

Abstract
Platelet-derived growth factor (PDGF) has been implicated in the pathogenesis of arterial atherosclerosis and venous neointimal hyperplasia. We examined the effects of PDGF isoforms on smooth muscle cells (SMCs) from arterial and venous origins in order to further understand the differential responsiveness of these vasculatures to proliferative stimuli. Serum-starved human arterial and venous SMCs exhibited very different proliferative responses to PDGF isoforms. Whereas, proliferation of arterial SMCs was strongly stimulated by PDGF-AA, venous SMCs showed no proliferative response to PDGF-AA, but instead demonstrated a significantly greater proliferative response to PDGF-BB than arterial SMCs. Part of this difference could be attributed to differences in PDGF receptors expression. There was a 2.5-fold higher (P <0.05) density of PDGF receptor-α (PDGF-Rα) and a 6.6-fold lower (P <0.05) density of PDGF-Rβ expressed on arterial compared to venous SMCs. Concomitant with an increased proliferative response to PDGF-AA in arterial SMCs was a marked PDGF-Rα activation, enhanced phosphorylation of ERK1/2 and Akt, a transient activation of c-Jun NH2-terminal kinase (JNK), and a significant reduction in expression of the cell-cycle inhibitor p27kip1. This pattern of signaling pathway changes was not observed in venous SMCs. No phosphorylation of PDGF-Rα was detected after venous SMC exposure to PDGF-AA, but there was enhanced phosphorylation of ERK1/2 and Akt in venous SMCs, similar to that seen in the arterial SMCs. PDGF-BB stimulation of venous SMC resulted in PDGF-Rβ activation as well as transactivation of epidermal growth factor receptor (EGF-R); transactivation of EGF-R was not observed in arterial SMCs. These results may provide an explanation for the differential susceptibility to proliferative vascular diseases of arteries and veins.
Keywords: PLATELET-DERIVED GROWTH FACTOR (PDGF), SIGNALING PATHWAYS, CELL PROLIFERATION, ARTERIAL AND VENOUS SMC
The proliferative phenotype of artery and veins is different from each other and appears to have clinically significant consequences. Coronary artery bypass grafting with the autologous saphenous vein or internal mammary artery is widely used to treat atherosclerotic coronary artery disease. Graft failure due to stenosis occurs much more frequently in saphenous vein grafts than internal mammary artery grafts [Loop et al., 1986; Motwani and Topol, 1998]. Graft stenosis is most frequently caused by neointimal hyperplasia, another type of vascular proliferative disease. Similarly, most neointimal hyperplasia occurs at the graft-venous anastomosis of hemodialysis arteriovenous grafts, resulting in 60% graft loss in the first year [US Renal Data System, 2007]. In contrast, atherosclerosis occurs in arteries but rarely in veins despite the exposure of both arteries and veins to the same systemic risk factors such as smoking and dyslipidemia. While hemodynamic and other local factors probably play a role in the different susceptibilities to atherosclerosis and neointimal hyperplasia between artery and veins, it is likely that the observed dissimilarities can be at least partially explained by intrinsic phenotypic differences between arterial and venous smooth muscle cells (SMCs). These phenotypic differences would include the activation of different mitogenic signaling pathways.
Platelet-derived growth factor (PDGF) is the most potent SMC mitogen released at the site of injury by platelets, endothelial cells, SMCs, and many other cell types [Heldin and Westermark, 1999; Raines, 2004]. The role for PDGF in the pathogenesis of arterial injury disorders, including atherosclerosis and post-angioplasty restenosis, has been well established. However, the effects of various PDGF isoforms and the differences between arterial and venous SMCs in their responses to these isoforms are largely unknown and may explain the clinically observed differences between the arterial and venous diseases. PDGF is a dimer composed of disulfide-linked polypeptides termed A-chain and B-chain. At least three isoforms (PDGF-AA, PDGF-AB, and PDGF-BB) are present in human platelets in comparable quantities [Hart et al., 1990]. The PDGF-C and PDGF-D chains were discovered more recently and their biology is not well established [Li et al., 2000; Bergsten et al., 2001; LaRochelle et al., 2001]. The role and expression of PDGF-C and PDGF-D in arteries and in atherosclerotic lesions remain unclear. Recently, Karvinen et al. [2009] reported that PDGF-C was strongly expressed in endothelial cells in both normal arteries and atherosclerotic lesions whereas PDGF-D was only weakly expressed in these cells. The functions of PDGF-CC and PDGF-DD have been explored and reviewed [Reigstad et al., 2005; Wagsater et al., 2009] and will not be further discussed in this paper.
PDGF stimulates cell proliferation by binding to PDGF receptor α (PDGF-Rα) or PDGF receptor β (PDGF-Rβ), causing receptor dimerization and autophosphorylation. Previous studies have demonstrated that PDGF-AA activates only αα receptor dimers, PDGF-AB activates both αα and αβ receptor dimers, whereas PDGF-BB activates αα, αβ, and ββ receptor dimers [Heldin et al., 1998]. Many studies have demonstrated the pathogenic roles of PDGF-AB, PDGF-BB, and PDGF-Rβ in the formation of atherosclerotic and neointimal hyperplasia lesions. However, their roles in the pathogenesis of venous diseases and the roles of PDGF-AA and PDGF-Rα in vascular disorders are largely unknown.
We have previously reported that the proliferation of human venous SMCs was more responsive to PDGF-AB stimulation compared to arterial SMCs [Li et al., 2006a]. In the present study, we investigated the proliferative effects of PDGF-BB and PDGF-AA on arterial and venous SMCs and identified differences in the underlying signaling pathways that led to different responses to these PDGF isoforms between arterial and venous SMCs. These results provide unique insights into the molecular basis of the varying susceptibility to proliferative disorders between arteries and veins.
METHODS
REAGENTS AND ANTIBODIES
All cell culture reagents were purchased from Cascade Biologics (Portland, OR), except fetal calf serum (FCS), which was obtained from Atlanta Biological (Lawrenceville, GA). Recombinant PDGF-AA, -AB, -BB, phycoerythrin (PE)-conjugated monoclonal antibodies directed against PDGF-Rα (FAB1264p) and PDGF-Rβ (FAB1263p), and the human phospho-RTK array kit were purchased from R&D Systems (Minneapolis, MN). Cell proliferation BrdU kits were obtained from Amersham Biosciences (Pittsburgh, PA). Antibodies directed against ERK1/2, phospho-ERK1/2, c-Jun NH2-terminal kinase (JNK) 1/2/3, phospho-JNK1/2/3, p38, phospho-p38, Akt, phospho-Akt (catalog #9102, #9101, #9252, #9251, #9212, #9211, #9272, and #9271, respectively), and the cell-cycle regulators, p21cip1 (#2946) and p27kip1 (#3688), were obtained from Cell Signaling Technologies (Beverly, MA). Goat anti-rabbit and goat anti-mouse antibodies conjugated with horseradish peroxidase were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Precast polyacrylamide gels, nitrocellulose membranes, and Immun-Star HRP Chemiluminescent Kits were purchased from Bio-Rad Laboratories (Hercules, CA). Complete Mini-Protease Inhibitor Cocktail was obtained from Roche Applied Science (Indianapolis, IN). The bicinchoninic acid (BCA) protein assay kit was purchased from Pierce Chemical (Rockford, IL).
