

Abstract
Foxp3+ regulatory T (Treg) cells play a critical role in immune homeostasis; however, the mechanisms to maintain their function remain unclear. Here, we report that the E3 ubiquitin ligase VHL is essential for Treg cell function. Mice with Foxp3-restricted VHL deletion displayed massive inflammation associated with excessive Treg cell interferon-γ (IFN-γ) production. VHL-deficient Treg cells failed to prevent colitis induction, but converted into Th1-like effector T cells. VHL intrinsically orchestrated such conversion under both steady and inflammatory conditions followed by Foxp3 downregulation, which was reversed by IFN-γ deficiency. Augmented hypoxia-inducible factor 1α (HIF-1α)-induced glycolytic reprogramming was required for IFN-γ production. Furthermore, HIF-1α bound directly to the Ifng promoter. HIF-1α knockdown or knockout could reverse the increased IFN-γ by VHL-deficient Treg cells and restore their suppressive function in vivo. These findings indicate that regulation of HIF-1α pathway by VHL is crucial to maintain the stability and suppressive function of Foxp3+ T cells.
Introduction
Regulatory T (Treg) cells are a unique subpopulation of CD4+ T cells that play a pivotal role in maintaining immune tolerance and preventing autoimmunity against self-antigens. The best-characterized population of Treg cells is manifested by the cell surface expression of CD25, the interlukin-2 (IL-2) receptor alpha chain (Sakaguchi, 2000). Treg cells can be divided into two types: the thymus-derived naturally occurring (tTreg) and the peripherally inducible Treg (pTreg) cells. The development and function of tTreg cells is determined by the transcription factor Foxp3 (Fontenot et al., 2003; Hori et al., 2003). Its mutation or deficiency is linked to systemic autoimmune diseases in both mice and humans (Bennett et al., 2001; Brunkow et al., 2001; Khattri et al., 2003; Wildin et al., 2001). Recently studies have documented that Treg cells can acquire specific transcriptional factors known to be essential for the differentiation and function of T helper (Th) cells and suppress different types of Th cell-mediated immune responses. For example, Treg cell lineage-specific suppression of Th1, Th2 and Th17 cells was demonstrated through specific transcription factors expressed in Treg cells including T-bet, IRF4 and STAT3, respectively (Chaudhry et al., 2009; Koch et al., 2009; Zheng et al., 2009). However, the molecular mechanisms underlying the maintenance of the Foxp3 expression and Treg cell plasticity remain largely unclear.
Inactivation or mutation of von Hippel–Lindau (VHL) gene in humans predisposes to the development of different tumors including those in kidney, retina, central nervous system, and the adrenal gland (Kaelin, 2008). It encodes two forms of 18 and 30 kDa and constitutes the essential component of the VHL E3 ubiquitin ligase complex with elongin B/C, cullin 2, and Ring box protein 1 (Rbx1) (Kamura et al., 1999; Stebbins et al., 1999). The most well documented substrate of the VHL complex is hypoxia-inducible factor 1α (HIF-1α), an oxygen sensor and transcription factor that controls the expression of various genes responsible for angiogenesis and glucose metabolism under low oxygen level (Semenza, 2007). Under normoxic conditions, HIF-1α is kept at low level, via the hydroxylation by prolyl hydroxylase domain (PHD) enzymes, the recognition and ubiquitination by VHL, followed by the degradation by the proteasome. Hypoxia reduces the activity of PHD enzymes, which leads to the accumulation of HIF-1α and the initiation of HIF-1α-dependent transcriptional program. Earlier studies documented that upregulation of HIF-1α is linked to the innate immunity via the NF-κB pathway (Rius et al., 2008), and is essential for myeloid cell-mediated inflammation (Cramer et al., 2003). Interesting, two recent studies have demonstrated that HIF-1α plays a critical role in the Th17/Treg cell balance (Dang et al., 2011; Shi et al., 2011). However, studies from other groups showed that hypoxia/HIF-1α pathway positively regulates Foxp3 induction (Ben-Shoshan et al., 2008; Clambey et al., 2012). One critical question remains whether the E3 ligase component VHL is involved in the regulation of Treg cells. To address this issue, we generated Vhlfl/fl Foxp3cre mice to selectively delete VHL expression in Treg cells and demonstrated that VHL plays an important role in maintaining Treg cell stability and function via modulating the HIF-1α pathway.
Results
VHL deficiency in Treg cells leads to Th1 cell-dominant autoimmunity
To examine the role of VHL in regulatory Foxp3+ T cells, we generated conditional VHL deletion by crossing Vhlfl/fl and Foxp3cre mice. The conditional knockout mice showed sign of illness, were not able to gain weight (Figures 1A), and eventually died at 6 to 11 weeks of age (Figure 1B). These mice showed increased infiltrates in the lung and liver (Figure 1C). A comprehensive biochemical analysis using mouse sera showed higher levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) and reduced glucose and creatinine concentration in Vhlfl/fl Foxp3cre mice (Figure S1A). Even though the numbers of T cells were not altered in conditional VHL knockout mice (Figure S1B), we found that activated/memory type-associated CD44highCD62Llow conventional CD4+ and CD8+ T cell population was notably increased in Vhlfl/fl Foxp3cre mice (Figures 1D and S1C). B cells also expressed higher level of CD69 (data not shown). Moreover, among effector T cells, Th1 cells were greatly induced in Vhlfl/fl Foxp3cre mice, but not Th17 cells (Figure 1E). Foxp3+ cells were slightly reduced in lymph nodes, but not in the spleen (Figures 1F and S1D). We further examined the distribution of Foxp3+, Th1 and Th17 cells from several organs and found that Foxp3 expression was reduced in lymph nodes, Peyer’s patches, small and large intestines, but not in the liver, lung and spleen (Figure 1G). Interestingly, Th1 cells from Vhlfl/fl Foxp3cre mice were increased in almost all organs regardless of the levels of Foxp3 expression, but Th17 cells were not altered, or only slightly changed. These results implied that VHL-deficient Foxp3+ cells are defective in their functions, thereby inducing the activation of conventional T cells, particularly Th1 cells, and subsequent inflammation.
Figure 1. Vhlfl/fl Foxp3cre mice develop autoimmunity.

(A) Growth curve of Vhlfl/fl Foxp3cre mice. Body weight was measured.
(B) Survival curve of Vhlfl/fl Foxp3cre mice is shown. **P<0.01 (Gehan-Breslow-Wilcoxon test).
(C) H&E stained liver and lung sections.
(D) Expression of CD44 and CD62L in CD4+ T cells from the spleen and lymph nodes. A representative dot plot (left panel) and combined plot (right panel) are shown.
(E) IFN-γ and IL-17 expression from CD4+ T cells. Cells were stimulated with PMA and ionomycin for 4 hrs before intracellular staining.
(F) Foxp3 expression from CD4+ T cells was assessed by flow cytometry. (G) Distribution of Foxp3, Th1 (IFN-γ+ IL-17−) and Th17 (IL-17+ IFN-γ−) cells from several organs was shown. Representative of at least three independent experiments. Combined plots for A (n=8~11), B (n=9), D (n=11~13), and G (n=4~8) are shown. Error bars indicate SEM. *P<0.05, **P<0.01, ***P<0.001 (Student’s t-test). See also Figure S1.
