeled sterols, and S. Andersson, M. S. Brown, J. L. Goldstein, H. H. Hobbs, D. F. Jelinek, and T. C. Südhof for advice and critical reading of the manuscript.
Footnotes
This research was supported by Grants HL-20948 (to D. W. R.) and GM-26105 (to U. F.) from the National Institutes of Health and Grant I-0971 (to D. W. R.) from the Robert A. Welch Foundation of Texas.
The abbreviation used is: CTX, cerebrotendinous xanthomatosis.
References
- 1.Myant NB. The Biology of Cholesterol and Related Steroids. Heinemann Medical Books; London: 1981. pp. 1–910. [Google Scholar]
- 2.Norton WT. In: Basic Neurochemistry. 2. Siegel GJ, Albers RW, Katzman R, Agranoff BW, editors. Little, Brown and Co; Boston: 1976. pp. 74–99. [Google Scholar]
- 3.Van Bogaert L, Scherer HJ, Epstein E. Une Forme Cerebrale De La Cholesterinose Generalisee. Masson; Paris: 1937. pp. 1–159. [Google Scholar]
- 4.Bjorkhem I, Skrede S. In: The Metabolic Basis of Inherited Disease. 6. Scriber CR, Beaudet AL, Sly WS, Valle D, editors. McGraw-Hill Book Co; New York: 1989. pp. 1283–1302. [Google Scholar]
- 5.Menkes JH, Schimschock JR, Swanson PD. Arch Neurol. 1968;19:47–53. doi: 10.1001/archneur.1968.00480010065004. [DOI] [PubMed] [Google Scholar]
- 6.Setoguchi T, Salen G, Tint GS, Mosbach EH. J Clin Invest. 1974;53:1393–1401. doi: 10.1172/JCI107688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oftebro H, Björkhem I, Skrede S, Schreiner A, Pedersen JI. J Clin Invest. 1980;65:1418–1430. doi: 10.1172/JCI109806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salen G, Shefer S, Cheng RW, Dayal B, Batta AK, Tint GS. J Clin Invest. 1979;63:38–44. doi: 10.1172/JCI109275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andersson S, Davis DL, Dahlbäck H, Jörnvall H, Russell DW. J Biol Chem. 1989;624:8222–8229. [PubMed] [Google Scholar]
- 10.Cali JJ, Russell DW. J Biol Chem. 1991;266:7774–7778. [PubMed] [Google Scholar]
- 11.Wikvall K. J Biol Chem. 1984;259:3800–3804. [PubMed] [Google Scholar]
- 12.Danielsson H. In: The Bile Acids, Chemistry, Physiology, and Metabolism. Nair PP, Kritchevsky D, editors. Plenum Publishing Corp; New York: 1973. pp. 1–32. [Google Scholar]
- 13.Björkhem I. In: Sterols and Bile Acids. Danielsson H, Sjövall SJ, editors. Elsevier Science Publishers B. V; Amsterdam: 1985. pp. 231–278. [Google Scholar]
- 14.Pastershank SP, Yip S, Sodhi HS. J Assoc Can Radiol. 1974;25:282–286. [PubMed] [Google Scholar]
- 15.Harlan WR, Jr, Still WJS. N Engl J Med. 1968;278:416–422. doi: 10.1056/NEJM196802222780803. [DOI] [PubMed] [Google Scholar]
- 16.Goldstein JL, Basu SK, Brown MS. Methods Enzymol. 1983;98:241–260. doi: 10.1016/0076-6879(83)98152-1. [DOI] [PubMed] [Google Scholar]
- 17.Lehrman MA, Schneider WJ, Südhof TC, Brown MS, Goldstein JL, Russell DW. Science. 1985;227:140–146. doi: 10.1126/science.3155573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lehrman MA, Russell DW, Goldstein JL, Brown MS. J Biol Chem. 1987;262:3354–3361. [PubMed] [Google Scholar]
- 19.Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, Mullis KB, Erlich HA. Science. 1988;239:487–491. doi: 10.1126/science.2448875. [DOI] [PubMed] [Google Scholar]
- 20.Sanger F, Nicklen S, Coulson AR. Proc Natl Acad Sci U S A. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zoller MJ, Smith M. Methods Enzymol. 1983;100:468–500. doi: 10.1016/0076-6879(83)00074-9. [DOI] [PubMed] [Google Scholar]
