

Abstract
Neurogenic orthostatic hypotension (nOH) is a fall in blood pressure on standing due to reduced norepinephrine release from sympathetic nerve terminals. nOH is a feature of several neurological disorders that affect the autonomic nervous system, most notably Parkinson disease (PD), multiple system atrophy, pure autonomic failure and other autonomic neuropathies. Droxidopa, an orally active synthetic amino acid that is converted to norepinephrine by the enzyme aromatic L-amino acid decarboxylase (dopa-decarboxylase), was recently approved by the FDA for the short-term treatment of nOH. It is presumed to raise blood pressure by acting at the neurovascular junction to increase vascular tone. This review summarizes the pharmacological properties of droxidopa, its mechanism of action, and the efficacy and safety results of clinical trials.
Keywords: Autonomic failure, Autonomic neuropathy, Blood pressure, Parkinson disease, Pure autonomic failure, Syncope, Multiple system atrophy, L-DOPS, Catecholamines, Dopamine-beta hydroxylase deficiency
1. INTRODUCTION
Symptomatic neurogenic orthostatic hypotension (nOH) is an orphan condition as it affects < 200,000 people in the US [1]. OH is a fall in blood pressure (BP) upon standing; the neurogenic prefix refers to the patients in whom OH is due to reduced norepinephrine release from sympathetic nerves, leading to defective vasoconstriction when upright [2].
OH is a very common problem, particularly in the frail elderly. It is due to a variety of medical conditions, such as intravascular volume depletion, severe anemia, use of antihypertensive therapies, and physical deconditioning. It usually resolves after the underlying cause is treated.
Neurogenic OH (nOH), in contrast, is a much less common and chronic condition. nOH is the result of a failure to increase sympathetic vasomotor nerve outflow and an inability to raise peripheral vascular resistance on standing [3,4]. nOH is a feature of several neurological disorders that affect autonomic neurons. These include neurodegenerative diseases associated with the abnormal deposition of the protein α-synuclein (i.e., synucleinopathies such as Parkinson disease), other peripheral neuropathies, high spinal cord injury and a handful of rare genetic diseases.
In patients with nOH, consistently BP falls each time the patient stands but the magnitude of the fall may vary. OH can impair perfusion to organs above the heart, most notably the brain, and results in symptoms of tissue hypoperfusion. Symptoms can be very disabling and have a profound impact on a patient’s quality of life [5]. OH increases the risk of falls and is an independent risk factor for mortality [6]. Treatment is complicated by the presence of supine hypertension, which occurs in up to 70% of patients with nOH [7,8].
Droxidopa is a prodrug, which is converted to norepinephrine, increases BP, and improves symptoms of nOH. It recently gained approval in the US for the treatment of symptomatic nOH. Here we review the history of droxidopa, its mechanism of action and the results of the clinical trials that led to its approval. We also provide practical advice on treating patients with this rare condition.
Definition
The definition of OH is based on expert consensus, first published in 1996 [9] and revised in 2011 [2]. It is defined as a fall of at least 20 mmHg in systolic BP or 10 mmHg in diastolic BP within 3 minutes of standing or upright tilt.
1.2 Symptoms
Not all patients with nOH are symptomatic. Symptoms of cerebral hypoperfusion emerge when BP standing falls below the lower limit of the cerebral autoregulatory range [10]. This usually occurs at a mean BP of 75 mmHg, corresponding to around 90/60 mmHg (systolic/diastolic) at heart level.[8] Symptoms disappear soon after the patient resumes sitting or lying and cerebral blood flow increases again. The chronic nature of nOH allows adaptive changes in cerebral autoregulatory mechanisms. Patients with nOH are frequently able to tolerate wide swings in BPs and often remain conscious at pressures that would otherwise induce syncope in healthy subjects [10].
Classical symptoms of nOH include lightheadedness, dizziness or feeling close to fainting. When the fall in BP is severe enough, consciousness is lost. In contrast to vasovagal (neurally-mediated) syncope, syncope in nOH occurs without signs of autonomic activation such as sweating, tachycardia, nausea or abdominal discomfort. After syncope, patients with nOH recover quickly and may be unaware of the event. Patients report that symptom severity varies day-to-day and indeed fluctuates throughout the day. Mornings tend to be most difficult as symptoms are aggravated by intravascular volume loss overnight [11]. Meals, particularly carbohydrate-rich, produce splanchnic vasodilatation and post-prandial hypotension (i.e., fall in BP within 2 hours of eating). Physical inactivity and cardiovascular deconditioning are common in patients with nOH, and, in turn, worsens symptom severity leading to a vicious cycle.
1.3 Causes
nOH occurs in diseases that affect the ability of sympathetic nerves to release norepinephrine upon standing, referred to as chronic autonomic failure [4]. nOH occurs in neurodegenerative disorders like Parkinson disease (PD), dementia with Lewy bodies, multiple system atrophy (MSA) and pure autonomic failure (PAF) [4]. These disorders are now classified as synucleinopathies because all share the abnormal deposition of α-synuclein in cells of the nervous system. nOH can also occur in patients with peripheral neuropathies such as those secondary to diabetes mellitus and amyloidosis, rare genetic diseases like dopamine β-hydroxylase deficiency [12] and familial dysautonomia [13]; and cervical-thoracic spinal cord injury. nOH is also prominent feature in some autoimmune diseases such as autonomic ganglionopathies due to antibodies against the nicotinic acetylcholine receptor, and paraneoplastic syndromes (Figure 1).
Figure 1. Localization of lesions in the sympathetic efferent pathways in different autonomic disorders.