VHL-deficient Treg cells showed impaired suppressive function in vivo
To investigate the effect of VHL deficiency on the biological function of Treg cells, we performed in vitro suppression assay. We prepared VHL-deficient Treg cell by sorting YFP+ cells and then confirmed the expression level of Foxp3 to an equivalent degree by intracellular staining (Figure S2A). VHL-deficient Foxp3+ cells were comparable to WT Foxp3+ cells in the suppression of proliferation of naïve T cells in vitro (Figure S2B). We further examined the suppressive activity in vivo by utilizing an adoptive transfer mouse model of colitis. VHL-deficient Treg cells failed to prevent the loss of body weight caused by naïve T cells in Rag1−/− mice (Figure 2A). Histologic examination of the distal colon indicated that lymphocyte infiltrates were increased and showed mucosa hyperplasia in mice adoptively transferred with VHL-deficient Treg cells (Figure 2B). VHL-deficient Treg cells were unable to inhibit the induction of T cells, particularly Th1 cells (Figures 2C and S2C–D). Whereas the frequency of Th1 cells was not changed, that of Th17 cells was increased from WT Treg cell-treated Rag1−/− mice (Figure S2F). While WT Treg cells still maintained the expression of Foxp3 and CD103, a marker for suppressive activity and migratory capacity to mucosal area, VHL-deficient Treg cells lost most Foxp3 expression at 8 weeks after adoptive transfer (Figures 2D and S2E). Our results demonstrated that VHL-deficient Treg cells have defects in suppressive activity in vivo with less stability of Foxp3 and a failure in the suppression of Th1 induction.
Figure 2. VHL deficiency leads to the loss of function by Treg cells.

(A) Clinical colitis progression was assessed by body weight loss. CD45.1+ naive T cells alone or together with sorted WT or VHL-deficient CD45.2+ Treg cells were adoptively transferred into Rag1−/− mice.
(B) Representative images of distal colon after H&E staining (left panel) and histological analysis (right panel).
(C) The absolute number of IFN-γ+IL-17−, IFN-γ+IL-17+ or IFN-γ−IL-17+ cells differentiated from transferred naïve T cells (CD45.1+). CD4+ T cells from the spleen, mesenteric lymph nodes (MLN) and large (L) intestines were examined.
(D) Expression level of Foxp3 and CD103 from transferred Treg cells (CD45.2+) was analyzed by intracellular staining. Representative of two independent experiments. Combined plots for A (n=4~5), B (n=4~5), C (n=4~5) and D (n=4~5) are shown. Error bars indicate SEM. *P<0.05, **P<0.01, ***P<0.001 (Student’s t-test). See also Figure S2.
Conversion into IFN-γ-producing Th1 cells by VHL ablation in Treg cells
To gain the insight into the impaired suppressive activity in vivo in Vhl−/− Treg cells, we examined the expression of key Treg cell markers including CD25, CTLA4, CD39, CD73, CD44, CD69 and GITR. Expression levels of those markers by VHL-deficient Treg cells were comparable to those from WT Treg cells (Figure S3A). Rather, the expression of CTLA4, GITR and CD39 were slightly increased in VHL-deficient Treg cells. We next examined the expression of helios and Nrp-1 in VHL-deficient Treg cells to distinguish different Treg cell subpopulations. We found that both helios+ and helios− Foxp3+ cells were reduced in Vhlfl/fl Foxp3cre mice. Similarly, Nrp-1+ and Nrp-1−Foxp3+ cells were decreased by VHL deficiency (Figure S3B). Interestingly, Foxp3−helios+ or Foxp3−Nrp+ T cells were increased in Vhlfl/fl Foxp3cre mice.
It is possible that the impaired suppression is caused by either defective proliferation or enhanced apoptosis of VHL-deficient Treg cells. Ki-67 expression was slightly increased in VHL-deficient Foxp3+ cells (Figure S3C). However, Foxp3− cells expressed higher level of Ki-67 from Vhlfl/fl Foxp3cre mice. This could be due to impaired Treg cell functions, which augmented non-Treg cells proliferation. Next, the homeostatic proliferation in vivo was assessed by an adoptive transfer of Violet-labeled Treg cells into Rag1−/− mice. VHL-deficient Treg cells did not show any defect in their proliferation in vivo (Figure S3D). However, they lost Foxp3 expression after cell division. To determine the apoptosis of Foxp3+ cells, Treg cells were stained with annexin V. Apoptotic VHL-deficient Treg cells are comparable to WT Treg cells (data not shown). In addition, to examine apoptosis of Treg cell in vitro, isolated Foxp3+ (YFP+) cells were activated and stained with annexin V and propidium iodide (PI). Annexin V+ apoptotic cells were rather decreased in VHL-deficient Foxp3+ cells (data not shown). Therefore, it is unlikely that the impaired functions of VHL-deficient Treg cells were caused by altered cell proliferation or enhanced cell death.
To explore whether the loss of function is due to the plasticity of VHL-deficient Foxp3+ cells, we screened the production of several inflammatory cytokines and chemokines after activation in vitro. Surprisingly, VHL-deficient Foxp3 cells were able to produce higher levels of cytokines and chemokines such as IFN-γ, IL-10, GM-CSF, IL-4, MIP-1α, MIP-1β, RANTES and TNFα (Figure 3A). VHL-deficient Foxp3+ cells were also able to produce large amounts of IFN-γ, but not IL-17 in vivo (Figures 3B and S3E). Similar to Th1 cells, VHL-deficient Foxp3+ cells displayed T-bet and CXCR3 expression (Figure S3F and S3G). We also observed that transferred VHL-deficient Foxp3+ cells in the colitis mice efficiently produced IFN-γ (Figures 3C and S2G). These results demonstrated that Treg cells had a tendency to become IFN-γ-producing effector T cells in vitro and in vivo in the absence of VHL.
Figure 3. Conversion of VHL-deficient Treg cells into IFN-γ-producing effectors.

(A) Cytokine and chemokine production from activated VHL-deficient Foxp3+ cells. Sorted Treg cells were activated by anti-CD3/28 antibodies for 48 hrs. The culture supernatants were harvested and analyzed by Bio-plex assay.
(B) IFN-γ and IL-17 expression in Foxp3+ T cells from Vhlfl/fl Foxp3cre mice. Cells were stimulated with PMA and ionomycin for 4 hrs before intracellular staining.
(C) The conversion of transferred Treg cells (CD45.2+) into IFN-γ or IL-17 expressing cells was determined from colitis mice. CD4+CD45.2+ Cells were analyzed by intracellular staining at 7 weeks after adoptive transfer. Combined plots for B (n=8~9) and C (n=4~5) are shown. Error bars indicate SEM. *P<0.05, **P<0.01 (Student’s t-test). Representative of two or three independent experiments are shown. See also Figure S3.
An intrinsic role of VHL in the conversion into IFN-γ-producing effector cells
To investigate whether the effect of VHL is Treg cell intrinsic, we generated mixed bone marrow (BM) chimeric mice by adoptively co-transferring WT (CD45.1) BM as well as BM (CD45.2) from Vhlfl/fl Foxp3cre mice into irradiated Rag1−/− mice. Unlike Vhlfl/fl Foxp3cre mice, BM mixed chimera mice did not show the activated CD44highCD62Llow phenotype by either CD45.2 or CD45.1 T cells (Figure 4A). However, the frequency of VHL-deficient Foxp3+ cells was much lower than the WT counterpart (Figure 4B). Nevertheless, VHL-deficient Foxp3+ cells were still able to express IFN-γ (Figure 4C). These data suggest that WT Foxp3+ cells could restore the impaired suppression by VHL-deficient Foxp3+ cells. Moreover, VHL-deficient Treg cells were still capable of producing IFN-γ even in the presence of functional WT Treg cells.