CELL CULTURE
Human arterial SMCs from four different donors and saphenous vein SMCs from one donor were purchased from Cambrex Bio Science (Chicago, IL). SMCs from saphenous veins were also isolated from two patients undergoing coronary artery bypass grafting, using a protocol approved by the Institutional Review Board at the University of Utah. Saphenous vein SMCs were isolated after manual removal of adventitial layers under a dissection microscope and enzymatic digestion of endothelial cells. The SMCs were identified by positive staining for smooth muscle α-actin.
All cells were cultured in a humidified 37°C incubator with 5% CO2 in Medium 231, 10% fetal bovine serum (FBS) and Smooth Muscle Growth Supplement (SMGS), supplemented with 5 μg/ml insulin, 0.5 ng/ml human epidermal growth factor, 2 ng/ml human basic fibroblast growth factor, 100 μg/ml streptomycin, 100 U/ml penicillin G, and 0.25 μg/ml amphotericin B. For all experiments, cells from passages 3–7 were used. Cells were grown to 70–80% confluence in the complete medium described above and then rendered quiescent by incubation with medium containing 0.5% FBS and no additional growth factors. The cells were kept quiescent for 48 h before the addition of PDGF isoforms.
ASSAY OF CELL PROLIFERATION IN RESPONSE TO VARIOUS PDGF ISOFORMS
SMCs were seeded on 96-well tissue culture plates at a density of 5 × 103 cells/well. PDGF-AA, -AB or, -BB was added to the quiescent cells at final concentrations of 0.1–100 ng/ml. DNA synthesis was measured by 5-bromo-2′-deoxyuridine (BrdU) incorporation 48 h after treatment. Briefly, after 48 h of incubation with PDGF, the BrdU labeling solution was added to the cells and incubated for another 12–24 h. The culture medium was then removed. The cells were fixed and the DNA denatured by the addition of fixative for 30 min at room temperature. Subsequently, horseradish peroxidase-labeled anti-BrdU monoclonal antibody was added and the plate incubated at room temperature for 90 min. The BrdU–antibody complexes were detected by a colorimetric reaction with the substrate (3,3′,5,5′-tetramethylbenzidine). The optical density was read at 450 nm in a microplate reader (Multiskan Ascent, Thermo Electron Corporation, San Jose, CA). All experiments were performed in triplicate wells and repeated in separate experiments.
QUANTITATIVE FLOW CYTOMETRY FOR PDGF RECEPTORS
Arterial and venous SMCs cultured in complete medium were stained with PE-conjugated anti-PDGF-Rα or anti-PDGF-Rβ monoclonal antibodies. The density of PDGF-Rα and PDGF-Rβ on cell surfaces was quantified by flow cytometry and expressed as antibodies bound per cell (ABC). Briefly, the cells were cultured to 80–90% confluence, detached with 0.5 mM EDTA (pH 8.0) in PBS, and washed with PBS (supplemented with 0.5% BSA). Cells were stained with PE-conjugated anti-PDGF-Rα or anti-PDGF-Rβ monoclonal antibodies for 30–45 min at 2–8°C. Cells were washed again and 1–2 μl of 7-amino-actinomycin D was added to assess viability. Cells were analyzed on a fluorescence-activated cell sorter (FACS) cytometer using CellQuest software (Becton–Dickinson, San Jose, CA). A QuantiBRITE PE Kit (BD Bioscience, Mississauga, ON, Canada) was used to calibrate the FL2 channel and to estimate the number of PE molecules bound per cell. By using the known ratio (1:1) of PE molecules to antibodies, the number of anti-PDGF antibodies bound per cell was calculated.
IMMUNOBLOTTING FOR ASSESSING SIGNALING PROTEIN ACTIVATION IN RESPONSE TO PDGF
Quiescent arterial and venous SMCs were stimulated with 50 ng/ml of PDGF-AA or PDGF-BB for 5 min. After PDGF stimulation, the cells were washed twice with ice-cold PBS and lysed using PBS containing 150 mM NaCl, 10 mM HEPES, 0.6% NP-40, pH 7.9, and one tablet of protease inhibitor cocktail per 10 ml of buffer that was prepared immediately before use. The lysates were incubated on ice for 30 min and then centrifuged at 10,000g for 10 min. Protein concentrations in the cell lysates were determined using the BCA protein assay. The phosphorylation states of receptor tyrosine kinases (RTKs) in the cell lysates were determined using a human phospho-RTK array kit. The array consisted of 42 different anti-RTK antibodies spotted in duplicate on nitrocellulose membranes. Cell lysates were diluted and incubated with the array membrane at 4°C overnight. After washing, the membrane was exposed to a pan-anti-phospho-tyrosine antibody conjugated to horseradish peroxidase (HRP). Phosphorylated tyrosines on activated receptors were detected by chemiluminescence on an X-ray film. Phospho-RTK array data were quantified using the ImageJ 1.41 software (http://rsb.info.nih.gov/ij/).
In other experiments, quiescent arterial or venous SMCs were stimulated with 50 ng/ml of PDGF-AA and PDGF-BB for 5 min (for the assessment of MAPK and Akt activation), variable durations (2, 5, 10, 15, 30, and 60 min for the assessment of phospho-JNK1/2) and 48 h (for the assessment of p27kip1 and p21cip1). The cell lysates were prepared as described above. Protein concentrations in the cell lysates were determined using the BCA protein assay. Equal amounts of the sample protein were subjected to SDS–polyacrylamide gel electrophoresis (PAGE) on precast 4–20% gradient gels. Following electrophoresis and transfer to nitrocellulose membranes, the blots were probed with antibodies against specific signaling proteins. The membranes were incubated with the primary antibody overnight at 4 °C, followed by HRP-conjugated secondary antibody for 45 min at room temperature. Chemiluminescence detection of the bound antibody was performed according to the manufacturer’s instructions. Densitometry of the band on images was quantified using the ImageJ 1.41 software.