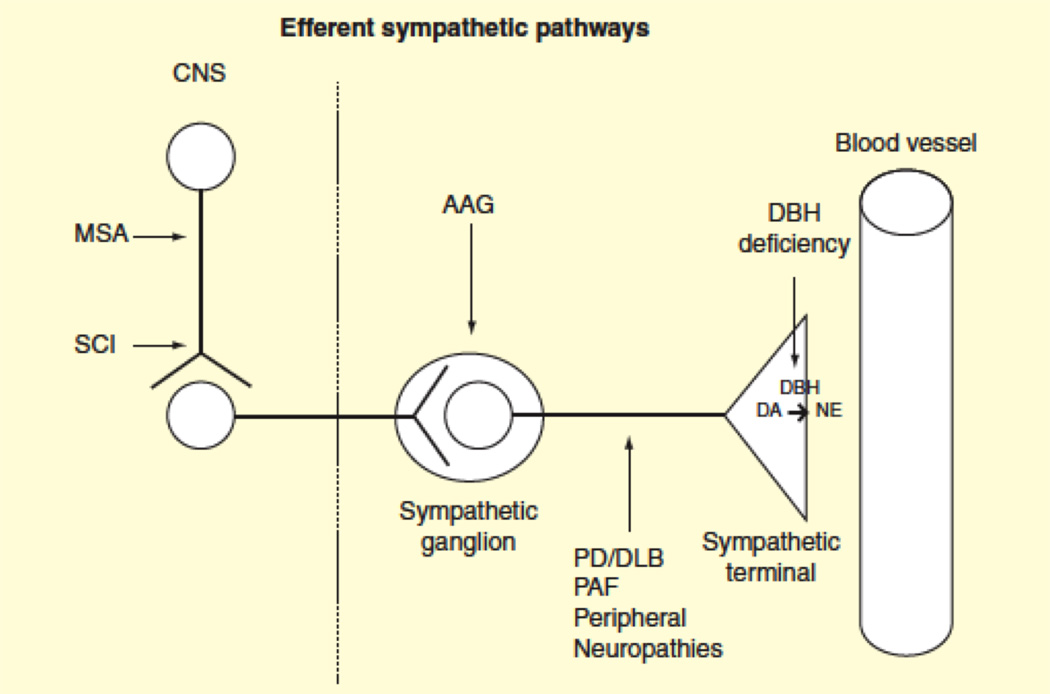
In multiple system atrophy (MSA), α-synuclein aggregates lead to neurodegeneration in the CNS, including brainstem areas that regulate cardiovascular autonomic function. In cervical-thoracic spinal cord injury (SCI) the connections between these brainstem areas and the efferent neurons are damaged. In patients with autoimmune autonomic ganglionopathy (AAG), antibodies against the ganglionic (nicotinic) acetylcholine receptor block the neurotransmission, leading to autonomic failure. Abnormal protein deposition in sympathetic postganglionic nerves takes place in Parkinson disease (PD), dementia with Lewy bodies (DLB), and pure autonomic failure (PAF). Other causes of peripheral neuropathies with autonomic involvement include familial amyloid polyneuropathy or Sjögren disease. In dopamine-beta hydroxylase (DBH) deficiency, dopamine (DA) is not converted to norepinephrine (NE) in the postganglionic sympathetic terminals.
1.4 Diagnosis
Identifying nOH requires BP readings while supine and upright, either during active standing or during a tilt-table test, to determine the presence of a 20/10 mmHg (systolic/diastolic) orthostatic fall. BP and heart rate should be measured with the patient supine for several minutes and after standing still for 3 minutes. The magnitude of the blood pressure fall and symptom severity vary at different times of the day; thus it may be necessary to re-test the patient in the morning when the orthostatic fall in pressure is more pronounced or after a meal if the history raises suspicions of post-prandial hypotension.
Confirming whether the OH is neurogenic may require autonomic testing including the BP response to the Valsalva maneuver and plasma norepinephrine levels. In patients with nOH, reduced sympathetic innervation causes heart rate to increase much less than expected considering the magnitude of the BP fall [5,14]. During the Valsalva maneuver, patients with nOH fail to show the classical BP “overshoot” after release of the strain (phase 4). An increase in plasma norepinephrine after 5–10 minutes of standing of less than 100% is indicative of defective baroreflex sympathetic activation and nOH (Table 1).
Table 1.
Features that distinguish non-neurogenic vs. neurogenic orthostatic hypotension
| Non-neurogenic orthostatic hypotension |
Neurogenic orthostatic hypotension |
|
|---|---|---|
| Frequency | Frequent (particularly in the elderly) | Less frequent |
| Onset | Variable | Usually chronic (Acute in some immune-mediated neuropathies and ganglionopathies) |
| Causes | Intravascular volume loss (e.g., dehydration, anemia) Inadequate vasoconstriction (e.g., varicose veins) Heart failure Physical deconditioning Medications |
Failure to increase sympathetic activity upon standing due to defective norepinephrine release |
| Outcome | Resolves when underlying cause is corrected | Chronic disorder |
| Sympathetic activity | Elevated | Low or absent |
| Increase in heart rate upon standing | Pronounced | Mild or absent |
| Blood pressure overshoot (phase 4) in Valsalva maneuver | Present | Absent |
| Increase in norepinephrine upon standing | Normal or enhanced | Blunted or absent |
| Additional symptoms of autonomic failure | No | Constipation Erectile dysfunction (men) Urinary abnormalities Sweating abnormalities |
| Concurrent neurological findings | None(if present, they are not related to OH) | None Parkinsonism Cerebellar signs Cognitive impairment Sensory neuropathy |
Since symptom severity can vary at particular times of the day it may be necessary to re-test the patient after a meal if the history raises suspicions of post-prandial hypotension. The use of 24-hour ambulatory BP monitoring can help in the diagnosis and management of nOH [15]. Patients with nOH typically have a reversal of the normal circadian blood pressure pattern with higher BP during the night. Nocturnal hypertension causes pressure natruiuresis and overnight volume depletion, worsening OH in the morning. In patients with chronic autonomic failure, while office readings of blood pressure in the supine position predict blood pressure throughout the night, in only one third of patients, BP in the standing position accurately predicts the severity of hypotension the day. This suggests that relying on clinic blood pressure values in the management of patients with chronic autonomic failure may result in under or over treatment. Thus, it is recommend that in addition to clinic readings, BP be assessed by ambulatory monitoring [16]. Ambulatory monitoring is also useful to tailor the use of short acting pressor agents to times when orthostatic hypotension is severe in patients that may remain seated for long periods of the day.
2. TREATMENT OF NEUROGENIC ORTHOSTATIC HYPOTENSION
Consensus guidelines for the treatment of nOH are lacking, although there are expert reviews [17,18]. There are no long-term studies showing the impact of treatment on survival, falls or quality of life. Up to 70% patients with nOH also have supine hypertension, which poses a therapeutic challenge. Increasing BP in the upright position can worsen hypertension when supine. Therefore, treatment of nOH requires careful consideration of the potential risks and benefits. The goal of treatment is to reduce symptom burden, prolong standing time, and improve physical capabilities. The steps in management include a) removing aggravating factors, b) implementing non-pharmacological measures and c) drug therapies (Figure 2).
Figure 2. Algorithm showing the therapeutic approach to patients with orthostatic hypotension.

Removal of aggravating factors and non-pharmacological measures must always precede the initiation of pharmacological agents.
2.1 Non-pharmacological approaches
Patient education is essential in the treatment of nOH. Patients should understand the effects of posture on BP and learn physical “tricks” to raise BP.
2.1.1 Removing aggravating factors
2.1.1.1 Drug-induced hypotension
Drugs that reduce intravascular volume, induce vasodilatation or block the activity of norepinephrine at the neurovascular junction exacerbate OH and worsen symptoms. Common offenders include diuretics, α-blockers prescribed for benign prostatic hypertrophy, phosphodiesterase-5 inhibitors like sildenafil used for erectile dysfunction, nitrates, centrally acting α2-agonists like clonidine, and tricyclic antidepressants. Levodopa and dopamine agonists may also lower blood pressure.