Figure 4. VHL plays an intrinsic role in Treg cell conversion.
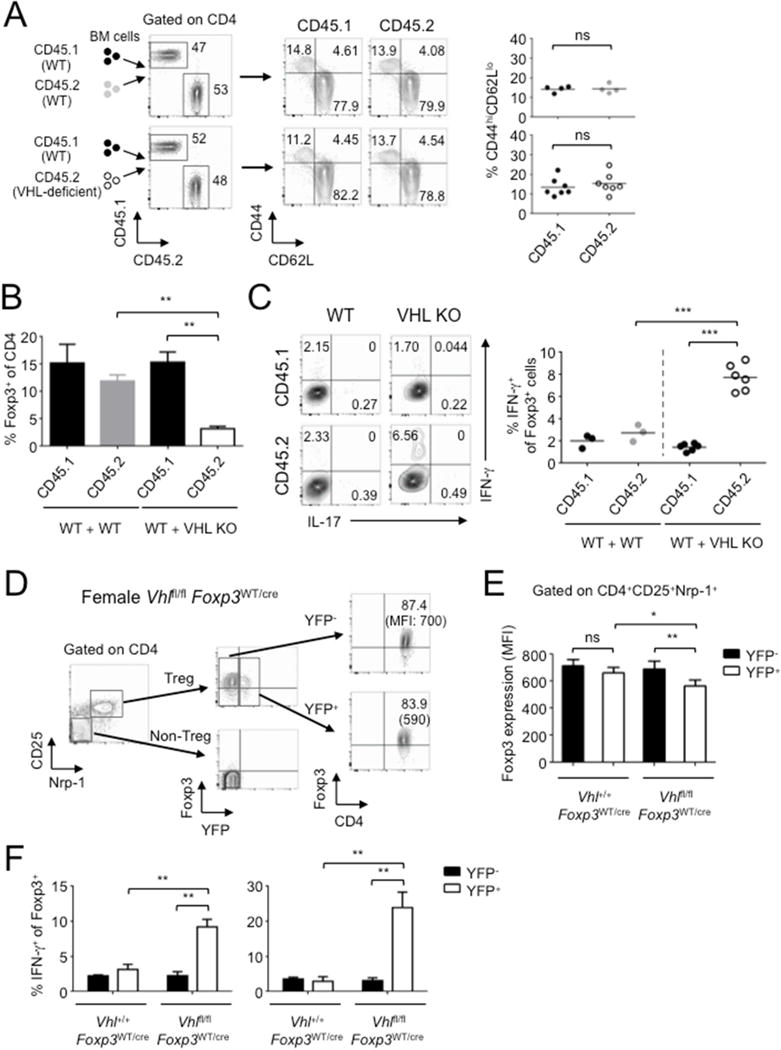
(A) Bone marrow (BM) cells from WT (CD45.1) and either WT (CD45.2) or Vhlfl/fl Foxp3cre (CD45.2) mice were adoptively co-transferred into irradiated Rag1−/− mice. Eight weeks after adoptive transfer, the expression of CD44 and CD62L by splenic CD4+ T cells was determined.
(B) Development of WT and VHL-deficient Foxp3+ T cells. Foxp3 expression was examined by intracellular staining.
(C) IFN-γ and IL-17 expression from WT and VHL-deficient Treg cells in the spleen from BM chimera mice. Foxp3+ cells were gated for analysis by FACS (left panel) and the percentage of IFN-γ+ T cells was calculated (right panel).
(D, E) Foxp3 expression by YFP− and YFP+ Treg cells in CD25+Nrp-1+ cells from Vhlfl/fl Foxp3WT/cre female mice. A combined plot for Foxp3 expression from YFP+ and YFP− Treg cells is shown in (E).
(F) IFN-γ and IL-17 expression by YFP− and YFP+Foxp3+ cells from the spleen of female Vhlfl/fl Foxp3WT/cre mice. Combined plots for A (n=4~7), B (n=4~7), C (n=3~6), E (n=4) and F (n=4) are shown. Error bars indicate SEM. *P<0.05, **P<0.01, ***P<0.001 (Student’s t-test). Representative of two independent experiments are shown. See also Figure S4.
One issue with the mixed BM chimeric mouse model was that the mice were lymphopenic in nature, which may affect the proper development/function of the Treg cells. To address this issue, we utilized female Vhlfl/fl Foxp3WT/cre heterozygote mice in which only half of the Treg cells carry a functional cre expression, and examined the intrinsic phenotype of VHL-deficient Treg cells. These mice did not develop inflammatory responses. Since most Treg cells from spleen are tTreg cells, we examined the Treg cells using anti-CD25 and anti-neuropilin-1 (Nrp-1) antibodies. Both YFP+ and YFP− cells still expressed Foxp3, with a slight reduction of Foxp3 in YFP+ population from Vhlfl/fl Foxp3WT/cre (Figures 4D and 4E). The frequency of YFP+ cells among total Foxp3+ T cells was reduced in lymph nodes, but not in the spleen, from Vhlfl/fl Foxp3WT/cre (Figure S4A). This suggests that under steady and lympho-replete conditions, VHL-deficient Treg cells are prone to lose Foxp3. However, IFN-γ production by YFP+ cells was much higher than that by YFP− cells from Vhlfl/fl Foxp3WT/cre mice (Figure 4F), suggesting that VHL-deficient Treg cells are able to undergo the conversion into IFN-γ-producing Th1-like cells even under non-inflammatory conditions.
We found that Ki-67 expression from VHL-deficient Treg cells was not affected in female Vhlfl/fl Foxp3WT/cre mice (Figure S4B), but that was reduced in mixed BM chimeric mice (Figure S4C). This may suggest that the proliferation of VHL-deficient Treg cells is regulated under certain non-inflammatory conditions. However, we still found that the emergence of Foxp3−Nrp-1+ or Foxp3−Helios+ populations from not only mixed BM chimera mice but also female Vhlfl/fl Foxp3WT/cre heterozygote mice (Figures S4D and S4E). We also observed increased Foxp3−Nrp-1+ population from VHL-deficient Treg cells after transfer into Rag1−/− mice (Figure S4F). The level of HIF-1α expression was higher in Nrp-1+Foxp3− population from Vhlfl/fl Foxp3WT/cre mice, but not from control Vhl+/+ Foxp3WT/cre mice, supporting that those populations had expressed Foxp3 (Figure S4G). These results support that the notion that the reduction of Foxp3+ cell numbers was mainly due to the conversion of VHL-deficient Treg cells into Th1-like cells. Expression of CD25 and CTLA4 from YFP+Foxp3+ cells was similar to YFP−Foxp3+ cells in female Vhlfl/fl Foxp3WT/cre heterozygote mice (Figure S4H), suggesting that the expression of Treg cell markers in VHL-deficient Foxp3+ cells was not due to inflammatory responses in conditional VHL knockout mice (Figure S3A). Taken together, the data suggested that the conversion of VHL-deficient Treg cells into IFN-γ-producing cells could impair Treg cell function and downregulate Foxp3 expression regardless of inflammatory responses.
