

Over the last several years, molecular analyses in acute myeloid leukemia (AML) have established clinical-pathologic and prognostic correlations that have better defined subgroups in this heterogeneous disease and influenced risk-adapted therapeutic approaches on recent clinical trials. Genetic markers such as FLT3 internal tandem duplications (FLT3-ITD), were first identified in adults1 and subsequently in children2 as conferring a poor prognosis. By contrast, mutations in nucleophosmin (NPM1)3,4 and more recently, CCAAT/enhancer-binding protein alpha (CEBPA)5 have been found to be associated with improved outcomes in both adult and pediatric studies. However, despite being initially described almost 20 years ago, the prognostic implications of mutations involving the RAS proto-oncogenes, remain controversial.6 The RAS gene family is comprised of three homologues, HRAS (11p15.5), KRAS (12p12.1), and NRAS (1p13.2), all of which function as membrane-associated guanosine nucleotide phosphate (GTP) binding proteins.7 Mutations in RAS genes are frequent in AML and serve as prototypic Class I lesions, initiating key downstream hyper-proliferative signal transduction pathways.8 NRAS mutations (NRASmut) are the most common, occurring in 10-20% of AML patients. RAS mutations have been linked to leukemic progression in myelodysplastic syndrome (MDS) and an inferior outcome in AML by some researchers;9-13 while other studies have reported no survival difference in AML patients harboring RAS mutations compared to those without RAS mutations.14,15 Still other reports have suggested the presence of a RAS mutation may actually improve survival and/or response to therapy.15 Here, we report on the incidence and prognostic significance of NRASmut in a large cohort of children with AML who were treated on two recent co-operative group studies.
Of the 1241 eligible patients with de novo AML enrolled on CCG-2961 and COG-AAML03P1, diagnostic DNA was available for 825 children. Demographics, laboratory and clinical characteristics, and outcome for patients with and without available specimens were comparable to the study population at large as reviewed in prior studies. 7 KRAS mutations were examined for 183 patients treated on CCG-2961 and only two mutations were identified, so no further analysis was conducted. HRAS mutations are rare in AML15 and thus not examined in this study (data not shown). Exons 1 and 2 of the NRAS gene were amplified, sequenced and evaluated for genomic alterations. Of the 825 specimens tested, 86 (10%) had a missense mutation in the amplified coding region. Mutations involved either codon 12 (n=52, 60%) or 13 (n=33, 38%) as has been previously described, with one unique mutation in codon 29. No mutations were identified in codon 61, a third codon previously reported to be involved in adult AML (Supplemental Table 1).16 In order to establish the somatic, disease-associated nature of these mutations, DNA extracted from remission specimens available from 12 patients with NRAS mutations was evaluated for the presence of NRAS mutations. We failed to demonstrate the presence of NRAS mutations in the remission specimens, demonstrating that the NRAS mutations are disease-associated mutations and not germline polymorphisms. Sex, age, and race were comparable between NRASmut positive and wild type patients. Similarly, there was no difference in frequency of organomegaly or extramedullary disease (central nervous system involvement or chloroma). Median diagnostic WBC (28.9 × 103 /μL vs. 21.6 × 103 /μL, p=0.19) and platelet counts (45 × 103 /μL vs. 48 × 103 /μL, p=0.97) at diagnosis were also similar between the two cohorts. Diagnostic bone marrow blast counts were lower in patients with NRASmut than in those without the mutation (64% vs. 71%, p=0.05) (Supplemental Table 1). NRAS mutational status was correlated with the French-American-British (FAB) Classification of AML subtypes17 for historic comparisons. AML FAB Classification subgroups were similarly distributed between patients with and without NRASmut, with 30% having M2 or M4 morphology (Figure 1A).
Figure 1. Morphologic and Genetic correlations of NRAS mutations in childhood AML.