2.1.1.2 Anemia
Anemia worsens OH by reducing blood viscosity and oxygen carrying capacity, and may also increase the amount of free nitric oxide, producing a powerful vasodilatory effect. nOH is associated with an increased incidence of anemia of chronic disease. Small case series show that raising red cell mass with erythropoietin therapy lessens the fall in BP on standing [19].
2.1.1.3 Dehydration
Intravascular volume depletion reduces circulating blood volume and worsens the fall in BP on standing. Many elderly patients are chronically volume depleted [20]. Patients should be aware of the diuretic effects of caffeine and alcohol, which is also a vasodilator.
2.1.1.4 Prolonged bed rest and physical deconditioning
Supine hypertension leads to pressure natriuresis and effective intravascular volume loss whenever a patient is laying flat. Even short periods of bed rest worsen nOH. Symptomatic burden can quickly lead to a reluctance to stand up and lack of physical activity. Physical immobility and skeletal muscle loss worsens the severity of OH, which leads to a “vicious cycle” of deconditioning [5].
2.1.2. Physical counter maneuvers
Physical counter-maneuvers are an essential component of managing nOH. Time should be spent making certain a patient understands the effect of gravitational fluid shifts on BP and symptoms. Patients should learn to change position gradually, and sit before standing. They should be made aware that Valsalva-like (straining with closed glottis) maneuvers produce an abrupt and frequently severe fall in BP. Straining during bowel movements is a common cause of syncope, hence constipation must be treated aggressively. Maneuvers like leg crossing, standing on tiptoes and buttock clenching effectively increase venous return, raise BP and lessen symptoms [21]. Good instructional videos are available [22].
2.1.3. Life-style changes
Symptoms of nOH are exacerbated in hot temperatures. Hot showers cause skin vasodilatation and should be avoided. Exercise is important but should be performed in the recumbent position (e.g., stationary bicycle, rowing machine, exercise with elastic bands for legs). Exercise in a pool is ideal, as the hydrostatic pressure of water counteracts gravity-induced fall in BP and increases orthostatic tolerance markedly.
2.1.4. Volume expansion
As vasoconstriction is defective when upright in patients with nOH, their BP standing is critically depended on intravascular volume. Volume expansion with salt and water is necessary, and fluid take should be 2–2.5L of water per day. Patients should be encouraged to increase salt intake by adding 1–2 teaspoon of salt to a healthy diet.
2.1.5. Acute drinking of water
In patients with nOH, drinking half a liter of water (16 oz or 1 pint) produces a marked increase in BP [23]. This can be used as a rescue measure because it is quick; the increase in BP occurs within 5–10 min and peaks in around 30 min, but it is short-lived. The increase in BP is thought to be due to a spinal sympathetic reflex [24].
2.1.6. Sleep with the head of the bed raised
Avoiding laying flat is the best way to get around supine hypertension. Raising the head of the bed by 15–22 cm (6–9 inches) creates a 30-degree incline. Sleeping with the head and torso elevated creates a minor gravitational stress throughout the night. This lowers BP and reduces overnight sodium and water excretion by the kidney. Nocturia improves, intravascular volume is higher upon awakening and symptoms are more tolerable during the morning hours. This simple intervention as a treatment for nOH dates back from 1940 [25].
2.1.7. Compression stockings
Compression stockings apply counter pressure to the lower limbs and abdomen, reducing venous pooling and capillary filtration [26]. High-waist stocking, that produce at least 15–20 mmHg, are an effective way to increase venous return. Patients with accompanying movement disorders struggle to put the stockings on – which limits their usefulness in everyday life. Abdominal binders appear to be a good alternative [26,27].
2.1.8. Small frequent meals
Eating results in blood pooling within the splanchnic circulation [28,29]. Normally, this is compensated for by increases sympathetic activity. In patients with nOH, however, vasoconstrictor nerve activity is deficient and many patients become severely hypotensive within 2 hours of eating [2,30]. Postprandial hypotension is worse after a carbohydrate-rich meal. Alcohol is also a powerful vasodilator and should be reserved for the evening, prior to laying supine.
2.2 Pharmacological approaches
While the above methods are effective, many patients with nOH still require pharmacological treatment to raise BP. This is achieved with two strategies: a) Expanding intravascular volume and b) Increasing peripheral vascular resistance.
2.2.1. Fludrocortisone
Fludrocortisone (9α-fluorocortisol) is a synthetic mineralocorticoid that increases renal sodium and water re-absorption, expands intravascular volume and increases blood pressure. Although it is not FDA-approved for this indication, a low dose of fludrocortisone (0.1–0.2 mg/day) is often used in the treatment of nOH. Although higher dosages are used they are rarely more effective and side effects are amplified. Clinical effects are seen after 1–2 weeks of treatment [18]. Frequent side effects include supine hypertension, hypokalemia and ankle edema [31]. Experimental data suggest it may also enhance the pressor effect of norepinephrine and angiotensin II. Long-term use exacerbates supine hypertension and produces end-organ target damage [32].
2.2.2. Midodrine
This selective α1-adrenoreceptor agonist does not cross the blood brain barrier and increases vascular resistance and BP. It was the first drug approved by the FDA for the treatment of nOH back in 1996 [33]. Midodrine raises BP within 40 minutes, has a peak effect 2-h post dose and its effects last for up to 4-hours. Starting dose is 2.5 mg and can be increased up to 10 mg [34]. Checking the BP in the sitting and standing position before and 1 hour after taking midodrine is helpful to titrate to optimal dosage. Patients can time dosages so that the peak effect can coincide with activities expected to exacerbate the fall in BP, such as exercising or walking. Patients should remain standing if possible or sitting after taking midodrine and should not dose it less than 3 hours before going to bed. Supine hypertension is the main safety concern, and the drug carries a boxed warning. Other side effects include pilomotor erection (“goose bumps”), skull pruritus, and, in rare cases, urinary retention.
2.2.3. Other medications
Erythropoetin (25–50 units/kg, subcutaneous, 3 times a week) in conjunction with iron supplements may be beneficial, especially in patients with nOH that have anemia of chronic disease [19]. Atomoxetine (18 mg) blocks the reuptake of norepinephrine and can be helpful to increase BP in some patients [35]. The long term safety and efficacy of these approaches have not been determined. Pyridostigmine, an inhibitor of cholinesterase, the enzyme that catalyzes the hydrolysis of acetylcholine and terminates its action, potentiates cholinergic neurotransmission in sympathetic ganglia. In a double blind study of 58 patients with nOH, 2 hours after taking 60 mg of pyridostigmine, upright systolic blood pressure was 4 mm Hg higher in the pyridostigmine group than in the placebo group [36]. The combination of 5 mg midodrine with 60 mg pyridostigmine was slightly more effective than pyridostigmine alone. Prostaglandin synthesis inhibitors (indomethacin, ibuprofen) [37] and the somastostatin analog octreotide [38] are sometimes effective in reducing postprandial hypotension but both agents may induce intolerable gastrointestinal side effects. The dopaminergic blocker metoclopramide increases BP in some patients with autonomic failure, but may aggravate or induce parkinsonian symptoms and is therefore contraindicated in PD. The long term safety and efficacy of these approaches have not been determined.