IFN-γ is responsible for impaired function of VHL-deficient Treg cells
Another important issue is whether IFN-γ affects impaired Treg cell function. To address this, we generated Ifng−/− Vhlfl/fl Foxp3cre mice. These double knockout mice showed reduced inflammatory phenotype in multiple organs (Figures 5A and S5A), and decreased T cell activation (Figure 5B), compared to conditional VHL knockout mice. The expression of Foxp3 was maintained as WT mice (Figure 5C). We further examined the suppressive activity of Ifng−/− Vhl−/− Treg cells in a murine colitis model. Ifng−/− Vhl−/− Treg cells suppressed autoimmunity in the colon and prevented from body weight loss (Figures 5D–G and S5B). Foxp3 expression was maintained in Ifng−/− Vhl−/− Treg cells in contrast to Vhl−/− Treg cells (Figure 5H). Similarly, Ifng−/− Treg cells were able to suppress colitis development in Rag1−/− mice (Figure S5C). These results implicate that IFN-γ is most likely responsible for the impaired suppressive activity in VHL-deficient Treg cells.
Figure 5. IFN-γ is responsible for inflammation by VHL deficiency.

(A) Histological images are shown from liver and lung of 7 week-old lfng−/− Vhlfl/fl Foxp3cre mice.
(B) CD62L, CD44 and Ki-67 expression from T cells in peripheral lymph nodes were assessed.
(C) Foxp3 expression among CD4+ T cells in peripheral lymph nodes was examined.
(D–H) Colitis development in Rag1−/− mice after the adoptive transfer of naïve T cells with either WT, Vhl−/− or Ifng−/− Vhl−/− Treg cells were shown. Histological images of distal colon
(D)histopathological scores (E), body weight (F), total colonic cell number (G) and Foxp3 expression from transferred Treg cells (CD4+CD45.2+) in the colon (H) were shown at 7 weeks after cell transfer.
(I) Colitis induction by VHL-deficient Foxp3+ cells alone. Sorted WT or VHL-deficient YFP+ Treg cells were adoptively transferred into Rag1−/− mice. Neutralizing αIFN-γ antibodies were administrated into the mice every week. The disease development was monitored for the changes of body weight. Combined plots for C (n=6), E (n=3~4), F (n=8), G (n=4), H (n=4) and I (n=3~5) are shown. Error bars indicate SEM. *P<0.05, **P<0.01, ***P<0.001 (Student’s t-test). Representative of two independent experiments are shown. See also Figure S5.
We next examined whether VHL-deficient Treg cells are inflammatory by themselves, since VHL-deficient Treg cells were able to convert into Th1-like effector cells. To address this possibility, only tTreg cells from WT or VHL-deficient mice were adoptively transferred into Rag1−/− mice to monitor body weight changes. We found that mice carrying VHL-deficient Foxp3+ cells did not gain body weight (Figure 5I). The progression of colitis was abrogated by injection with neutralizing anti-IFN-γ antibodies (Figures 5I and S5D–E). Importantly, the phenotypes such as Foxp3 loss and IFN-γ induction from VHL-deficient Treg cells were partially reversed by neutralizing anti-IFN-γ antibodies (Figures S5F and S5G). To examine whether IFN-γ produced by VHL-deficient Treg cells affects Th1 differentiation from naïve T cells, we utilized conditioned medium from cultured VHL-deficient Treg cells in vitro. The cultured supernatants from Vhl−/− Treg cells were efficient to induce Th1 polarization, whereas those from WT or Ifng−/− Vhl−/− Treg cells were not (Figure S5H). This implied that VHL-deficient Treg cells could induce Th1 immune responses by producing IFN-γ.
Increased HIF-1α promotes Treg cell conversion into IFN-γ+ effector T cells
It has been well established that VHL deficiency induces the accumulation of HIF-1α, and the subsequent upregulation of HIF-1α-inducible genes (Kaelin, 2008). We confirmed the increased expression of HIF-1α in VHL-deficient Foxp3+ cells (Figures 6A and S4G). HIF-1α serves as a key transcription factor for anaerobic glycolysis, angiogenesis and erythoropoiesis (Semenza, 2007). The data derived from quantitative real-time PCR showed that the expression of all glycolysis enzyme genes examined were greatly upregulated in VHL-deficient Foxp3+ cells (Figure 6B). To further examine whether the glycolytic reprogramming is required for the conversion of VHL-deficient Foxp3+ cells, we tested whether 2-DG, an inhibitor of hexokinase, can reverse the effect of VHL deficiency. The production of inflammatory cytokines and chemokines from VHL-deficient Foxp3+ cells was blocked by 2-DG (Figures 6C and 6D). These results implied that glycolytic pathway is necessary for the conversion of VHL-deficient Treg cells into effector T cells.
Figure 6. The role of glycolytic pathway for the conversion of Treg cells.

(A) HIF-1α expression in Treg cells in the spleen from Vhlfl/fl Foxp3cre mice. The MFI was measured by flow cytometry (left panel) and the values were calculated and a combined plot for A (n=4) is shown (right panel).
(B) Gene expression of glycolytic enzymes by VHL deficiency. qRT-PCR was performed to determine mRNA levels of the indicated genes. Sorted YFP+ Treg cells from the spleen and lymph nodes were used.
(C) WT or VHL-deficient pTreg cells were activated with anti-CD3/28 antibodies and IL-2 in presence of 2-DG (0.5 mM) for 48 hrs. Expression of IFN-γ and IL-4 was examined after stimulation with PMA and ionomycin for 4 hrs.
(D) Cell culture supernatants from (C) were harvested and analyzed for the production of cytokines and chemokines by Bio-plex. Error bars indicate SEM. *P<0.05 (Student’s t-test). Representative of two or three independent experiments are shown.
A recent study reported that HIF-1α plays an important role in inducing RORγt transcription by binding to Rorc gene in Th17 cell differentiation (Dang et al., 2011). However, VHL-deficient Treg cell did not show the induction of RORγt mRNA expression, but the T-bet and IFN-γ expression was highly enhanced from activated VHL-deficient Treg cells (Figures 7A) as well as freshly isolated VHL-deficient Treg cells (data not shown). Since Th17 cells are enriched in intestines, due to a Th17 skewing condition in vivo, we examined IL-17 expression by intestinal Treg cells. As expected, WT Foxp3+ cells from intestines expressed certain level of IL-17, whereas VHL-deficient Foxp3+ cells expressed less IL-17 and more IFN-γ (Figure S3E). To further test that the accumulation of HIF-1α by VHL deficiency is responsible the effect of Treg cell conversion, we next examined the effect of DMOG, a chemical inhibitor for PHD and found that it caused a similar effect as VHL deficiency on Treg cells (Figure 7A). It has been shown that HIF-1α induces IFN-γ expression by binding on the IFN-γ promoter in macrophages (Acosta-Iborra et al., 2009). We next examined whether IFN-γ gene could be a direct target of HIF-1α in Foxp3+ cells. We found that there are several putative hypoxia-responsive element (HRE) regions in the mouse Ifng gene (Figure 7B). Chromatin immunoprecipitation (ChIP) assay was performed to determine whether HIF-1α binds onto HRE regions in the Ifng gene. We found that of the potential HIF-1α binding sites, I, II, III, and VII HRE regions were detected to be responsible for HIF-1α binding (Figure 7C). The pattern of HIF-1α binding was very similar to that with histone H3 (trimethyl K4) antibodies, which supported the notion that these regions were in transcriptionally active states. To further verify HIF-1α-dependent transcriptional regulation, we utilized the HIF inhibitor, chetomin, which inhibits HIF binding to the transcriptional coactivator p300, thereby attenuating HIF-dependent transcription (Kung et al., 2004). We found that HIF inhibitor was able to block the induction of IFN-γ expression by VHL-deficient Treg cells (data not shown).