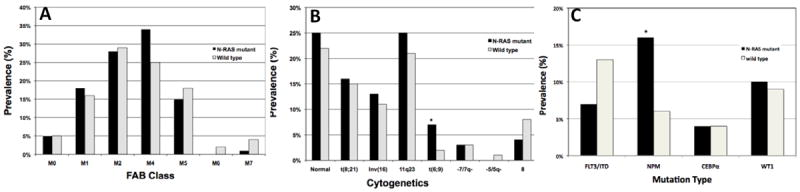
A. Distribution of FAB AML subtype of pediatric patients who underwent NRAS mutational analysis. B. Distribution of cytogenetic abnormalities of patients with and without NRASmut. C. Distribution of mutations in FLT3-ITD, NPM, CEBPA and WT1 genes in patients with and without NRASmut.
*denotes statistically significant difference
The presence of NRASmut was correlated with specific cytogenetic and molecular alterations in our study population. NRASmut were similarly distributed among the major cytogenetic groups (Figure 1B). Cytogenetic information was available on 563/901 (62.5%) of the de novo patients treated on CCG-2961. On COG-AAML03P1, 316/340 (93%) of patients had available cytogenetic data of which 185/316 (59%) were centrally reviewed. Of the 86 NRASmut patients, 68 had cytogenetic information available for further comparisons of whom 17 (25%) had a normal karyotype and another 17 (25%) had abnormalities of chromosome 11q23. These frequencies were comparable to those found with wild-type NRAS. Deletions or monosomies of chromosome 7 [-7/del(7q)] were rare in patients with NRASmut (3%) and no patient with NRASmut had monosomy 5 or del (5q) (Figure 1B). NRASmut has been previously found to correlate with abnormalities of chromosomes 3 [inv(3)/t(3;3), t(3;5)]15,16 and 16 [inv(16)/t(16;16)],16 however this was not found in our study. Seven percent of NRASmut were associated with t(6;9)(p23;q24) compared with only 2% of wild type NRAS (p=0.014). However, the presence of NRASmut had no impact on the poor prognosis of patients with t(6;9)(p23;q24), who demonstrated 3 year overall survival (OS) of 0% vs. 38 ± 34% (p=0.696) and event-free survival (EFS) of 0% vs. 16 ± 32% (p=0.986) for NRASmut and wild type patients, respectively. t(6;9)(p23;q24) translocations were frequently associated with the poor prognostic marker FLT3-ITD, even in NRASmut patients (2/5 patients with t(6;9) and NRASmut; 7/9 with t(6;9) and wild type NRAS), in which FLT3-ITD was uncommon (see below). Correlation of NRASmut with common mutations in AML was also determined. FLT3-ITD appeared to be less common in those with NRASmut (7% vs. 13%, p=0.115); while NPM1 mutations were significantly more common in those with NRASmut (16% vs. 6%, p=0.003) compared to wild type. There was no association observed between NRASmut and CEBPA or WT1 mutations (Figure 1C). Five patients with NRASmut and WT1 mutations had a third abnormality (t(8;21), 2 patients; inv(16), 2 patients; and t(9;11), 1 patient). Overall, 77 of 86 patients with NRASmut (90%) had at least one additional mutation or cytogenetic abnormality (Supplemental Figure 1).
NRASmut status was correlated with specific risk group. NRASmut was more prevalent in those with low risk AML (p=0.028) as defined by the presence of the core binding factor (CBF) group [t(8;21)(q22;q22) or inv(16)t(16;16)], CEBPA, or NPM1 mutations (NPMc+). However, this association appeared to be in large part due to the significant association of NRAS mutations with NPMc+. There was no significant correlation with individual CBF abnormalities, with comparable numbers of patients demonstrating t(8;21)(q22;q22) with and without NRASmut (16% vs. 15%, p=0.80) and similarly for abnormalities of chromosome 16 (13% vs. 11%, p=0.63). In contrast, NRASmut was inversely related with high-risk disease; only 4% of those with NRASmut had monosomy 7, monosomy 5/del(5q), or FLT3-ITD with a high allelic ratio compared with 15% of patients without the mutation (p=0.007, Supplemental Table 1).