STATISTICAL ANALYSIS
Results are reported as means ± SD. Comparison between two groups was performed using the two-tailed Student’s t-test as implemented in Microsoft Excel. Statistically significant results are defined as P <0.05.
RESULTS
PDGF-STIMULATED CELL PROLIFERATION
Arterial SMCs and venous SMCs were separately incubated with incremental concentrations of PDGF-AA, PDGF-AB, or PDGF-BB. Consistent with our previous report [Li et al., 2006a], PDGF-AB stimulated the proliferation of both arterial and venous SMCs although the magnitude was greater in venous SMCs than in arterial cells at maximal concentrations (50 and 100 ng/ml) of PDGF-AB (Fig. 1B). Similar patterns were observed with PDGF-BB-stimulated proliferation in arterial and venous SMCs (Fig. 1C). In contrast, PDGF-AA significantly increased proliferation in arterial SMCs, but had no effect on the proliferation of venous SMCs (Fig. 1A).
Fig. 1.

PDGF-induced proliferation of arterial smooth muscle cells (ASMC, open circles; n = 6) and venous smooth muscle cells (VSMC, closed circles; n = 6), as assessed by the bromo-deoxyuridine incorporation assay. All three PDGF isoforms significantly stimulated the proliferation of arterial SMC. In contrast, PDGF-AA exerted no mitogenic effect on venous SMC (A), although PDGF-AB (B), or PDGF-BB (C) stimulated greater DNA synthesis in venous SMCs at high concentrations (50 and 100 ng/ml), compared to arterial SMCs. *P <0.05, arterial SMCs compared to venous SMCs.
DENSITIES OF PDGF RECEPTOR ISOFORMS ON ARTERIAL AND VENOUS SMCS
Arterial and venous SMCs cultured in complete medium were stained with PE-conjugated anti-PDGF-Rα or anti-PDGF-Rβ monoclonal antibodies. The densities of PDGF-Rα and PDGF-Rβ on cell surfaces were quantified by flow cytometry. While both cell types expressed PDGF-Rα, the expression was significantly higher on arterial SMCs than on venous SMCs (9,914 ± 4,262 vs. 3,915 ± 1,764 molecules/cell; P <0.05) (Fig. 2). In contrast, PDGF-Rβ expression was significantly higher on venous SMCs than on arterial SMCs (73,220 ± 29,320 vs. 11,090 ± 5,478 molecules/cell; P <0.05). The densities of PDGF-Rα and PDGF-Rβ were similar on arterial SMCs, but the density of PDGF-Rβ was much higher than that of PDGF-Rα on venous SMCs.
Fig. 2.

Expression of PDGF receptor isoforms (PDGF-Rα and PDGF-Rβ) on arterial smooth muscle cells (ASMCs, open bars) and venous smooth muscle cells (VSMCs, solid bars). Cells were cultured in full medium, without additional growth factors. Receptors were quantified by flow cytometry. Each bar represents the means ± SD of four observations. The number of PDGF-Rα receptors was similar to that of PDGF-Rβ receptors on arterial SMCs. The number of PDGF-Rα receptors on venous SMCs was twofold lower (*P <0.05) than that on arterial SMCs, while PDGF-Rβ expression on venous SMCs was 6.6-fold greater (*P <0.05) than that on arterial SMCs.
ACTIVATION OF PDGF RECEPTORS
In arterial SMCs, PDGF-AA activated PDGF-Rα, as assessed by receptor phosphorylation, but not PDGF-Rβ, while PDGF-BB activated both PDGF-Rα and PDGF-Rβ (Fig. 3). In contrast, PDGF-AA failed to activate PDGF-Rα after 5 min exposure in venous SMCs. This is consistent with the unresponsiveness of venous SMC proliferation to PDGF-AA stimulation. The phosphorylation of PDGF-Rα in response to PDGF-BB stimulation was similar in arterial and venous SMCs (pixel density: 8,771 ± 2,178 vs. 7,888 ± 1,084; P >0.05). PDGF-Rβ activation by PDGF-BB, however, was significantly higher in venous SMCs than in arterial SMCs (pixel density: 46,328 ± 10,533 vs. 21,511 ± 2,751; P <0.05) (Fig. 3), in accordance with the higher PDGF-Rβ expression on venous SMCs (Fig. 2).
Fig. 3.

Differential phosphorylation of PDGF receptors (PDGF-R) and EGF receptors (EGF-R) in arterial smooth muscle cells (ASMCs) and venous SMCs (VSMCs) in response to two PDGF isoforms. Cells were incubated for 5 min with either PDGF-AA (50 μg/ml) or PDGF-BB (50 μg/ml), then lysed. The cell lysate was subsequently subjected to analysis in duplicate in a phospho-RTK immunoblot array containing 42 RTKs. A: Representative image of a phospho-RTK immunoblot array. The regions on the blot where various phospho-receptors were visualized are indicated by arrows and specific receptor labels (phosphorylated PDGF-Rα labeled as p-PDGF-Rα, phosphorylated PDGF-Rβ labeled as p-PDGF-Rβ, and phosphorylated EGF-R labeled as p-EGF-R). The unlabeled dots shown in the four corners of each sub-panels in panel A are internal controls. B: Means ± SD of chemiluminescent signals (quantified as image pixel density) from three separate phospho-RTK array experiments. The asterisk indicates a statistically significant (*P <0.05) difference between ASMC versus VSMC. Phospho-PDGF-Rα increased in ASMC, but not in VSMC, in response to PDGF-AA. PDGF-AA had no effect on phosphorylation of PDGF-Rα or PDGF-Rβ in VSMC. Upon PDGF-BB stimulation, the phosphorylation of PDGF-Rα increased to a similar degree in ASMC and VSMC. The increase in phosphorylation of PDGF-Rβ was high in ASMC, but was even greater in VSMCs (*P <0.05). PDGF-BB transactivated EGF-R in VSMC but not in ASMC.
TRANSACTIVATION OF EGF RECEPTOR IN VENOUS SMCS BY PDGF-BB
In addition to activating PDGF-Rs, PDGF-BB also caused activation of the epidermal growth factor receptor (EGF-R) on venous SMCs. In contrast, PDGF-BB did not activate EGF-R in arterial SMCs (Fig. 3). PDGF-AA, which binds only to PDGF-Rα, did not transactivate EGF-R in either cell type.