Figure 7. HIF-1α directly binds to IFNG gene in VHL-deficient Treg cells.

(A) qRT-PCR was performed for determining mRNA levels of the indicated genes. Sorted YFP+ Treg cells from spleen and lymph nodes were activated with anti-CD3/28 antibodies and IL-2 in presence of a PHD inhibitor, DMOG, for 48 hrs.
(B) Structural schematics of IFNG gene with putative hypoxia response elements (HREs). Blue boxes for exon and red boxes for HRE were shown. Consensus sequences for putative HRE are in red.
(C) Binding of HIF-1α and histone H3 (trimethyl K4) on the IFNG gene. Sorted YFP+ Treg cells were activated with anti-CD3/28 antibodies and IL-2 for 48 hrs. Relative binding activity was normalized by binding in WT cells.
(D) Effect of shRNA knockdown for HIF1-α on IFN-γ and Glut1 expression from VHL-deficient Treg cells. shRNAs were expressed in pTreg cells by retroviral transduction. Infected Treg cells (mAmetrine+YFP+) were further sorted and activated with anti-CD3/28 antibodies and IL-2 for 48 hrs.
(E) Histology of liver and lung from 8 week-old Vhlfl/fl Hif1afl/fl Foxp3cre mice with H&E staining.
(F) CD62L, CD44 and Ki-67 expression in splenic T cells were determined from Vhlfl/fl Hif1afl/fl Foxp3cre mice.
(G) IFN-γ expression from splenic CD4+ T cells was examined after cells were activated with PMA and ionomycin for 4 hrs.
(H) Colitis induction was examined by weight change after an adoptive transfer of naïve T cells (CD45.1) and either WT, Vhl−/− or Vhl−/− Hif1a−/− Treg cells (CD45.2) into Rag1−/− mice.
(I) Foxp3 expression from transferred Treg cells (CD4+CD45.2+) was determined by flow cytometry. Combined plots for A (n=4), G (n=4~8), H (n=4~5) and I (n=3) are shown. Error bars indicate SEM. *P<0.05, **P<0.01, ***P<0.001 (Student’s t-test). Representative of two or three independent experiments are shown. See also Figure S6 and S7.
To further confirm a causal role of HIF-1α in mediating VHL function, we performed shRNA-mediated knockdown assay in Treg cells. Knockdown of HIF-1α abolished the induction of IFN-γ and Glut1 gene expression by VHL-deficient Treg cells (Figure 7D). To investigate the effect of HIF-1α overexpression on IFN-γ and Glut1 expression, a hydroxylation-defective triple mutated P402A/P577A/N813A HIF-1α (HIF-1α-TM) was used for enhancing their stability. Overexpression of HIF-1α was able to upregulate IFN-γ expression by WT Treg cells (Figure S6A). To confirm the role of HIF-1α in VHL-deficient Treg cells in vivo, we generated conditional HIF-1α/VHL double knockout mice. HIF-1α deficiency in VHL-deficient Treg cells was able to completely restore impaired Treg cell function by histopathological examination (Figure 7E). We also found T cell activation and Th1 induction from Vhlfl/fl Foxp3cre mice were suppressed by HIF-1α deletion (Figures 7F and G). We did not see any difference between WT and Hif1afl/fl Foxp3cre mice (Figure S6B–F). Moreover, we tested whether Vhl−/− Hif1a−/− Treg cells are functional in a colitis model. As expected, Vhl−/− Hif1a−/− Treg cells were suppressive in colitis developments in contrast to Vhl−/− Treg cell (Figures 7H and S6G–H). Hif1a−/− Treg cells were comparable to WT Treg cells in suppression of colitis induction (data not shown). Foxp3 expression from Vhl−/− Hif1a−/− Treg cells, but not Vhl−/− Treg cell, was stable at 8 weeks after the adoptive transfer (Figure 7I). Of note, we found that HIF-2α expression in Treg cells was much lower than HIF-1α (Figure S6I) and the effect of HIF-2α might be minimal. Moreover, we tested whether hypoxia induces the conversion of Treg cells. WT mice showed more IFN-γ and CXCR3 expression from both Foxp3+ and Foxp3− cells after exposure to hypoxia in vivo (Figure S14), suggesting the conversion of Foxp3+ cells into Th1-like cells is induced under hypoxic conditions. Thus, our data demonstrated that VHL-deficient Treg cells express IFN-γ through both transcriptional activation by the binding of accumulated HIF-1α to IFN-γ gene and HIF-1α-induced glycolytic metabolic shift.
Discussion
Here we employ a genetic approach to selectively delete VHL in Treg cells and show that loss of VHL in Treg cells causes massive multiple tissue inflammation and activation of both Foxp3− and Foxp3+ CD4+ T cells. VHL-deficient Treg cells produce excessive amounts of IFN-γ, with only slight effect on IL-17 production. Loss of VHL in these cells results in the defective inhibition of normal CD4+ T cells, and the conversion into Th1-like pathogenic T cells. In addition, we show that VHL-deficient Treg cells exhibit an increased expression of HIF-1α, and the subsequent upregulation of glycolytic enzymes. At the molecular level, we show that HIF-1α directly binds to the IFNG gene. Our study thus indicates VHL as a critical regulator of Treg cell stability and function by controlling the HIF-1α pathway to produce IFN-γ.
The present study does not seem to reconcile with the previous two studies describing a reciprocal role of HIF-1α in Th17/Treg cell balance (Dang et al., 2011; Shi et al., 2011), since we found that the primary function of VHL is the regulation of IFN-γ production by Treg cells, whereas the effect on IL-17 production is quite marginal, with only distinguishable difference in intestinal Treg cells. This discrepancy may imply a differential role of HIF-1α in either Treg cell differentiation as revealed by the two previous studies, or the function and/maintenance in established Treg cells as presented in the current study. The increased IFN-γ expression is dependent on the HIF-1α-glycolytic pathway, since we found that the expression of HIF-1α and the HIF-1α-dependent glycolytic enzymes was augmented in VHL-deficient Treg cells. More importantly, the increased IFN-γ production was repressed by the incubation with glycolytic inhibitor 2-DG. Since HIF-1α is the most well documented target for VHL E3 ligase complex and HIF-1α plays a critical role in the glycolytic metabolic reprograming (Kaelin, 2008; Semenza, 2007), our results indeed support the critical role of VHL in HIF-1α-glycolytic pathway in controlling Treg cell function. In addition, the demonstration of HIF-1α binding to the IFN-γ gene indicates that VHL regulates IFN-γ gene expression via both an indirect mechanism (glycolytic reprogramming) and a direct mechanism (transcriptional modification). It has been reported that the pyruvate kinase M2, which is upregulated in VHL−/− Treg cells, acts as co-activator for HIF-1α to recruit p300 to activate transcriptional activation of its target genes (Luo et al., 2011). Although the lack of a significant effect of VHL on IL-17 suggests that the VHL is not essential for the conversion of Treg cell into Th17 cells, HIF-1α itself may possess a unique role in regulating IL-17 expression, which is independent of the VHL-mediated degradation.