Response to induction chemotherapy and clinical outcome was assessed for patients with and without NRASmut. Patients with NRASmut had similar complete remission (CR) rates after one course of induction chemotherapy as patients without the mutation (85% vs. 80%, p=0.268). The actuarial OS at 5 years from study entry was 47±13% for the NRASmut patients compared to that of 55±4%, for those without the mutation (p=0.364, Supplemental Figure 2A), with a corresponding EFS of 38±11% and 44±4% (p=0.55, Supplemental Figure 2B) for those with and without NRASmut, respectively. Those in CR had a similar relapse risk (RR) regardless of the presence of NRASmut (39±12% vs. 36±4%, p=0.973, Supplemental Figure 2C) with an actuarial disease-free survival (DFS) rate of 44±13% vs. 52±4%, p=0.257, respectively (Supplemental Figure 3D).
As NRASmut were more prevalent in patients with low risk disease, we considered whether NRASmut might have a clinical impact in specific risk groups in AML. The presence of NRASmut did not affect clinical outcome in patients with low-risk disease; the OS rate from study entry for those with NRASmut was 59±20% vs. 73±6% for those without NRASmut, p=0.114. For those who achieved an initial CR, the RR was 25±16% vs. 24±7% for those with and without NRASmut, p=0.956, with a corresponding DFS rate of 52±19% vs. 61±8%, p=0.190. Next we examined whether NRASmut could delineate prognosis in two heterogenous subgroups, specifically, patients with standard-risk AML and those with normal karyotypes. Within the standard-risk group, NRASmut had no effect on the OS rate (42±20% vs. 51±6%, p=0.669), EFS rate (31±17% vs. 40±6%, p=0.692), RR (44±21% vs. 42±7%, p=0.712), or DFS rate (41±20% vs. 51±7%, p=0.616). Similarly, the OS rate (24±35% vs. 55±9%, p=0.356), EFS rate (24±25% vs. 45±10%, p=0.230), RR (47±27% vs. 39±10%, p=0.707), and DFS rate (28±24% vs. 51±11%, p=0.106) were comparable between patients with normal karyotypes with or without the mutation (Supplemental Table 2). As there were only 3 patients with high-risk disease who carried an NRASmut, meaningful clinical assessment of NRASmut could not be performed for this cohort, but all 3 patients with NRASmut and other high-risk features either failed to achieve remission (N=1) or had a relapse after achieving an initial CR (N=2), with no survivors.
This current retrospective study represents an evaluation of a large pediatric cohort specifically focused on the incidence and impact of NRASmut on outcome. We identified NRAS mutations in 10% of children with AML treated on two consecutive cooperative clinical trials. NRASmut were found predominantly at codon 12 in keeping with other reports15,16 and also at codon 13, although none were present at codon 61, a third commonly mutated location. Bacher et al.16 identified codon 61 mutations specifically in adult AML samples with inv(16)/t(16;16) or inv(3)/t(3;3) and found these cytogenetic abnormalities were over-represented in the NRASmut positive population. A high frequency of NRASmut in AML samples harboring inv(16)/t(16;16) has also been observed in other adult studies.15,18 We did not find a higher frequency of abnormalities of chromosome 16 between patients with NRASmut and those with wild-type NRAS (p=0.63), which is consistent with our prior data2 and likely accounts for the absence of codon 61 abnormalities.
By extension, the higher frequency of low risk AML that we observed in NRASmut patients cannot be attributed to a higher frequency in CBF AML as seen in adults, but rather appears to be a result of an association with NPM1 mutations. NRASmut is a proto-typical Class I “proliferation/survival lesion”, which has been proposed to require collaboration with Class II “differentiation/self-renewal” lesions to induce leukemogenesis.7,8,19 CBF abnormalities represent Class II lesions. CEBPA mutations, are also good risk Class II lesions that are frequently found in normal karyotype AML.3 However, we observed no relationship between CEBPA mutations and NRASmut, which is consistent with adult data.19 We did, however, detect a significantly higher frequency of NRASmut in patients with NPM1 mutations (p=0.0003). NPM1 mutations represent another good risk Class II lesion. This correlation has not been previously described in adults,19 however it appeared responsible for the association with low risk AML that we observed. The presence of more than one Class I lesion is uncommon in AML,19 thus it is in keeping with prior studies15 that NRASmut and the high risk FLT3-ITD were rarely found together in the same patient. Other high risk features such as monosomy 5 or 7 or del (5q) were similarly uncommon in NRASmut patients. Taken together, these data suggest that this dual class paradigm of leukemogenesis remains valid in NRAS-mediated AML, although collaboration with distinct Class II abnormalities may distinguish adult and pediatric disease.