ACTIVATION OF MAPK AND AKT PATHWAYS BY PDGF
In addition to differences in PDGF-R expression levels and activation, differences in proliferative responses to PDGF might also be due to differences in signal transduction pathways between arterial and venous SMCs. Members of the mitogen-activated protein kinase (MAPK) family and Akt have been shown to be among the most critical signaling molecules for PDGF responses. The MAPK family includes extracellular signal-regulated kinases (ERK1/2), c-Jun NH2-terminal kinases (JNK1/2/3), and p38 kinase. We therefore compared the phosphorylation of ERK1/2, JNK1/2/3, p38 and Akt in response to PDGF-AA, PDGF-AB, or PDGF-BB between arterial and venous SMCs to determine if differences in downstream signaling pathways might account for differences in proliferation induced by the various PDGF isoforms.
After 5 min of stimulation, PDGF-BB induced similar MAPK (ERK1/2, JNK1/2/3, and p38) and Akt activation in arterial and venous SMCs (Fig. 4). Although no phosphorylation of PDGF-Rα was detected after venous SMC exposure to PDGF-AA (Fig. 3A), enhanced phosphorylation of ERK1/2 and Akt in venous SMCs was observed, which was similar to that seen in arterial SMCs (Fig. 4). PDGF-AA, which only binds PDGF-Rα, activated JNK1 in arterial SMCs, but not in venous SMCs (Fig. 4). PDGF-AB and PDGF-BB, which binds both PDGF-Rα and PDGF-Rβ, induced similar JNK1 phosphorylation in arterial and venous SMCs. JNK2/3 was not activated in either cell type.
Fig. 4.

Phosphorylation of proteins in the MAPK and Akt pathways in arterial smooth muscle cells (ASMC) and venous smooth muscle cells (VSMC) in response to various PDGF isoforms. Cells were incubated for 5 min with either PDGF-AA (“AA”, 50 μg/ml), AB (“AB”, 50 μg/ml), or PDGF-BB (“BB”, 50 μg/ml), then lysed. The cell lysate was subsequently subjected to SDS–PAGE and immunoblotting using the respective specific antibodies against the phosphorylated proteins. A: Representative blots from three separate experiments are shown. “C” indicates control or unstimulated quiescent cells. B: Means ± SD of chemiluminescent signals (quantified as image pixel density) from three separate experiments. PDGF-AA increased the level of phosphorylated JNK1 (p-JNK1) in ASMC, but not in VSMC. Other than JNK-1, there were no substantial differences in the levels of phosphorylated ERK1/2, p38, or Akt upon stimulation by PDGF-AA, PDGF-AB, or PDGF-BB between ASMC and VSMC. Phosphorylation of JNK 2/3 was not observed in either cell type after any PDGF isoform treatment.
We also studied the time course of phosphorylation of JNK1/2/3 in response to PDGF-AA or PDGF-BB in both cell types. After PDGF-AA stimulation, there was a transient activation of JNK1 at 5 min in arterial SMCs. In contrast, there was no JNK1/2/3 activation in venous SMCs for up to 60 min (Fig. 5). With PDGF-BB stimulation, phosphorylation of JNK1 was observed at 2 min and persisted for at least 15 min in arterial SMCs. The phosphorylation of JNK1 was also observed at 2 min in venous SMCs and persisted for up to 60 min (Fig. 5). JNK2/3 was not activated in either cell type at any time point examined.
Fig. 5.
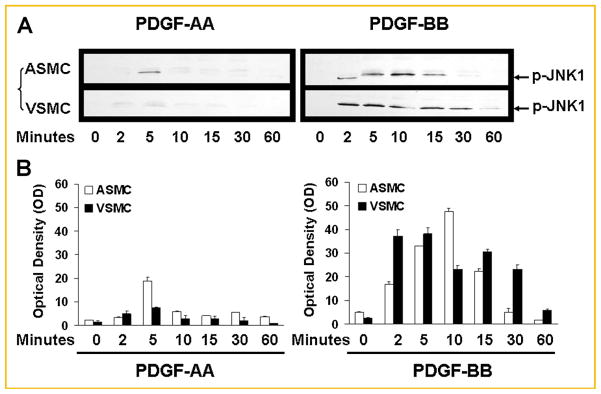
Time course of changes in phosphorylation of JNK1/2/3 in arterial smooth muscle cells (ASMC) and venous smooth muscle cells (VSMC) in response to two PDGF isoforms. Cells were incubated for the indicated durations with either PDGF-AA (“AA”, 50 μg/ml) or PDGF-BB (“BB”, 50 μg/ml), then lysed. The cell lysate was subsequently subjected to SDS–PAGE and immunoblotting using a specific antibody against phosphorylated JNK (p46/p-JNK1 and p54/p-JNK2/3). A: Representative blots from three separate experiments are shown. “C” indicates control or unstimulated quiescent cells. B: Means ± SD of chemiluminescent signals (quantified as image pixel density) from three separate experiments. PDGF-AA stimulated transient activation of JNK1 in ASMC, but not in VSMC. PDGF-BB stimulated JNK1 phosphorylation in both ASMC and VSMCs, although the activation of JNK1 in VSMC was sustained longer than that in ASMC. There was no JNK2/3 phosphorylation observed in either ASMC or VSMC at any time point.
REGULATION OF CYCLIN-DEPENDENT KINASE (CDK) INHIBITORS (p27KIP1/p21CIP1) BY PDGF
In addition to activation of MAPK pathways, the expression of downstream regulators of cell proliferation, p27kip1 and p21cip1, was studied. The protein level of p27kip1, a cyclin-dependent kinase (CDK) inhibitor, in the lysate of quiescent arterial SMCs was high, and was down-regulated at 48 h by the exposure of cells to PDGF-AA, PDGF-AB, or PDGF-BB (Fig. 6). In contrast, the level of p27kip1 was high in quiescent venous SMCs and was down-regulated at 48 h by PDGF-AB and PDGF-BB, but not by PDGF-AA (Fig. 6). These results are consistent with the observation that PDGF-AA effectively induced the proliferation of arterial SMCs, but not venous SMCs (Fig. 1). In contrast to p27kip1, the protein level of p21cip1 was similarly high in unstimulated control and arterial SMCs treated with any of the three PDGF isoforms. In venous SMCs, the expression of p21cip1 was low in quiescent cells. Exposure of venous SMCs to PDGF-AA did not change p21cip1 expression, but PDGF-AB and PDGF-BB both up-regulated p21cip1 expression (Fig. 6).
Fig. 6.