It is interesting to note the differential levels of Foxp3 expression in VHL-deficient Treg cells among different tissues, with normal Foxp3 expression in the spleen, lung, liver, and much less expression in lymph nodes, lymphoid tissues from the intestine under normal conditions. This may correlate with the oxygen levels among different locations, since lymph nodes and intestine are known to be more hypoxic (reviewed in (Sitkovsky and Lukashev, 2005)). In a sense, the data do not support the hypothesis that VHL acts as an E3 ligase to promote VHL-mediated Foxp3 degradation, as previously proposed (Dang et al., 2011). Rather, it indicates that VHL is not essential for Foxp3 expression per se, since we found that VHL−/− Treg cells still lost (or even more) their Foxp3 expression under inflammatory conditions, whereas Foxp3 expression is slightly reduced in steady state (in female Vhlf/f Foxp3WT/cre mice). Therefore, it is likely that the loss of Foxp3 expression by VHL deficiency could be accelerated by secondary inflammatory effects. Importantly, the reduced Foxp3 expression in hypoxic tissues does not necessarily correlates with the alteration of IFN-γ production. Therefore, these results suggest the existence of alternative compensatory mechanisms for HIF-1α to induce Foxp3 degradation in the absence of the E3 ligase component VHL. One potential candidate is the SIAH E3 ligases that are responsible for the degradation of PHD, the upstream regulator and oxygen sensor of HIF pathway (Nakayama et al., 2004). Or otherwise, the upregulated glycolytic metabolism in VHL-deficient Treg cells may prime them to lose Foxp3 upon secondary inflammatory stimulation. It should be noted that previous studies have also documented that hypoxia-HIF plays a positive role in Foxp3 induction (Ben-Shoshan et al., 2008; Clambey et al., 2012). However, these findings are not supported by other studies (Dang et al., 2011; Shi et al., 2011) and even our current study. One postulation is that the amounts of HIF-1α are tightly controlled; either excessive increase or decrease may disrupt the normal metabolic homeostasis, which may result in different or even opposite genetic phenotypes.
It may be multifactorial for the explanation of impaired suppression by VHL-deficient Treg cells in vivo. We favor the possibility that they produce inflammatory cytokines and chemokines to induce inflammatory immune responses and this may facilitate to boost the activation of other immune cells, together with the loss of Foxp3 in VHL-deficient Treg cells in vivo. In addition, localization of Treg cells could be another critical factor for their function in vivo. VHL-deficient Treg cells could upregulate CXCR3 on their surface similar to Th1 cells. VHL-deficient Treg cells may thus have different localization/migration capacities, which can affect their suppressive activities. Loss of VHL in Treg cells may also affect their homeostasis, which can contribute to the inflammatory responses observed in Vhlfl/fl Foxp3cre mice. Indeed, metabolic reprogramming is important in homeostasis of lymphocyte. Glut1-dependent metabolic changes is required for proliferation both T and B cells (Caro-Maldonado et al., 2014; Jacobs et al., 2008). HIF-1α is responsible for proliferation or prolonged survival of lymphocytes (Sueoka et al., 2013; Zou et al., 2013). On the contrary, HIF-1α also promotes cell death in hypoxic T cells (Carraro et al., 2007). It is likely that homeostasis is partially regulated by VHL, because the proliferation of VHL-deficient Treg cells was reduced in mixed BM chimera mice. However, experimental homeostatic proliferation of VHL-deficient Treg cells was intact in Rag1−/− mice after an adoptive transfer in short-term period. These results postulate that homeostasis may be apt to be regulated by secondary effects such as the microenvironments. Nevertheless, whether VHL plays a direct role in Treg cell homeostasis or an indirect role by affecting IFN-γ production and/or Foxp3 expression awaits further investigation.
The acquisition of other transcription factors for Th cells by Treg cells has been recently well documented, such that the expression of T-bet is required for Treg cell to control Th1 response, Irf-4 for the suppression of Th2, Stat3 for the prevention of Th17 response (Chaudhry et al., 2009; Koch et al., 2009; Zheng et al., 2009). Here we show that loss of the E3 ligase VHL results in the expression of T-bet and T-bet-driven IFN-γ production, which attributes to the inflammatory responses in VHL-deficient mice. Though T-bet or IFN-γ is required for Treg cell function (Koch et al., 2009; Koenecke et al., 2012), the present study supports the previous observations that IFN-γ production by Treg cells induces Th1 responses and tissue damages (Lu et al., 2010; Oldenhove et al., 2009; Ouyang et al., 2012). In addition, the loss of Foxp3 expression under stress or inflammatory conditions may further render these mutant Treg cells to become pathogenic T cells to cause auto-inflammatory responses. Although the issue of Treg cell conversion into pathogenic Treg cells remains an issue of debate (Miyao et al., 2012; Murai et al., 2009; Rubtsov et al., 2010; Zhou et al., 2009), the current study may suggest that VHL is involved in the stability/maintenance of Treg cells. Deletion of IFN-γ was effective in enhancing Foxp3 stability in this study. It is likely that VHL ablation induced HIF-1α-dependent production of inflammatory cytokines, which could deteriorate Treg cell stability, followed by the reduction of Foxp3 expression under inflammatory conditions as well as non-inflammatory conditions. Obviously, the detailed mechanism underlying whether HIF regulation by VHL is involved in the maintenance of Treg cell stability and pathogenic Treg cell-mediated inflammatory responses awaits further investigation.
The present study establishes a critical role of HIF regulation by VHL in Treg cell regulation. Several previous studies have documented hypoxia and HIF expression in T cell activation, TCR-induce signaling transduction, Th1/Th2 differentiation (reviewed in (Sitkovsky and Lukashev, 2005)), suggesting that this pathway functions under normal physiological as well as inflamed conditions. It can be envisioned that dynamic changes of the glycolytic reprogramming due to the alteration of oxygen tension in normal local microenvironments or inflamed/damaged tissues will have either beneficial or harmful consequences in T cells. In addition, it is known that tumor tissues are hypoxic, which renders tumor cell grow and expand in low oxygen environment (the Warburg effect) (Kim and Dang, 2006). Such hypoxic condition may also affect the T helper and CD8+ T cells, and probably Treg cells, which together contribute to the mechanisms of tumor immune response or evasion. The molecular understanding of VHL-regulated HIF pathway has enabled the development of small molecule inhibitor to potentially harness the bioenergetics for cancer therapy (Buckley et al., 2012), and further elucidation of T cell metabolism will have translational implication for immunological/inflammatory diseases and immune-based cancer treatment.
Experimental Procedures
Mice
Vhlfl/fl, Hif1afl/fl, Ifng−/−, Rag1−/−, and CD45.1 congenic mice were purchased from Jackson Laboratories. Foxp3-cre-YFP mice were from A. Rudensky (Rubtsov et al., 2008). Vhlfl/fl, Hif1afl/fl and Ifng−/− mice were crossed with Foxp3-cre-YFP mice to generate Vhlfl/fl Foxp3cre, Vhlfl/fl Foxp3WT/cre, Vhlfl/fl Foxp3cre, Ifng−/− Vhlfl/fl Foxp3cre or Vhlfl/fl Hif1afl/fl Foxp3cre in the animal facility at La Jolla Institute for Allergy and Immunology under an approved protocol. Six to eight week-old mice were used for the experiments.