2.3. Unmet needs of currently available therapies
Symptomatic nOH is a disabling disorder. Severely affected patients are unable to stand but for a few seconds, making it impossible to perform even simple activities of daily living. The risk of falls and injuries is increased and patients can become socially isolated due to the burden of symptoms [39]. Success with available agents is only partial in severe cases, with many patients with nOH continuing to have severe symptoms. Exercise becomes intolerable, which inevitably leads to physical deconditioning and muscle atrophy, which worsen the fall in BP. Short acting pressor agents are an important therapeutic tool to break this cycle and improve mobility. New treatment options are clearly needed.
3. DROXIDOPA
Droxidopa (Northera®) (L-threo-3,4-dihydroxyphenyl-serine or L-DOPS) is a synthetic catechol-amino acid that after oral administration is converted to the naturally occurring sympathetic neurotransmitter norepinephrine. It has been approved in Japan for the treatment of nOH since 1989. In 2014, the US Food and Drug Administration (FDA) approved droxidopa (Northera®) as a treatment for nOH. Having a second drug available to treat nOH was an important milestone in the management of this rare disease.
3.1. History
Droxidopa was first synthesized in 1919 by German chemists [40], who speculated that the compound was a catecholamine precursor. In the late 1940’s, Blaschko and colleagues in England showed that droxidopa could be converted to norepinephrine when incubated with Streptococcus faecalis [41]. Blaschko went on to show that the synthesis of norepinephrine required a single decarboxylation step and was dependent on the enzyme aromatic amino acid decarboxylase (AAAD) [42,43]. Schmiter low showed that rabbits fed with droxidopa had increased urinary norepinephrine output [44]. Thirty-years later, the slow-onset long-lasting pressor effect of oral droxidopa was demonstrated in rats [45]. The pressor effect was enhanced in sympathectomized rats, suggesting a potential role in the treatment of nOH in humans with autonomic failure.
A clinical report on droxidopa in patients with nOH was published in 1989 [46], and showed 600 mg increased plasma norepinephrine and BP in patients with familial amyloid polyneuropathy. Droxidopa was used to treat nOH caused by dopamine β-hydroxylase (DBH) deficiency, a rare genetic disorder in which patients lack the enzyme to convert dopamine to norepinephrine. Since the synthesis of norepinephrine from droxidopa does not require DBH (Figure 3), norepinephrine was replenished and symptoms of nOH improved dramatically [47,48]. Subsequent studies in Japan led to its approval in 1989 for the treatment of nOH in Parkinson disease (PD), multiple system atrophy (MSA) and familial amyloid polyneuropathy [49–52]. In 2000, this approval was expanded to include post-dialytic symptomatic hypotension [53].
Figure 3. Metabolism of catecholamines including droxidopa.

Both L-DOPA and droxidopa (L-DOPS) are converted to dopamine and norepinephrine by the same enzyme, dopa decarboxylase or L-aromatic amino acid decarboxylase (AAAD).
3.2 Chemistry
Droxidopa is structurally similar to the precursor of dopamine L-DOPA (L-3,4-dihydroxyphenylalanine) with the exception of an added hydroxyl group (Figure 3). Both substrates are converted to biologically active catecholamines by the enzyme aromatic aminoacid decarboxylase (AAAD) in a single decarboxylation step. AAAD is ubiquitously expressed throughout the body.
DOPS has four stereoisomers [54], but only the L-isoform (droxidopa) is converted to active L-norepinephrine. Studies with racemic mixtures show that the D stereoisomer competitively inhibits the decarboxylation of the L stereoisomer, and reduces the rate of L-norepinephrine formation [55]. Only the pure L isoform of 3,4-threo-dihydroxyphenylserine (droxidopa, Northera ®) is used for treatment. Droxidopa is slightly soluble in water, has a molecular weight of 213.19 and its molecular formula is C9H11NO5.
3.3 Pharmacodynamics
Based on an average elimination half-life of 2.5 h, droxidopa could be administered every 4 hours during the day. Its pressor effect, however, is through its conversion to norepinephrine. The half-time for the decline in plasma norepinephrine is about 9 h (Figure 4). After a single oral dose, a pressor effect is apparent after one hour, peaks at 3–4 h, and lasts for around 6 h (Figure 5). The mechanism of the pressor effect is not fully understood but a threshold norepinephrine plasma level required to exert a significant pressor effect in patients with nOH is ~700 pg/ml (Figure 6). Droxidopa has little effect on heart rate either supine or standing (Figure 7) [56].
Figure 4. Plasma levels of droxidopa and norepinephrine after droxidopa administration in 8 subjects with autonomic failure.

Time 0 represents the time of droxidopa administration. The peak droxidopa level was attained 3 hours after dosage. Droxidopa remained detectable for at least 22 hours. Plasma norepinephrine peaked 6 hours after droxidopa administration. Plasma levels of norepinephrine remained significantly elevated for at least 46 hours.Modified from: Kaufmann H, et al. Circulation 2003;108(6):724-8.
Figure 5. Effects of droxidopa and placebo on mean blood pressure standing.

Administration of droxidopa in 19 patients with neurogenic orthostatic hypotension significantly increased their blood pressure (BP) after standing 3-minutes. Pressor effect began 1 h after droxidopa administration and lasted 6 h while standing. The peak standing BP occurred 3.5 h after droxidopa administration. Asterisks denote statistical significance (p<0.05). Modified from: Kaufmann H, et al. Circulation 2003; 108(6): 724-8.
Figure 6. Changes in systolic blood pressure versus plasma concentration of norepinephrine after droxidopa administration.

The threshold norepinephrine plasma level required to exert a pressor effect in 8 patients with neurogenic orthostatic hypotension was ~700 pg/mL. The regression curve represents the best fit (r2=0.914, p<0.005). Modified from: Kaufmann H, et al. Circulation 2003; 108(6): 724-8.
Figure 7. Supine and standing heart rate in patients taking droxidopa and placebo in subjects enrolled in the Study 301.