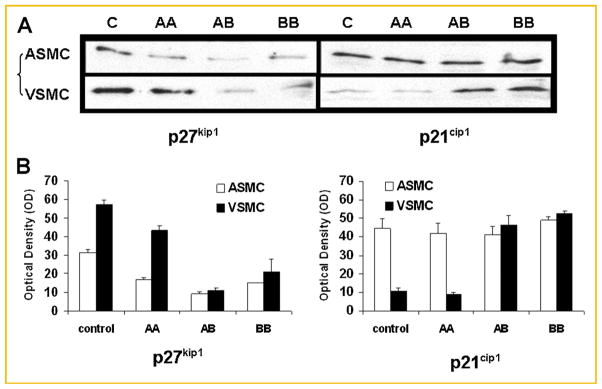
Protein expression of cell-cycle inhibitors, p27kip1 and p21cip1 in arterial smooth muscle cells (ASMC) and venous smooth muscle cells (VSMC) in response to various PDGF isoforms. Cells were incubated for 48 h with either PDGF-AA (“AA”, 50 μg/ml) or PDGF-BB (“BB”, 50 μg/ml), then lysed. The cell lysate was subsequently subjected to SDS–PAGE and immunoblotting using specific antibodies against either p27kip1 or p21cip1. A: Representative blots from two separate experiments are shown. “C” indicates control or unstimulated quiescent cells. B: Means ± SD of chemiluminescent signals (quantified as image pixel density) from two separate experiments. Protein levels of p27kip1 were high in quiescent ASMC and down-regulated by PDGF-AA, PDGF-AB, or PDGF-BB. Protein levels of p27kip1 were also high in quiescent VSMC and down-regulated by PDGF-AB or PDGF-BB, but not significantly affected by PDGF-AA. The level of p21cip1 expression was similar in the control and ASMC treated with various PDGF isoforms. In VSMC, the expression of p21cip1 was barely detectable in quiescent cells, which was not altered by PDGF-AA. However, p21cip1 expression was increased substantially by PDGF-AB and PDGF-BB in VSMC.
DISCUSSION
The clinical literature indicates that vein coronary bypass grafts are more prone to neointimal hyperplasia than artery grafts. Similarly, the vein-graft anastomosis is more prone to the development of hyperplasia than the artery-graft anastomosis in hemodialysis AV grafts. The development of rational therapies to prevent vascular graft failure requires a better understanding of the molecular basis for these differences between arteries and veins, which was the major goal of the studies described herein. Because of the known importance of PDGF in the pathogenesis of vascular hyperplasia and our previous observations that PDGF isoforms have differential effects on the proliferation in arterial and venous SMCs [Li et al., 2006a], it seemed logical to explore differences in the PDGF receptors and signal transduction pathways between the arterial and venous SMCs.
A major finding of the present study is the different receptor densities of PDGF-Rα and PDGF-Rβ between arterial SMCs and venous SMCs and how these differences might potentially explain the differences in proliferative responses by these cells. These results are consistent with and extend previous findings from other laboratories. Yang et al. [1998] reported high PDGF-Rβ mRNA expression in the internal mammary artery, saphenous vein and aorta; in contrast, PDGF-Rα mRNA expression was low in all these blood vessels. More recently, Weiss et al. [2007] demonstrated that PDGF-Rα protein expression was higher in SMCs from the internal mammary artery than SMCs from the saphenous vein, while PDGF-Rβ protein expression was lower in the arterial SMCs than venous SMC. Both studies reported lower PDGF-Rα expression in venous SMC compared to arterial SMC, but did not explore the PDGF-Rα signaling pathways with regard to differences in PDGF regulation of SMC proliferation. Using flow cytometry to measure PDGF-Rα and PDGF-Rβ in our study, we have extended these earlier works by showing differences in surface expression of PDGFR-α and PDGFR-β on primary cultured cells. We found that the density of PDGF-Rα and PDGF-Rβ were equivalent on the surface of arterial SMCs. On venous SMCs, the density of PDGF-Rα was approximately 39% of that on arterial SMCs, while the density of PDGF-Rβ was 6.6-fold higher than that on arterial SMCs (Fig. 2). Importantly, venous SMCs showed the higher PDGF-Rβ density (Fig. 2), which coincided with a higher level of PDGF-Rβ activation (Fig. 3) and a higher proliferation rate in response to PDGF-BB (Fig. 1), compared to arterial SMCs.
A second important observation of the present study is the ability of PDGF-BB to transactivate EGF-R, in addition to activating PDGF-Rα and PDGF-Rβ, in venous SMCs (Fig. 3). In contrast, no transactivation of EGF-R by PDGF-BB was observed in arterial SMCs. The transactivation of EGF-R appears to be specific for PDGF-BB, since PDGF-AA was devoid of this property in either arterial or venous SMCs. EGF-R transactivation has been demonstrated through other receptor tyrosine kinases, including PDGF-Rβ [Hackel et al., 1999]. PDGF-BB has been shown to stimulate the formation of PDGF-Rβ/EGF-R heterodimers in rat aortic SMCs and disruption of these heterodimers by antioxidants or a small molecule Src inhibitor decreased ERK1/2 activation by PDGF-BB [Saito et al., 2001]. These data suggest that PDGF-Rβ/EGF-R heterodimers represent a novel signaling complex which likely plays an important role in PDGF signal transduction in venous SMCs. In contrast, we did not observe the transactivation of EGF-R by PDGF-BB in human aortic SMCs (Fig. 3). The discrepancy between the previously reported study and the present study might be due to species differences (rat vs. human), or the different method used to detect phosphorylated EGF-R or other factors. To the best of our knowledge, the present study is the first to report a difference in EGF-R transactivation by PDGF between arterial and venous SMCs. These results are significant because this differential EGF-R transactivation might contribute, at least in part, to the greater proliferation induced by PDGF-BB in venous SMCs versus arterial SMCs. Moreover, several small-molecule inhibitors of EGF-R kinase activity are currently used to treat various forms of cancer and might have clinical application to prevent venous SMC hyperplasia in grafts.