NRASmut did not affect the outcome in pediatric AML, as we found similar rates of CR and relapse, as well as 5-year EFS and OS rates in patients with the mutated and wild-type NRAS. These findings were consistent even when only patients with standard-risk or normal karyotype disease were considered. Although NRASmut may not provide prognostic information in AML, the role of this mutation in activation of the RAS/MEK/ERK signal transduction pathway is well documented20 and provides a key regulatory step in the transformation of myeloid progenitors and the evolution of AML. Murine studies have demonstrated that interrupting the RAS signal transduction pathway may induce cellular apoptosis in AML cells harboring the NRASmut, and thus interruption of the signaling pathway may have clinical utility.21 Risk-adapted therapy based on the prognostic implications of recurrent molecular lesions represents the most significant advance in both adult and pediatric AML management in recent years.
Pediatric AML remains a unique subset of myeloid diseases that are distinct from their adult counterparts and worthy of independent evaluation. Our study has demonstrated similarities with that of Bowen et al.15 regarding the lack of association of NRASmut with FLT3-ITD, but also highlighted differences with a novel association with NPM1 mutations, t(6;9) translocations and absence of association with CBF leukemias. Given lack of clinical significance for NRAS mutations, implementation of NRAS mutations for risk based therapy allocation in pediatric AML cannot be justified at this time. However as this mutation remains an attractive target for directed therapy, NRAS mutation profiling should be performed and documented as part of correlative research protocols.
Acknowledgments
Project Funding: AAML03P1 and CCG 2961 are funded by grants U10 CA98543 & U10 CA98413; Myeloid Leukemia Reference Laboratory is supported by U10 CA114766.
Footnotes
Conflict of Interest Statement
Dr. Craig Hurwitz is employed by Reatta Pharma and Dr. Janet Franklin is employed by Amgen Pharmaceuticals. The authors have no other disclosures or conflicts of interest.
References
- 1.Thiede C, Steudel C, Mohr B, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99:4326–4335. doi: 10.1182/blood.v99.12.4326. [DOI] [PubMed] [Google Scholar]
- 2.Zwaan CM, Meshinchi S, Radich JP, et al. FLT3 internal tandem duplication in 234 children with acute myeloid leukemia: prognostic significance and relation to cellular drug resistance. Blood. 2003;102:2387–2394. doi: 10.1182/blood-2002-12-3627. [DOI] [PubMed] [Google Scholar]
- 3.Falini B, Mecucci C, Tiacci E, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352:254–266. doi: 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]
- 4.Cazzaniga G, Dell’Oro MG, Mecucci C, et al. Nucleophosmin mutations in childhood acute myelogenous leukemia with normal karyotype. Blood. 2005;106:1419–1422. doi: 10.1182/blood-2005-03-0899. [DOI] [PubMed] [Google Scholar]
- 5.Ho PA, Alonzo TA, Gerbing RB, et al. Prevalence and prognostic implications of CEBPA mutations in pediatric acute myeloid leukemia (AML): a report from the Children’s Oncology Group. Blood. 2009;113:6558–6566. doi: 10.1182/blood-2008-10-184747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Janssen JW, Steenvoorden AC, Lyons J, et al. RAS gene mutations in acute and chronic myelocytic leukemias, chronic myeloproliferative disorders, and myelodysplastic syndromes. Proc Natl Acad Sci U S A. 1987;84:9228–9232. doi: 10.1073/pnas.84.24.9228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Frohling S, Scholl C, Gilliland DG, Levine RL. Genetics of myeloid malignancies: pathogenetic and clinical implications. J Clin Oncol. 2005;23:6285–6295. doi: 10.1200/JCO.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 8.Gilliland DG, Tallman MS. Focus on acute leukemias. Cancer Cell. 2002;1:417–420. doi: 10.1016/s1535-6108(02)00081-8. [DOI] [PubMed] [Google Scholar]