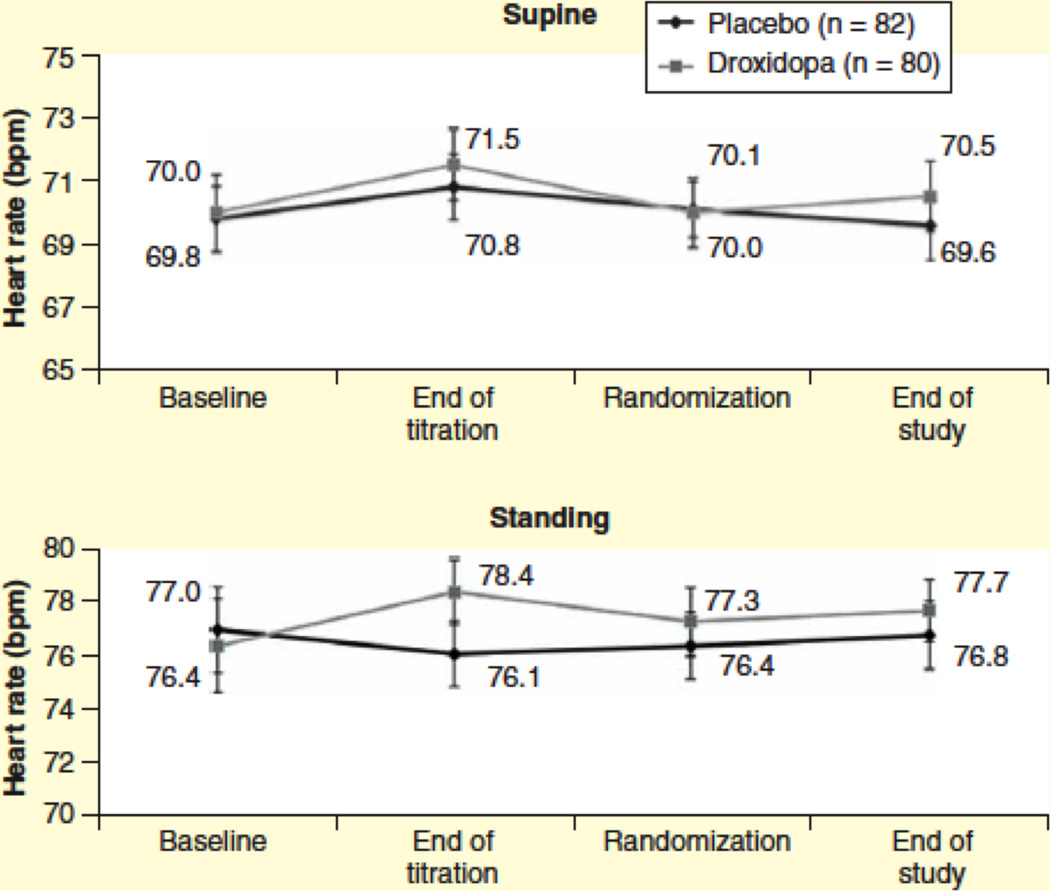
There were no statistically significant differences.
Three mechanisms of action have been proposed (Figure 8):
Central stimulator of sympathetic activity. Droxidopa crosses the blood brain barrier and is metabolized in the CNS to norepinephrine by the enzyme AAAD, raising the possibility that it may function as a central stimulator of sympathetic outflow [44,54]. This mechanism, however, is largely defunct, since the pressor effect of droxidopa is blocked by high doses of carbidopa [57], which inhibits AAAD in the periphery, but not in the brain.
Circulating hormone. AAAD is ubiquitously expressed in tissues, including the kidney, gastrointestinal tract and liver. Strong evidence indicates that norepinephrine production after droxidopa administration occursin non-neuronal tissues. Norepinephrine then exerts its effects as a circulating vasoconstrictor hormone. Indeed, patients with nOH and extensive degeneration of sympathetic neurons (such as those with PAF, PD or DLB) retain the ability to convert droxidopa into norepinephrine and increase their BP [58].
Peripheral sympathetic neurotransmitter. Droxidopa may convert into norepinephrine within sympathetic terminals and the newly synthesized norepinephrine is then released as a neurotransmitter upon neuronal activation. This mechanism of action has been shown in patients with DBH deficiency [47,48]. In these patients, sympathetic neurons lack DBH but are otherwise intact. The absence of DBH creates a blockage in the catecholaminergic pathways that prevents the formation of norepinephrine from dopamine, and sympathetic nerve terminals release dopamine instead of norepinephrine. Infusing tyramine, an agent that releases the catecholamine content from sympathetic neurons, raises circulating norepinephrine in normal subjects but only raises dopamine in patients with DBH deficiency [48]. The synthesis of norepinephrine from droxidopa bypasses the enzyme DBH; administering droxidopa, therefore, overcomes the enzymatic defect. After droxidopa administration in patients with DBH deficiency infusion of tyramine increases circulating norepinephrine. This is strong evidence indicating that norepinephrine is synthesized from droxidopa intraneuronaly, stored in vesicles and released by sympathetic terminals [47,48]. Additional evidence of intraneuronal conversion of droxidopa to norepinephrine comes from comparing plasma norepinephrine levels following droxidopa administration in patients with MSA and Lewy body disorders (PD, DLB and PAF). Volume distribution curves show that norepinephrine levels remains elevated for longer periods in patients with intact peripheral sympathetic neurons (MSA) compared to those with profound loss of sympathetic neurons (Lewy body disorders) [58]. This suggests that intraneuronal conversion of droxidopa and vesicular storage of norepinephrine within sympathetic terminals occurs in MSA and prolongs the biological effect. In patients with widespread loss of sympathetic terminals, the synthesis of norepinephrine from droxidopa mostly occurs in extra neuronal tissues [58] (Figure 9).
Figure 8. Possible mechanisms of actions of droxidopa.
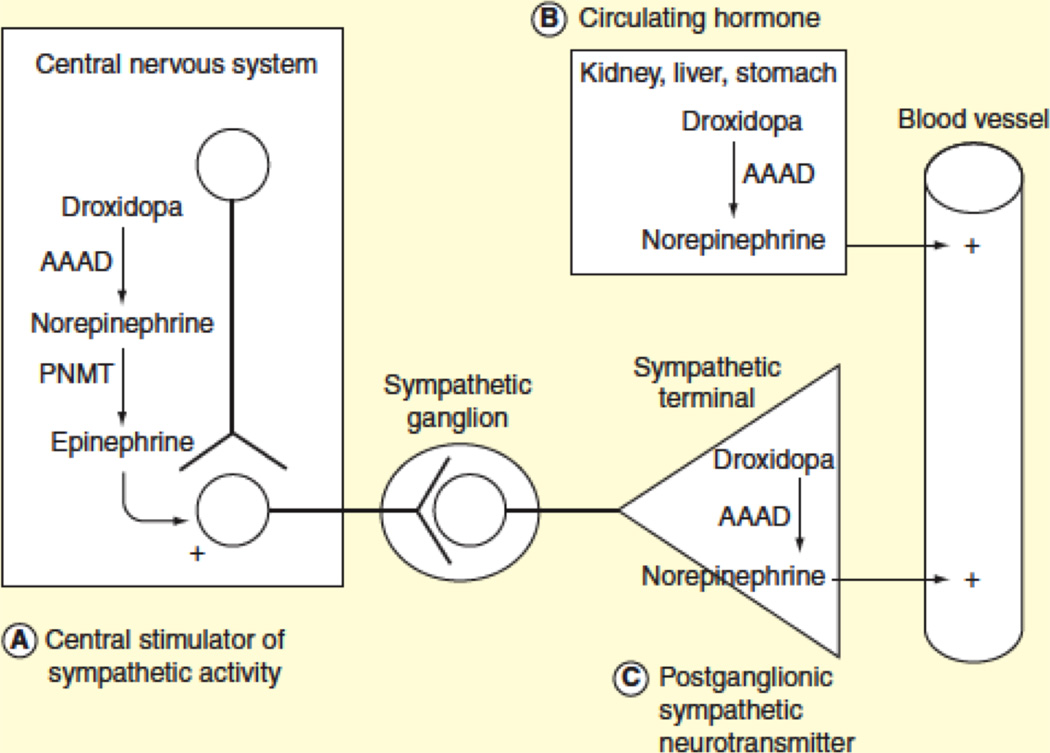
As central stimulator of sympathetic activity (A), circulating hormone (B) or peripheral (postganglionic) sympathetic neurotransmitter (C).