A finding that has revealed important insight into PDGF signaling in vascular SMCs is that PDGF-AA failed to induce venous SMC proliferation, while significantly stimulating proliferation in arterial SMCs (Fig. 1). Interestingly, venous SMCs were found to express PDGF-Rα, although the expression was reduced approximately twofold compared to arterial SMCs (Fig. 2). More importantly, PDGF-AA stimulated phosphorylation of PDGF-Rα in arterial SMCs, but not in venous SMCs (Fig. 3). There are at least two potential explanations for the ineffectiveness of PDGF-AA to significantly activate PDGF-Rα in venous SMC. First, the relatively low number of PDGF-Rα on venous SMC may reflect the lower intrinsic autophosphorylation capacity. It is also conceivable that, in venous SMC, the majority of PDGF-Rα is present as heterodimers with PDGF-Rβ. Under these circumstances, PDGF-AA, which can activate only PDGF-Rα homodimers, would autophosphorylate merely a small fraction of PDGF-Rα. Second, the activation of PDGF-Rα is rapid, yet weak, after PDGF-AA stimulation. Although we observed phosphorylation of PDGF-Rα in venous SMC at 2 min after PDGF-AA stimulation (data not shown), the level was barely detectable. Thus, these differences in sensitivity towards PDGF-AA might contribute to the higher propensity of the arterial wall to undergo cellular proliferation, a crucial event in the pathogenesis of atherosclerosis. This notion is compatible with previous studies which suggested that the PDGF A-chain contributes to SMC proliferation and the phenotype in arterial proliferative disease. For example, Nilsson et al. [1985] showed that normal, growth-arrested arterial SMC did not express PDGF A-chain mRNA, whereas cultured arterial SMC and SMC in atherosclerotic plaques express PDGF A-chain mRNA and secrete PDGF-AA protein. Kruppel-like zinc-finger transcription factor 5 (KLF5) has been established as a transcription factor that alters when arterial SMCs transform from the contractile to the synthetic phenotype; as a result of stimulation by angiotensin II, the PDGF A-chain is expressed [Shindo et al., 2002]. PDGF-A mRNA was absent in non-transplanted hearts, but a significant correlation was found between PDGF-A mRNA up-regulation and the degree of arterial intimal thickening that occurred in the rat heterotopic cardiac allograft [Hachida et al., 1998]. More recently, Li et al. [2006b] reported that stent-based delivery of an antisense oligodeoxynucleotide targeting the PDGF-A chain effectively inhibited neointimal formation after stent implantation in the porcine coronary artery. Blockade of PDGF-Rα reduced intimal SMC accumulation in rat models of transplant arteriosclerosis [Hachida et al., 1999]. Collectively, these studies strongly suggest that PDGF-AA and PDGF-Rα play an important role in arterial injury disorders and thus represent potential targets for therapies to prevent arterial SMC hyperplasia. In contrast, our results in venous SMCs suggest that therapies targeting PDGF-AA and the PDGF-Rα are unlikely to be useful in treating SMC hyperplasia in veins.
PDGF-AA induced the phosphorylation of ERK1/2 and Akt, similar to that seen in arterial SMCs (Fig. 4). The similar activation of these important mitogenic pathways in both cell types was unexpected, since PDGF-AA failed to significantly activate PDGF-Rα or induce proliferation in venous SMCs. Therefore, lysates from venous SMCs exposed to PDGF-AA were screened using the phosphotyrosine kinase array. The results of these experiments showed no enhancements in phosphorylation of any of the 42 receptor tyrosine kinases (Fig. 3). Thus, it is unclear through which receptor the ERK1/2 and Akt downstream signaling pathways were activated in response to PDGF-AA in venous SMCs. Interestingly, exposure of venous SMCs to PDGF-BB, which stimulated robust proliferation in these cells (Fig. 1), caused essentially the same degree of activation of ERK1/2 and Akt as did PDGF-AA (Fig. 4). Thus, it appears that in venous SMCs, activation of the ERK1/2 and Akt pathways is not sufficient to stimulate a proliferative response. These data are consistent with other reports in different cell types. Sachinidis et al. [1993] reported that PDGF-AB and PDGF-BB stimulated a marked DNA synthesis whereas PDGF-AA induced only a slight increase of DNA synthesis in rat aortic SMC. PDGF-AB and PDGF-BB caused a strong stimulation of PDGF receptors autophosphorylation, whereas the effect of PDGF-AA was undetectable. Nonetheless, all three PDGF isoforms induced equivalent amounts of the early growth response genes, such as c-fos, egr-1, and c-myc mRNA. The authors concluded that the induction of early response genes in rat SMC by PDGF-AA is not sufficient to stimulate DNA synthesis. It was further speculated that little to no receptor phosphorylation is required to link receptor activation to the induction of c-fos, egr-1, and c-myc. Choudhury et al. [2000] demonstrated that all three PDGF isoforms (PDGF-AA, PDGF-AB, and PDGF-BB) stimulated PLCγ1 and PI 3 kinase in glomerular mesangial cells. However, PDGF-AA did not induce DNA synthesis or migration of mesangial cell even with a subsequent increase in PLC and PI 3 kinase activation. These previous reports, along with the data presented herein, suggest that transient, low degree of PDGFR-α phosphorylation in venous SMC can induce marked phosphorylation of ERK1/2 and Akt, but this level of activation is not sufficient to elicit cell proliferation.
One potentially important difference between PDGF-AA stimulation of venous and arterial SMCs that might account for the different proliferative responses is activation of the JNK1 pathway. Unlike arterial cells, PDGF-AA failed to significantly stimulate the phosphorylation of JNK1 in venous SMCs (Fig. 4). Thus, activation of JNK1, either alone or in combination with the ERK1/2 and Akt pathways, may be required to elicit a proliferative response to PDGF-AA. While the JNK pathway has been shown to play an important role in apoptosis, its association with other cellular process and pathologic conditions has only recently been recognized. Celada and co-workers demonstrated that JNK1, but not JNK2, played a predominant role in the induction of pro-inflammatory mediators in response to lipopolysaccharide and tumor necrosis factor-α in macrophages [Sanchez-Tillo et al., 2007]. Work by Yu et al. [2000] revealed that, in NIH3T3 cells, JNK1 was activated downstream of PDGFR-α but not PDGFR-β and that inhibition of JNK1 activity using a dominant-negative JNK1 mutant markedly enhanced PDGF-BB-mediated cell growth. The same group further showed that the PDGFR-α/JNK1 pathway is critical for PDGF-mediated apoptosis as well as PDGF-induced expression of p21 in NIH3T3 cells [Yu et al., 2003]. Both studies suggest that PDGFR-α may have an antagonistic function for PDGFR-β signaling. To the best of our knowledge, the present study was the first one that investigated the role of PDGFR-α/JNK1 pathways in venous SMC. This observation is important, since the loss of PDGFR-α-induced negative signaling in venous SMC may contribute to the greater proliferation induced by PDGF-BB in venous SMCs versus arterial SMCs. This hypothesis needs further confirmation.