- 22.Esser V, Limbird LE, Brown MS, Goldstein JL, Russell DW. J Biol Chem. 1988;263:13282–13290. [PubMed] [Google Scholar]
- 23.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 24.Russell DW, Schneider WJ, Yamamoto T, Luskey KL, Brown MS, Goldstein JL. Cell. 1984;37:577–585. doi: 10.1016/0092-8674(84)90388-x. [DOI] [PubMed] [Google Scholar]
- 25.Barton DE, Yang-Feng TL, Mason AJ, Seeburg PH, Francke U. Genomics. 1989;5:91–99. doi: 10.1016/0888-7543(89)90091-8. [DOI] [PubMed] [Google Scholar]
- 26.Nebert DW, Nelson DR, Coon MJ, Estabrook RW, Feyereisen R, Fujii-Kuriyama Y, Gonzalez FJ, Guengerich FP, Gunsalus IC, Johnson EF, Loper JC, Sato R, Waterman MR, Waxman DJ. DNA Cell Biol. 1991;10:1–14. doi: 10.1089/dna.1991.10.1. [DOI] [PubMed] [Google Scholar]
- 27.Alonso MA, Barton DE, Francke U. Immunogenetics. 1988;27:91–95. doi: 10.1007/BF00351081. [DOI] [PubMed] [Google Scholar]
- 28.Southern EM. J Mol Biol. 1975;98:503–517. doi: 10.1016/s0022-2836(75)80083-0. [DOI] [PubMed] [Google Scholar]
- 29.Feinberg AP, Vogelstein B. Anal Biochem. 1983;132:6–13. doi: 10.1016/0003-2697(83)90418-9. [DOI] [PubMed] [Google Scholar]
- 30.Morel Y, Picado-Leonard J, Wu DA, Chang CY, Mohandas TK, Chung B, Miller WL. Am J Hum Genet. 1988;43:52–59. [PMC free article] [PubMed] [Google Scholar]
- 31.Solish SB, Picado-Leonard J, Morel Y, Kuhn RW, Mohandas TK, Hanukoglu I, Miller WL. Proc Natl Acad Sci U S A. 1988;85:7104–7108. doi: 10.1073/pnas.85.19.7104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lyon MF, Kirby MC. Mouse Genome. 1990;87:28–37. [Google Scholar]
- 33.Gonzales FJ. Pharmacol Rev. 1989;40:243–288. [PubMed] [Google Scholar]
- 34.Tuls J, Geren L, Millet F. J Biol Chem. 1989;264:16421–16425. [PubMed] [Google Scholar]
- 35.Cooper DN, Youssoufian H. Hum Genet. 1988;78:151–155. doi: 10.1007/BF00278187. [DOI] [PubMed] [Google Scholar]
- 36.Björkhem I, Fausa O, Hopen G, Oftebro H, Pedersen JI, Skrede S. J Clin Invest. 1983;71:142–148. doi: 10.1172/JCI110742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Skrede S, Björkhem I, Kvittingen EA, Buchmann MS, Lie SO, East C, Grundy S. J Clin Invest. 1986;78:729–735. doi: 10.1172/JCI112633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miki H, Takeuchi H, Yamada A, Nishioka M, Matsuzawa Y, Hamamoto I, Hiwatashi A, Ichikawa Y. Clin Chim Acta. 1986;160:255–263. doi: 10.1016/0009-8981(86)90192-0. [DOI] [PubMed] [Google Scholar]
- 39.Berginer VM, Abeliovich D. Am J Med Genet. 1981;10:151–157. doi: 10.1002/ajmg.1320100209. [DOI] [PubMed] [Google Scholar]
- 40.Koopman BJ, Wolthers BG, van der Molen JC, van der Slik W, Waterreus RJ, van Spreeken A. J Inherited Metab Dis. 1988;11:56–75. doi: 10.1007/BF01800057. [DOI] [PubMed] [Google Scholar]
- 41.Salen G, Meriwether TW, Nicolau G. Biochem Med. 1975;14:57–74. doi: 10.1016/0006-2944(75)90020-4. [DOI] [PubMed] [Google Scholar]
- 42.Salen G, Vladimir MD, Shore V, Horak I, Horak E, Tint GS, Shefer S. N Engl J Med. 1987;316:1233–1238. doi: 10.1056/NEJM198705143162002. [DOI] [PubMed] [Google Scholar]
- 43.Glomset JA. J Lipid Res. 1968;9:155–167. [PubMed] [Google Scholar]
- 44.Mahley RW. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 45.Church GM, Gilbert W. Proc Natl Acad Sci U S A. 1984;81:1991–1995. doi: 10.1073/pnas.81.7.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jelinek DF, Andersson S, Slaughter CA, Russell DW. J Biol Chem. 1990;265:8190–8197. [PMC free article] [PubMed] [Google Scholar]