Figure 9. Different plasma norepinephrine (NE) responses to droxidopa (L-DOPS) despite similar L-DOPS and dihydroxyphenylglycol (DHPG) levels, in multiple system atrophy (MSA) and pure autonomic failure (PAF).

The increment in plasma NE after L-DOPS would be derived mainly from non-neuronal cells; however, in MSA, release of NE stored in sympathetic nerves would result in delayed decline of plasma NE. COMT = catechol-O-methyltransferase; LAAAD = L-aromatic-amino-acid decarboxylase; MAO = monoamine oxidase; NAAT = neutral amino acid transporter; NET = cell membrane nor- epinephrine transporter; U-2 =uptake-2 transporter; VMAT = vesicular monoamine transporter. Modified from: Goldstein DS, et al. Clin Auton Res 2004; 14(6): 363-8.
3.4 Pharmacokinetics and metabolism
3.4.1. Absorption
In preclinical studies droxidopa had a circulating half-time of 1–4 h, with a bioavailability of 90%, independent of age or gender. In humans, peak plasma concentrations (Cmax) are reached by 1–4 h post-dose (mean of approximately 3 h) and decline mono-exponentially with a half-time of 2 to 3 h. Norepinephrine and its neuronal metabolite DHPG also peak at about 3 hours but decline multi-exponentially with an initial half-time for the decline in plasma norepinephrine of about 9 h. Hence, after L-DOPS administration plasma levels of norepinephrine decline much more slowly [58]. Peak droxidopa and norepinephrine plasma concentrations coincide with the increase in BP. In rats with renal impairment, the area under the curve of circulating droxidopa levels was increased. High-fat meals have only moderate impact on droxidopa exposure with Cmax and area under the plasma concentration-time curve (AUC) decreasing by 35% and 20%, respectively. The Cmax are delayed by approximately 2 hours with a high-fat meal.
3.4.2. Distribution
After systemic administration of droxidopa, the highest tissue concentration occurs in the liver and kidney [59]. Preclinical and clinical studies measuring norepinephrine and its metabolites in plasma and cerebrospinal fluid (CSF) after droxidopa reveal that relatively little circulating droxidopa enters the CNS [60,61]. In humans, CSF levels of norepinephrine do increase, especially in patients concurrently treated with carbidopa [61,62]. Droxidopa exhibits plasma protein binding of 75% at 100 ng/mL and 26% at 10,000 ng/ml. The estimated apparent volume of distribution of droxidopa is about 200 L in humans.
3.4.3. Metabolism
The metabolism of droxidopa occurs within the catecholamine pathways. Droxidopa has three fates: a) conversion to its major metabolite 3-O-methyl-DOPS by catechol-O-methyl-transferase (COMT), b) conversion to protocatechualdehyde by DOPS aldolase and the desired path c) conversion to norepinephrine by AAAD. It is unknown if the metabolites 3-O-methyl-DOPS and protocatechualdehyde contribute to the pharmacological effect. Plasma droxidopa level decline mono-exponentially, with a half-time of 2–3 h while norepinephrine decline multi-exponentially with an initial half time for the decline in plasma of about 9 h. Plasma levels of norepinephrine remain elevated, which is notable given that the half-life of norepinephrine is 1–2 min, indicating that droxidopa is taken up by neuronal and non-neuronal tissues, and continues to be converted into norepinephrine, which is then released into the circulation [59].
3.4.4. Excretion
Droxidopa has a mean elimination half-life of 2–3 hours. The major route of elimination is renal. Studies in animals showed that ~75% of the radiolabeled dose is excreted in urine within 24 hours of oral dosing.
4. CLINICAL EFFICACY
4.1. Difficulties in clinical trials in nOH
Performing clinical trials in orphan diseases with adequate power is challenging. Assembling a large cohort of patients requires multiple treatment centers in several countries. An added difficulty is the “background noise” caused by wide BP variability of patients with nOH. Symptom severity can vary from day-to-day, throughout the day and are affected by meals, temperature, dehydration and physical activities. The patient-reported outcomes have limitations in patients with nOH and reliance on symptoms alone may not always be an accurate indicator of tissue hypoperfusion. Symptoms of nOH can be non-specific, including fatigue and difficultly concentrating and may mimic the levodopa “off” state in PD patients. Conversely, patients may have difficultly distinguishing symptoms of nOH from other causes of lightheadedness [8]. Furthermore, nOH is frequently associated with progressive neurodegenerative disorders; failure to demonstrate long-term improvement could conceivably be due to worsening of the underlying neurodegenerative disorder rather than failure of the active agent.
4.2. Phase II studies
Initial studies with droxidopa in patients with nOH were conducted with a racemic mixture of the D- and L-stereoisomers, and showed conflicting results [54]. In 1984, a placebo-controlled study using racemic DL-threo-DOPS failed to show improvement in 6 patients with nOH (due to diabetic autonomic neuropathy and PAF). Only 2% of the administered DL-DOPS was converted to biologically active L-norepinephrine [63]. A later study in patients with nOH showed that DL-DOPS did increase supine and upright BP as well as norepinephrine [64]. An early, small (n=6), open-label study performed in the US showed that droxidopa increased upright BP in patients with MSA, but less in PAF [65]. A subsequent, better-powered double blind, randomized, placebo-controlled study showed that a single dose of droxidopa (200 to 2,000 mg) effectively increased BP and improved symptoms in 19 patients with nOH due to MSA and PAF [57]. An open-label European study showed that droxidopa (100 to 300 mg three times daily) improved symptoms of nOH in patients with MSA and PAF after 6-weeks of treatment [66] (Table 2).
Table 2.