Regulation of the cell cycle is also a crucial component of cellular responses to mitogens. Progress through the cell cycle, and subsequently cell proliferation, is inhibited by the cyclin-dependent kinase (CDK)-2 inhibitor, p27kip1. PDGF-AA down-regulated the protein expression of p27kip1 in arterial SMCs (Fig. 6). In contrast, the protein level of p27kip1 was unchanged in the presence of PDGF-AA in venous SMCs, compared with unstimulated cells. The lack of an effect of PDGF-AA on p27kip1 correlated with the inability of PDGF-AA to stimulate venous SMC proliferation (Fig. 1), suggesting that p27kip1 plays a dominant role relative to ERK1/2 and Akt pathways in regulating venous SMC proliferation. The protein levels of another CDK inhibitor, p21cip1, remained unchanged in arterial SMCs after exposure to any PDGF isoform. In contrast, p21cip1 levels markedly increased in venous SMC in response to PDGF-AB and PDGF-BB. This paradoxical effect is likely due to the more complex role of p21cip1 in cell cycle progression compared to p27kip1. Increased levels of p21cip1 have been associated with increased, as well as decreased, DNA synthesis [Okamoto et al., 2004]. Since an increase in venous SMC proliferation occurred despite a significant increase in p21cip1 levels in response to PDGF-AB or PDGF-BB (Fig. 6), it is reasonable to postulate that the cell cycle inhibitory activity of p21cip1 was insufficient to overcome the pro-proliferative activities of other cell-cycle regulatory proteins. An important concluding observation from these findings is that the signaling pathways regulating proliferation in venous and arterial SMC respond very differently to the same mitogens, and that this probably contributes to the different hyperplastic responses seen in different vascular beds exposed to similar mitogenic stimuli.
In summary, venous SMCs are inherently more proliferative in response to PDGF-BB than arterial SMC, and this difference is likely due to the greater PDGF-Rβ expression and transactivation of EGF-R by PDGF-BB in venous SMC. On the other hand, arterial SMCs, but not venous SMCs, responded to the pro-proliferative effects of PDGF-AA, presumably via activation of the PDGF-Rα/JNK1/p27kip1 pathway. To the best of our knowledge, this is the first detailed comparison of PDGF-Rα signaling pathways in response to PDGF-AA stimulation between arterial and venous SMCs. These findings provide a plausible molecular basis for the varying susceptibility between arteries and veins to different proliferative vascular diseases. Understanding these differences in molecular mechanisms would facilitate the development of specific pharmacologic strategies that may selectively inhibit arterial or venous SMC proliferation.
Acknowledgments
Grant sponsor: National Heart, Lung and Blood Institute; Grant number: R01-HL67646; Grant sponsor: Medical and Research Services of the Department of Veterans Affairs, Dialysis Research Foundation; Grant sponsor: National Kidney Foundation of Utah and Idaho.
This work was supported by the National Heart, Lung and Blood Institute (R01-HL67646), Medical and Research Services of the Department of Veterans Affairs, Dialysis Research Foundation and the National Kidney Foundation of Utah and Idaho.
References
- Bergsten E, Uutela M, Li X, Pietras K, Ostman A, Heldin CH, Alitalo K, Eriksson U. PDGF-D is a specific, protease-activated ligand for the PDGF beta-receptor. Nat Cell Biol. 2001;3:512–516. doi: 10.1038/35074588. [DOI] [PubMed] [Google Scholar]
- Choudhury GG, Grandaliano G, Jin DC, Katz MS, Abboud HE. Activation of PLC and PI 3 kinase by PDGF receptor alpha is not sufficient for mitogenesis and migration in mesangial cells. Kidney Int. 2000;57:908–917. doi: 10.1046/j.1523-1755.2000.00907.x. [DOI] [PubMed] [Google Scholar]
- Hachida M, Zhang X, Lu H, Hoshi H, Furutani Y, Matsuoka R, Koyanagi H. Association between the degree of platelet-derived growth factor-A chain mRNA expression and coronary arteriosclerosis in the transplanted heart. Heart Vessels. 1998;13:24–29. doi: 10.1007/BF02750640. [DOI] [PubMed] [Google Scholar]
- Hachida M, Zhang X, Lu H, Hoshi H, Koyanagi H. Effects of immunosuppressants on platelet-derived growth factor-A chain mRNA expression and coronary arteriosclerosis in rat cardiac allografts. Jpn Circ J. 1999;63:303–308. doi: 10.1253/jcj.63.303. [DOI] [PubMed] [Google Scholar]
- Hackel PO, Zwick E, Prenzel N, Ullrich A. Epidermal growth factor receptors: Critical mediators of multiple receptor pathways. Curr Opin Cell Biol. 1999;11:184–189. doi: 10.1016/s0955-0674(99)80024-6. [DOI] [PubMed] [Google Scholar]
- Hart CE, Bailey M, Curtis DA, Osborn S, Raines E, Ross R, Forstrom JW. Purification of PDGF-AB and PDGF-BB from human platelet extracts and identification of all three PDGF dimers in human platelets. Biochemistry. 1990;29:166–172. doi: 10.1021/bi00453a022. [DOI] [PubMed] [Google Scholar]
- Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79:1283–1316. doi: 10.1152/physrev.1999.79.4.1283. [DOI] [PubMed] [Google Scholar]
- Heldin CH, Ostman A, Ronnstrand L. Signal transduction via platelet-derived growth factor receptors. Biochim Biophys Acta. 1998;1378:F79–F113. doi: 10.1016/s0304-419x(98)00015-8. [DOI] [PubMed] [Google Scholar]
- Karvinen H, Rutanen J, Leppanen O, Lach R, Levonen AL, Eriksson U, Yla-Herttuala S. PDGF-C and -D and their receptors PDGFR-alpha and PDGFR-beta in atherosclerotic human arteries. Eur J Clin Invest. 2009;39:320–327. doi: 10.1111/j.1365-2362.2009.02095.x. [DOI] [PubMed] [Google Scholar]