- 9.Hirai H, Kobayashi Y, Mano H, et al. A point mutation at codon 13 of the N-ras oncogene in myelodysplastic syndrome. Nature. 1987;327:430–432. doi: 10.1038/327430a0. [DOI] [PubMed] [Google Scholar]
- 10.Hirai H, Okada M, Mizoguchi H, et al. Relationship between an activated N-ras oncogene and chromosomal abnormality during leukemic progression from myelodysplastic syndrome. Blood. 1988;71:256–258. [PubMed] [Google Scholar]
- 11.Lubbert M, Mirro J, Jr, Kitchingman G, et al. Prevalence of N-ras mutations in children with myelodysplastic syndromes and acute myeloid leukemia. Oncogene. 1992;7:263–268. [PubMed] [Google Scholar]
- 12.Misawa S, Horiike S, Kaneko H, et al. Significance of chromosomal alterations and mutations of the N-RAS and TP53 genes in relation to leukemogenesis of acute myeloid leukemia. Leuk Res. 1998;22:631–637. doi: 10.1016/s0145-2126(98)00056-3. [DOI] [PubMed] [Google Scholar]
- 13.Paquette RL, Landaw EM, Pierre RV, et al. N-ras mutations are associated with poor prognosis and increased risk of leukemia in myelodysplastic syndrome. Blood. 1993;82:590–599. [PubMed] [Google Scholar]
- 14.Nakagawa T, Saitoh S, Imoto S, et al. Multiple point mutation of N-ras and K-ras oncogenes in myelodysplastic syndrome and acute myelogenous leukemia. Oncology. 1992;49:114–122. doi: 10.1159/000227023. [DOI] [PubMed] [Google Scholar]
- 15.Bowen DT, Frew ME, Hills R, et al. RAS mutation in acute myeloid leukemia is associated with distinct cytogenetic subgroups but does not influence outcome in patients younger than 60 years. Blood. 2005;106:2113–2119. doi: 10.1182/blood-2005-03-0867. [DOI] [PubMed] [Google Scholar]
- 16.Bacher U, Haferlach T, Schoch C, Kern W, Schnittger S. Implications of NRAS mutations in AML: a study of 2502 patients. Blood. 2006;107:3847–3853. doi: 10.1182/blood-2005-08-3522. [DOI] [PubMed] [Google Scholar]
- 17.Bennett JM, Catovsky D, Daniel MT, et al. Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French-American-British Cooperative Group. Ann Intern Med. 1985;103:620–625. doi: 10.7326/0003-4819-103-4-620. [DOI] [PubMed] [Google Scholar]
- 18.Boissel N, Leroy H, Brethon B, et al. Incidence and prognostic impact of c-Kit, FLT3, and Ras gene mutations in core binding factor acute myeloid leukemia (CBF-AML) Leukemia. 2006;20:965–970. doi: 10.1038/sj.leu.2404188. [DOI] [PubMed] [Google Scholar]
- 19.Schlenk RF, Dohner K, Krauter J, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1909–1918. doi: 10.1056/NEJMoa074306. [DOI] [PubMed] [Google Scholar]
- 20.Zuber J, Tchernitsa OI, Hinzmann B, et al. A genome-wide survey of RAS transformation targets. Nat Genet. 2000;24:144–152. doi: 10.1038/72799. [DOI] [PubMed] [Google Scholar]
- 21.Kim WI, Matise I, Diers MD, Largaespada DA. RAS oncogene suppression induces apoptosis followed by more differentiated and less myelosuppressive disease upon relapse of acute myeloid leukemia. Blood. 2009;113:1086–1096. doi: 10.1182/blood-2008-01-132316. [DOI] [PMC free article] [PubMed] [Google Scholar]