Phase II clinical studies with L-DOPS and DL-DOPS in patients with neurogenic OH
| Study | Subjects (N) | Active agent |
Design | Results |
|---|---|---|---|---|
| Hoeldtke et al, 1984 [63] | 6 (2 PAF and 4 autonomic diabetic neuropathy) | DL-DOPS | Open label crossover study using a single dose of DL-DOPS (600 or 800 mg) and single dose of placebo in separate days. | No change in supine or upright BP after DL-DOPS |
| Kaufmann et al, 1991 [65] | 6 (2 PAF; 4 MSA) | L-DOPS | Open label dose titration studyfollowed by 2 days of open label administration (700–1000 mg/day) | L-DOPS increased supine BP in MSA and PAF, and standing BP only in MSA. |
| Freeman et al, 1999 [64] | 10 (6 MSA; 4 PAF) | DL-DOPS | Randomized, double-blind, placebo-controlled, crossover study using a single dose of 1000 mg of DL-DOPS and single dose of placebo | DL-DOPS significantly increased supine and upright BP. |
| Mathias et al, 2001 [66] | 32 (26 MSA; 6 PAF) | L-DOPS | Open label incremental study (4 weeks) followed by a 6-week maintenance study (100–300 mg twice/day) | L-DOPS significantly reduced the fall in systolic BP upon standing and the symptoms of OH. |
| Kaufmann et al, 2003 [57] | 19 (11 MSA; 8 PAF) | L-DOPS | Single blind dose titration study followed by a 3-day double blind, placebo-controlled, crossover trial. | L-DOPS significantly increased supine and standing BP for several hours and improved symptoms of OH. After L-DOPS, BP increases were associated with increases in plasma NE levels. |
4.3 Phase III studies
4.3.1. Dose optimization and enrichment design
Because of the variable pressor response in patients with nOH, phase III trials had a dose optimization period with a forced upward titration from 100 to 600 mg three times daily. Dose titration lasted a maximum of 14 days with patients receiving escalating dosages in 100 mg three times daily-increments until a) becoming asymptomatic (with a score of 0 in the item 1 of the Orthostatic Hypotension Questionnaire [OHQ] [see below] [67]), b) developing sustained supine hypertension >180/110 mmHg, c) reaching the maximum dose of 600 mg three/times day; or d) patient complains of intolerable side effects. Responders were defined as patients that had at least a 1-point improvement in item 1 of the OHQ (dizziness/lightheadedness) in conjunction with a 10 mmHg or more increase in standing systolic BP.
4.3.2. End-points
The phase III clinical trials of droxidopa involved more than 600 patients with nOH. The primary end-point in each trial was a change in symptom severity score measured with the orthostatic hypotension questionnaire (OHQ). The OHQ is a self-reported, validated symptom assessment tool made up of two components [67]: a) the OH symptom assessment (OHSA) scale, with 6 separate items, and b) the OH daily activity scale (OHDAS) with 4 individual items. The questionnaire was developed specifically for patients with nOH. The OHQ uses a scale from 0 (no symptoms/no interference) to 10 (worst possible/complete interference) and asks the patient to rate their symptoms for the previous week. Each of the 10 individual items within the OHQ can be used individually or as a composite summing together the total score of all items (Figure 10). Both item 1 of the OHSA scale (symptoms of dizziness/lightheadedness), and the composite overall OHQ score were used as primary end-points in phase III clinical trials. Secondary outcomes were measurement of BP after 3 minutes of standing and clinicians’ ratings and patients’ self-ratings on the Clinical Global Impression (CGI) severity and improvement scales. The CGI severity is a 7-point scale scored from 1 (no symptoms) to 7 (severe symptoms), and the CGI improvement is a 7-point scale scored from 1 (very much improved) to 7 (very much worse).
Figure 10. Orthostatic Hypotension Questionnaire (OHQ).

The orthostatic hypotension questionnaire (OHQ) is a validated symptom assessment tool made up of two components: a) the OH symptom assessment (OHSA) scale, with 6 separate items, and b) the OH daily activity scale (OHDAS) with 4 individual items. The OHQ uses a scale from 0 (no symptoms/no interference) to 10 (worst possible/complete interference) and asks the patient to rate their symptoms for the previous week.
4.3.3. Results
4.3.3.1. Double-blind randomized placebo-controlled short term clinical studies
Study 301 was a double blind, randomized, placebo-controlled, parallel-group study conducted at 94 US, Canadian and European sites. The trial enrolled 162 patients with nOH due to PD, MSA, PAF or non-diabetic autonomic neuropathy [56]. Following an open-label dose titration phase (starting at 100mg and escalating to 600 mg three times/day), “responders” (162 patients, 62% of enrolled) proceeded to a washout period and were then randomized to receive placebo or droxidopa for 1-week (double-blind). Differences between groups were compared on day 7. Compared with placebo, patients randomized to droxidopa had better improvement in overall OHQ scores (p<0.003) and standing systolic BP that was 7 mmHg higher (p<0.001). Differences between groups were modest, partly because the placebo group failed to worsen back to their original state, suggesting a possible carry over effect of droxidopa requiring a longer washout period to disappear.
Study 302 was a double-blind, placebo-controlled, randomized withdrawal trial including 181 patients with nOH due to PD, MSA, PAF or non-diabetic autonomic neuropathy [68]. Study 302 began with an open label titration and 1 week treatment phase. After completing the open-label treatment phase, responders were randomized to withdraw to placebo or continue taking droxidopa. Differences between groups were assessed 2 weeks after randomization. The mean dose of droxidopa was 386±178 mg (three times/day), slightly lower than in study 301 (p>0.33). After withdrawal, there were no significant differences in symptoms of dizziness/lightheadedness (OHQ item 1), the primary outcome measure, between droxidopa and placebo patients. This was because placebo patients experienced continuing relief of the dizziness/lightheadedness score and standing SBP at the end of the study compared with baseline. This again raised the possibility of a carryover effect of droxidopa. Substantial carryover effects have been observed for levodopa, which like droxidopa undergoes decarboxylation to become a neurotransmitter. Alternatively, patients may have improved simply by their participation in the study, e.g., by better adherence to non-pharmacological treatment recommendations [68]. Post-hoc analysis of the overall composite score for OH symptoms and activities (OHQ composite), however, did show a significant improvement with droxidopa (p=0.026). Standing systolic BP was not different between the two treatment groups. Patients’ self-ratings of clinical global impression (CGI) of symptom severity showed a significant difference favoring droxidopa (p=0.008).
Study 306 was originally designed to evaluate the clinical efficacy of droxidopa over an 8-week double-blind period [69]. In a preplanned interim efficacy analysis, the initial 51 subjects (study nOH306A) [70] showed no significant difference across groups in the change in OHQ composite score, the trial primary endpoint. Exploratory analyses suggested efficacy for dizziness/lightheadedness score and led to a change in the trial’s primary efficacy measure while data for subsequent subjects remained blinded. The subsequent 171 enrolled patients formed study nOH306B, multi-center, double blind, randomized, placebo-controlled, parallel-group study restricted to patients with symptomatic nOH associated with PD and conducted exclusively in the US [69]. Subjects were randomized initially and underwent up to 2 weeks of double-blind titration with either droxidopa or placebo. Patients taking droxidopa had a significant improvement in symptoms of dizziness/lightheadedness (OHQ item 1) compared with placebo after 1 week of treatment (p = 0.018) [69]. At that time, patients taking droxidopa also had significantly higher systolic BPs in the standing position compared with placebo (p=0.032).