- LaRochelle WJ, Jeffers M, McDonald WF, Chillakuru RA, Giese NA, Lokker NA, Sullivan C, Boldog FL, Yang M, Vernet C, Burgess CE, Fernandes E, Deegler LL, Rittman B, Shimkets J, Shimkets RA, Rothberg JM, Lichenstein HS. PDGF-D, a new protease-activated growth factor. Nat Cell Biol. 2001;3:517–521. doi: 10.1038/35074593. [DOI] [PubMed] [Google Scholar]
- Li X, Ponten A, Aase K, Karlsson L, Abramsson A, Uutela M, Backstrom G, Hellstrom M, Bostrom H, Li H, Soriano P, Betsholtz C, Heldin CH, Alitalo K, Ostman A, Eriksson U. PDGF-C is a new protease-activated ligand for the PDGF alpha-receptor. Nat Cell Biol. 2000;2:302–309. doi: 10.1038/35010579. [DOI] [PubMed] [Google Scholar]
- Li L, Blumenthal DK, Masaki T, Terry CM, Cheung AK. Differential effects of imatinib on PDGF-induced proliferation and PDGF receptor signaling in human arterial and venous smooth muscle cells. J Cell Biochem. 2006a;99:1553–1563. doi: 10.1002/jcb.20993. [DOI] [PubMed] [Google Scholar]
- Li Y, Fukuda N, Kunimoto S, Yokoyama S, Hagikura K, Kawano T, Takayama T, Honye J, Kobayashi N, Mugishima H, Saito S, Serie K. Stent-based delivery of antisense oligodeoxynucleotides targeted to the PDGF A-chain decreases instent restenosis of the coronary artery. J Cardiovasc Pharmacol. 2006b;48:184–190. doi: 10.1097/01.fjc.0000246940.91191.1f. [DOI] [PubMed] [Google Scholar]
- Loop FD, Lytle BW, Cosgrove DM, Stewart RW, Goormastic M, Williams GW, Golding LA, Gill CC, Taylor PC, Sheldon WC, Proudfit WL. Influence of the internal-mammary-artery graft on 10-year survival and other cardiac events. N Engl J Med. 1986;314:1–6. doi: 10.1056/NEJM198601023140101. [DOI] [PubMed] [Google Scholar]
- Motwani JG, Topol EJ. Aortocoronary saphenous vein graft disease: Pathogenesis, predisposition, and prevention. Circulation. 1998;97:916–931. doi: 10.1161/01.cir.97.9.916. [DOI] [PubMed] [Google Scholar]
- Nilsson J, Sjolund M, Palmberg L, Thyberg J, Heldin CH. Arterial smooth muscle cells in primary culture produce a platelet-derived growth factor-like protein. Proc Natl Acad Sci USA. 1985;82:4418–4422. doi: 10.1073/pnas.82.13.4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Kato S, Arima N, Fujii T, Morimatsu M, Imaizumi T. Cyclin-dependent kinase inhibitor, p21Waf1, regulates vascular smooth muscle cell hypertrophy. Hypertens Res. 2004;27:283–291. doi: 10.1291/hypres.27.283. [DOI] [PubMed] [Google Scholar]
- Raines EW. PDGF and cardiovascular disease. Cytokine Growth Factor Rev. 2004;15:237–254. doi: 10.1016/j.cytogfr.2004.03.004. [DOI] [PubMed] [Google Scholar]
- Reigstad LJ, Varhaug JE, Lillehaug JR. Structural and functional specificities of PDGF-C and PDGF-D, the novel members of the platelet-derived growth factors family. FEBS J. 2005;272:5723–5741. doi: 10.1111/j.1742-4658.2005.04989.x. [DOI] [PubMed] [Google Scholar]
- Sachinidis A, Schulte K, Ko Y, Meyer zu Brickwedde MK, Hoppe V, Hoppe J, Vetter H. The induction of early response genes in rat smooth muscle cells by PDGF-AA is not sufficient to stimulate DNA-synthesis. FEBS Lett. 1993;319:221–224. doi: 10.1016/0014-5793(93)80550-e. [DOI] [PubMed] [Google Scholar]
- Saito Y, Haendeler J, Hojo Y, Yamamoto K, Berk BC. Receptor heterodimerization: Essential mechanism for platelet-derived growth factor-induced epidermal growth factor receptor transactivation. Mol Cell Biol. 2001;21:6387–6394. doi: 10.1128/MCB.21.19.6387-6394.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Tillo E, Comalada M, Xaus J, Farrera C, Valledor AF, Caelles C, Lloberas J, Celada A. JNK1 is required for the induction of Mkp1 expression in macrophages during proliferation and lipopolysaccharide-dependent activation. J Biol Chem. 2007;282:12566–12573. doi: 10.1074/jbc.M609662200. [DOI] [PubMed] [Google Scholar]
- Shindo T, Manabe I, Fukushima Y, Tobe K, Aizawa K, Miyamoto S, Kawai-Kowase K, Moriyama N, Imai Y, Kawakami H, Nishimatsu H, Ishikawa T, Suzuki T, Morita H, Maemura K, Sata M, Hirata Y, Komukai M, Kagechika H, Kadowaki T, Kurabayashi M, Nagai R. Kruppel-like zinc-finger transcription factor KLF5/BTEB2 is a target for angiotensin II signaling and an essential regulator of cardiovascular remodeling. Nat Med. 2002;8:856–863. doi: 10.1038/nm738. [DOI] [PubMed] [Google Scholar]
- US Renal Data System. 2007 USRDS annual report. Minneapolois: United States Renal Data System; 2007. [Google Scholar]
- Wagsater D, Zhu C, Bjorck HM, Eriksson P. Effects of PDGF-C and PDGF-D on monocyte migration and MMP-2 and MMP-9 expression. Atherosclerosis. 2009;202:415–423. doi: 10.1016/j.atherosclerosis.2008.04.050. [DOI] [PubMed] [Google Scholar]
- Weiss S, Frischknecht K, Greutert H, Payeli S, Steffel J, Luscher TF, Carrel TP, Tanner FC. Different migration of vascular smooth muscle cells from human coronary artery bypass vessels. Role of Rho/ROCK pathway. J Vasc Res. 2007;44:149–156. doi: 10.1159/000099141. [DOI] [PubMed] [Google Scholar]
- Yang Z, Oemar BS, Carrel T, Kipfer B, Julmy F, Luscher TF. Different proliferative properties of smooth muscle cells of human arterial and venous bypass vessels: Role of PDGF receptors, mitogen-activated protein kinase, and cyclin-dependent kinase inhibitors. Circulation. 1998;97:181–187. doi: 10.1161/01.cir.97.2.181. [DOI] [PubMed] [Google Scholar]
- Yu J, Deuel TF, Kim HR. Platelet-derived growth factor (PDGF) receptor-alpha activates c-Jun NH2-terminal kinase-1 and antagonizes PDGF receptor-beta-induced phenotypic transformation. J Biol Chem. 2000;275:19076–19082. doi: 10.1074/jbc.M910329199. [DOI] [PubMed] [Google Scholar]
- Yu J, Liu XW, Kim HR. Platelet-derived growth factor (PDGF) receptor-alpha-activated c-Jun NH2-terminal kinase-1 is critical for PDGF-induced p21WAF1/CIP1 promoter activity independent of p53. J Biol Chem. 2003;278:49582–49588. doi: 10.1074/jbc.M309986200. [DOI] [PubMed] [Google Scholar]