Cell isolation and sorting
Cells were isolated from the spleen and lymph nodes by grinding with 70 μm Nylon Mesh (Fisher Scientific). For isolation of cells from intestines, tissues were cut into pieces (3–4 cm) and washed with HBSS medium containing 5 mM EDTA for 5 times to remove epithelial cells. Then, tissues were digested with type III collagenase (Worthington) for 1 hr at 37°C on a rotator. Splenic and lymph node cells were used for isolation of CD4+ T cells by using CD4+ isolation Kit II (Miltenyi Biotech). Enriched CD4+ T cells were further sorted for either YFP+ (Foxp3+) T cells or CD25−CD44−CD62L+ naïve T cells by FACSAria (BD Bioscience). Purity of sorted cells was >99%.
Flow cytometry
Isolated lymphocytes from tissues were stained with αCD44 (IM7), αCD62L (MEL-14), αCD25 (PC61), αCD69 (H1.2F3), αCD39 (24DMS1), αCD73 (TY/11.8), αCTLA4 (UC10-4B9), αGITR (DTA-1), αCD4 (RM4-5), αCD8α (53-6.7) and αNrp-1 (3E12) antibodies. For CXCR3 expression, cells were stained with αCXCR3 (220803) antibodies and further incubated with biotin anti-rat IgG2a (MRG2a-83), followed by staining with PE-streptavidin and αCD4 antibodies. For intracellular staining, cells were fixed and permeabilized with Fixation/Permeabilization buffer (eBioscience), and then further stained with αFoxp3 (FJK-16s), αKi-67 (B56), αHelios (22F6) and αHIF-1α (241812) antibodies. For intracellular staining for cytokines, cells were stimulated with PMA (10 ng/ml) and ionomycin (1 μM) in presence of GolgiStop (BD Bioscience) for 4 hrs at 37°C, and then permeabilized and stained with αIFN-γ (XMG1.2) and αIL-17 (TC11-18H10.1) antibodies. Stained cells were analyzed by FACSCantoII or LSRII (BD Bioscience).
Colitis induction
Naive (CD4+CD25−CD44−CD62L+) T cells from the spleen and lymph nodes of CD45.1 congenic mice were sorted. Foxp3+ (CD45.2+CD4+YFP+) T cells from Vhl+/+ Foxp3cre, Vhlfl/fl Foxp3cre, Ifng−/− Vhlfl/fl Foxp3cre or Vhlfl/fl Hif1afl/fl Foxp3cre mice were prepared by cell sorting. For colitis suppression by Treg cells, naïve T cells (4 × 105) together with WT or KO Treg cells (1 × 105) were adoptively co-transferred i.p. into Rag1−/− mice. For the colitis induction by Treg cell only, sorted WT or VHL-deficient YFP+ cells (2 × 105) were transferred i.p. into Rag1−/− mice. For injection of anti-IFN-γ antibodies into Rag1−/− mice, the mice were received 0.3 mg of neutralizing antibodies every week for 7 weeks. To monitor colitis development, body weight was measured up to 50 days. The large intestine was processed for tissue sectioning and H&E staining.
Mixed bone marrow chimera mice
Bone marrow cells (5 × 106) were obtained from tibia and fibula from WT (CD45.1) and Vhlfl/fl Foxp3cre mice (CD45.2) were co-transferred i.v. into irradiated Rag1−/− mice. Mice were sacrificed at 8 weeks after BM transfer.
Homeostatic proliferation of Treg cells
Sorted Treg cell (YFP+) cells were prepared by labeling with CellTrace Violet. Violet-labeled Treg cells (2 × 105) were adoptively transferred i.v. into Rag1−/− mice. To assess the proliferation, mice were sacrificed at day 8 after an adoptive transfer.
qRT-PCR
Isolation of mRNA from cells was performed by using RNeasy mini kit (Qiagen). cDNAs were synthesized by reverse transcriptase (Superscriptase III, Invitrogen). Quantitative PCR was performed on LightCycler 480. Expression level was normalized by cyclophilin A or β-actin. The primers used in qRT-PCR were listed in Supplementary Table 1.
Chromatin immunoprecipitation (ChIP) assay
Activated pTreg cells with anti-CD3/28 antibodies and IL-2 for 48 hrs were fixed with 1% formaldehyde for 10 min at RT. Then, cells were lyzed in lysis buffer and sonicated by using E220 Focused-ultrasonicator (Covaris). DNA-protein complex was immunoprecipitated with anti-HIF-1α antibodies (H1alpha67, Novus Biologicals) or anti-Histone H3 (tri methyl K4) antibodies (ab8580, Abcam) for overnight. Immunoprecipitated complexes were reverse-crosslinked and DNA was eluted. Quantitative PCR was performed and analyzed. PCR primers for qPCR were listed in Supplementary Table 2.
Statistical analysis
Gehan-Breslow-Wilcoxon test was used for Kaplan-Meier survival curves. The other data were analyzed by paired or unpaired t-test using GraphPad Prizm 5.
Supplementary Material
Acknowledgments
We thank A. Rudensky for providing the Foxp3-cre mice, and H-s. Jin, J. Lopez, and other members in the laboratory for technical help and advice. This work is supported by NIH grants (RO1AI62969, RO1AI78272, and PO1AI089624) from NIAID.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplemental information
Supplemental information includes 7 figures, 2 tables, and Supplemental experimental procedures and can be found with this article online.
Author contributions
J.L., and Y.-C.L conceptually conceived this project, and J.L performed the experiments, analyzed the data, and wrote the manuscript. C.E and Y.P helped some experiments. Y.-C.L supervised this study and helped manuscript writing.
References
- Acosta-Iborra B, Elorza A, Olazabal IM, Martin-Cofreces NB, Martin-Puig S, Miro M, Calzada MJ, Aragones J, Sanchez-Madrid F, Landazuri MO. Macrophage oxygen sensing modulates antigen presentation and phagocytic functions involving IFN-gamma production through the HIF-1 alpha transcription factor. Journal of immunology. 2009;182:3155–3164. doi: 10.4049/jimmunol.0801710. [DOI] [PubMed] [Google Scholar]
- Ben-Shoshan J, Maysel-Auslender S, Mor A, Keren G, George J. Hypoxia controls CD4+CD25+ regulatory T-cell homeostasis via hypoxia-inducible factor-1alpha. European journal of immunology. 2008;38:2412–2418. doi: 10.1002/eji.200838318. [DOI] [PubMed] [Google Scholar]
- Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE, Saulsbury FT, Chance PF, Ochs HD. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nature genetics. 2001;27:20–21. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nature genetics. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- Buckley DL, Van Molle I, Gareiss PC, Tae HS, Michel J, Noblin DJ, Jorgensen WL, Ciulli A, Crews CM. Targeting the von Hippel-Lindau E3 ubiquitin ligase using small molecules to disrupt the VHL/HIF-1alpha interaction. Journal of the American Chemical Society. 2012;134:4465–4468. doi: 10.1021/ja209924v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro-Maldonado A, Wang R, Nichols AG, Kuraoka M, Milasta S, Sun LD, Gavin AL, Abel ED, Kelsoe G, Green DR, Rathmell JC. Metabolic reprogramming is required for antibody production that is suppressed in anergic but exaggerated in chronically BAFF-exposed B cells. J Immunol. 2014;192:3626–3636. doi: 10.4049/jimmunol.1302062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carraro F, Pucci A, Pellegrini M, Pelicci PG, Baldari CT, Naldini A. p66Shc is involved in promoting HIF-1alpha accumulation and cell death in hypoxic T cells. J Cell Physiol. 2007;211:439–447. doi: 10.1002/jcp.20951. [DOI] [PubMed] [Google Scholar]
- Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, Rudensky AY. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science. 2009;326:986–991. doi: 10.1126/science.1172702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, de Zoeten EF, Cambier JC, Stenmark KR, Colgan SP, Eltzschig HK. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E2784–2793. doi: 10.1073/pnas.1202366109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang EV, Barbi J, Yang HY, Jinasena D, Yu H, Zheng Y, Bordman Z, Fu J, Kim Y, Yen HR, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell. 2011;146:772–784. doi: 10.1016/j.cell.2011.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nature immunology. 2003;4:330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299:1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- Jacobs SR, Herman CE, Maciver NJ, Wofford JA, Wieman HL, Hammen JJ, Rathmell JC. Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol. 2008;180:4476–4486. doi: 10.4049/jimmunol.180.7.4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG., Jr The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nature reviews. Cancer. 2008;8:865–873. doi: 10.1038/nrc2502. [DOI] [PubMed] [Google Scholar]
- Kamura T, Koepp DM, Conrad MN, Skowyra D, Moreland RJ, Iliopoulos O, Lane WS, Kaelin WG, Jr, Elledge SJ, Conaway RC, et al. Rbx1, a component of the VHL tumor suppressor complex and SCF ubiquitin ligase. Science. 1999;284:657–661. doi: 10.1126/science.284.5414.657. [DOI] [PubMed] [Google Scholar]
- Khattri R, Cox T, Yasayko SA, Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol. 2003;4:337–342. doi: 10.1038/ni909. [DOI] [PubMed] [Google Scholar]
- Kim JW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer research. 2006;66:8927–8930. doi: 10.1158/0008-5472.CAN-06-1501. [DOI] [PubMed] [Google Scholar]
- Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. 2009;10:595–602. doi: 10.1038/ni.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenecke C, Lee CW, Thamm K, Fohse L, Schafferus M, Mittrucker HW, Floess S, Huehn J, Ganser A, Forster R, Prinz I. IFN-gamma production by allogeneic Foxp3+ regulatory T cells is essential for preventing experimental graft-versus-host disease. J Immunol. 2012;189:2890–2896. doi: 10.4049/jimmunol.1200413. [DOI] [PubMed] [Google Scholar]
- Kung AL, Zabludoff SD, France DS, Freedman SJ, Tanner EA, Vieira A, Cornell-Kennon S, Lee J, Wang B, Wang J, et al. Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer cell. 2004;6:33–43. doi: 10.1016/j.ccr.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Lu LF, Boldin MP, Chaudhry A, Lin LL, Taganov KD, Hanada T, Yoshimura A, Baltimore D, Rudensky AY. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell. 2010;142:914–929. doi: 10.1016/j.cell.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, Cole RN, Pandey A, Semenza GL. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell. 2011;145:732–744. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyao T, Floess S, Setoguchi R, Luche H, Fehling HJ, Waldmann H, Huehn J, Hori S. Plasticity of Foxp3(+) T cells reflects promiscuous Foxp3 expression in conventional T cells but not reprogramming of regulatory T cells. Immunity. 2012;36:262–275. doi: 10.1016/j.immuni.2011.12.012. [DOI] [PubMed] [Google Scholar]
- Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, Kronenberg M. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nature immunology. 2009;10:1178–1184. doi: 10.1038/ni.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Frew IJ, Hagensen M, Skals M, Habelhah H, Bhoumik A, Kadoya T, Erdjument-Bromage H, Tempst P, Frappell PB, et al. Siah2 regulates stability of prolyl-hydroxylases, controls HIF1alpha abundance, and modulates physiological responses to hypoxia. Cell. 2004;117:941–952. doi: 10.1016/j.cell.2004.06.001. [DOI] [PubMed] [Google Scholar]
- Oldenhove G, Bouladoux N, Wohlfert EA, Hall JA, Chou D, Dos Santos L, O’Brien S, Blank R, Lamb E, Natarajan S, et al. Decrease of Foxp3+ Treg cell number and acquisition of effector cell phenotype during lethal infection. Immunity. 2009;31:772–786. doi: 10.1016/j.immuni.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W, Liao W, Luo CT, Yin N, Huse M, Kim MV, Peng M, Chan P, Ma Q, Mo Y, et al. Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature. 2012;491:554–559. doi: 10.1038/nature11581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, Johnson RS, Haddad GG, Karin M. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453:807–811. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, Benoist C, Rudensky AY. Stability of the regulatory T cell lineage in vivo. Science. 2010;329:1667–1671. doi: 10.1126/science.1191996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubtsov YP, Rasmussen JP, Chi EY, Fontenot J, Castelli L, Ye X, Treuting P, Siewe L, Roers A, Henderson WR, Jr, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S. Regulatory T cells: key controllers of immunologic self-tolerance [In Process Citation] Cell. 2000;101:455–458. doi: 10.1016/s0092-8674(00)80856-9. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factor 1 (HIF-1) pathway. Science’s STKE : signal transduction knowledge environment. 2007;2007:cm8. doi: 10.1126/stke.4072007cm8. [DOI] [PubMed] [Google Scholar]
- Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. The Journal of experimental medicine. 2011;208:1367–1376. doi: 10.1084/jem.20110278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitkovsky M, Lukashev D. Regulation of immune cells by local-tissue oxygen tension: HIF1 alpha and adenosine receptors. Nature reviews. Immunology. 2005;5:712–721. doi: 10.1038/nri1685. [DOI] [PubMed] [Google Scholar]
- Stebbins CE, Kaelin WG, Jr, Pavletich NP. Structure of the VHL-ElonginC-ElonginB complex: implications for VHL tumor suppressor function. Science. 1999;284:455–461. doi: 10.1126/science.284.5413.455. [DOI] [PubMed] [Google Scholar]
- Sueoka E, Sueoka-Aragane N, Sato A, Ide M, Nakamura H, Sotomaru Y, Taya C, Yonekawa H, Kitagawa T, Kubota Y, et al. Development of lymphoproliferative diseases by hypoxia inducible factor-1alpha is associated with prolonged lymphocyte survival. PLoS One. 2013;8:e57833. doi: 10.1371/journal.pone.0057833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, Levy-Lahad E, Mazzella M, Goulet O, Perroni L, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nature genetics. 2001;27:18–20. doi: 10.1038/83707. [DOI] [PubMed] [Google Scholar]
- Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, Corcoran L, Treuting P, Klein U, Rudensky AY. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature. 2009;458:351–356. doi: 10.1038/nature07674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martinez-Llordella M, Ashby M, Nakayama M, Rosenthal W, Bluestone JA. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nature immunology. 2009;10:1000–1007. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou J, Li P, Lu F, Liu N, Dai J, Ye J, Qu X, Sun X, Ma D, Park J, Ji C. Notch1 is required for hypoxia-induced proliferation, invasion and chemoresistance of T-cell acute lymphoblastic leukemia cells. J Hematol Oncol. 2013;6:3. doi: 10.1186/1756-8722-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.