The pooled results of 306A and 306b (composite 306) showed that patients taking droxidopa had a significant improvement in symptoms of dizziness/lightheadedness (OHQ item 1) compared with placebo after 1 week of treatment (p < 0.01) [69] (Figure 11)
Figure 11. Changes of OHSA item 1 from baseline in study 306 overall.

** p< 0.01 versus placebo, ANCOVA. BL = baseline; SE = standard error. From: Hauser RA, et al. Mov Disord 2015; 30(5): 646–654.
4.3.3.2. Long term clinical studies
Long-term efficacy of droxidopa has not been documented. Study 303 was a long-term continuation of studies 301 and 302 and featured a withdrawal design. A total of 75 patients with nOH received droxidopa for a further 3 months and entered into a 2-week randomized withdrawal phase. This study did not show statistically significant difference between treatment arms, however it is important to note that this was an exploratory study not designed to generate statistical significance. Potential carryover of droxidopa may have influenced the results, as patients on placebo did not return to their baseline symptom score or blood pressure standing within the two-week withdrawal phase. Moreover, this was an exploratory study not powered to reach significance.
In Study 306 overall, differences in change in OHQ item 1 scores from baseline to maintenance weeks 2, 4, and 8 showed numerical trends favoring droxidopa that approached statistical significance (p=0.077 at week 8) [69].
Table 3 summarizes the results of these and other phase III clinical studies with droxidopa in patients with nOH.
Table 3.
Phase III clinical studies with droxidopa (L-DOPS) in patients with neurogenic OH
| Study | Subjects (N) | Design | Enrichment design | L-DOPS dosage | Length | Titration | Primary efficacy endpoints | Results |
|---|---|---|---|---|---|---|---|---|
| 301 [56] | 162 (PD, MSA, PAF, NDAN, DBHD) | Multicenter, multinational, double blind, randomized controlled trial parallel group induction design | Yes | 100–600 mg three times daily | Initial open label titration, 1-week washout, 7-day randomized placebo controlled phase | Open label – before randomization | OHQ composite score at 1 week | Compared with placebo, patients on droxidopa had a significant improvement in overall OHQ scores (p<0.003); and standing BP that was 7 mmHg higher (p<0.001) |
| 302 [68] | 101 (PD, MSA, PAF, NDAN, DBHD) | Multicenter, double blind, randomized controlled trial with withdrawal design | Yes | 100–600 mg three times daily | Initial open label titration, 7-day open label treatment, 2-week of double blind placebo controlled phase | Open label – before randomization | OHQ Item 1 (dizziness / lightheadedness) at 2 weeks | No differences in the OHQ item 1. Significant improvement of the overall OHQ composite score on droxidopa than placebo (p=0.013). Standing systolic BP was not different between the two groups. Patient self-reported symptom severity showed a significant improvement with droxidopa (p=0.008). |
| 306A [70] 306B [69] |
51 PD (306A) 171 PD (306 B) |
Multicenter, double blind, randomized controlled trial parallel group induction design | No | 100–600 mg three times daily | 2-week double-blind titration; 8 weeks double blind maintenance treatment | Double blind – after randomization | OHQ composite score at 8 weeks (306A) OHQ Item 1 at 1 week (306B) |
For the initial 51 subjects (Study 306A), the primary efficacy measure (OHQ composite score) did not show significant change versus placebo at week 8. For the subsequent 171 subjects (Study 306B) patients taking droxidopa had a significant improvement in symptoms of dizziness/lightheadedness (OHQ item 1) compared to placebo after 1 week (p = 0.018); and higher systolic BP standing (p=0.032). |
| 303 (Unpublished) | 75 (PD, MSA, PAF, NDAN, DBHD) | Multicenter, open-label extension to 301 and 302 studies | No | 100–600 mg three times daily | 3-month open label treatment; 2 week placebo-controlled withdrawal phase; after the controlled phase of the trial some patients continued on open-label droxidopa for up to 2 years | No | OHQ composite score | No significant differences between treatment arms. |
| 304 (Unpublished) | 255 (PD, PAF, NDAN, DBHD) | Long term, open-label safety extension to 301, 302 and 306 studies | No | 100–600 mg three times daily | Up to 1–2 years | No | Safety measures | N/A |
| 305 (Unpublished) |
18 (PD, PAF, NDAN, DBHD) | Multicenter, open-label extension to 301 and 304 studies | No | 100–600 mg three times daily | An off drug 24-hour ambulatory BP assessment was compared to a 4–6 week on-drug 24-hour ambulatory BP assessment | No | Mean 24-hour systolic and diastolic BP | Patients on droxidopa had a significantly higher mean 24-hour systolic (p=0.027) and diastolic (p=0.003) BP. |
4.3.3.3. Concomitant medications
Inhibition of AAAD with high dose carbidopa can abolish the pressor effect of droxidopa by preventing its peripheral conversion to norepinephrine. This was demonstrated in early phase II studies [57], with a 200 mg single dose of carbidopa administered concomitantly with droxidopa. In clinical practice, however, the carbidopa dose routinely given to patients with PD treated with L-DOPA is considerably smaller than that (25–50 mg three times/day) and does not appear to block the pressor effect of droxidopa. No dedicated drug-drug interaction studies have been performed for droxidopa, however. Post-hoc analysis of study 301 suggests a lesser pressor effect with carbidopa usage [56]. Subgroup analysis is hampered by most of the non-carbidopa users having no underlying movement disorder (PAF and non-diabetic autonomic neuropathies). Study 306 featured only patients with PD of which 90% were taking carbidopa/levodopa. Despite the use of carbidopa, a pressor effect was demonstrated in the short term, which was not sustained, and suggests a need to re-titrate the dose of droxidopa.
Dopamine agonists, amantadine derivatives, and MAO-B inhibitors did not an affect droxidopa. Norepinephrine reuptake inhibitors (e.g., venlafaxine) may potentiate the pressor effect of droxidopa and should be used with caution.
4.4. Post-marketing surveillance
During post-marketing surveillance in Japan, anecdotal cases of a symptom complex resembling malignant hyperthermia/neuroleptic malignant syndrome have been reported in patients with MSA [71,72] and one patient with progressive supranuclear palsy [73].
5. SAFETY AND TOLERABILITY
Clinical experience with droxidopa shows that it is safe and well tolerated. The most commonly observed adverse reactions associated with treatment were headache, dizziness, nausea, and hypertension. The most common adverse reactions leading to discontinuation of droxidopa were hypertension and nausea. In the long-term, open-label extension studies, a total of 422 patients, mean age 65 years, were treated with droxidopa for a mean total exposure of approximately one year. The commonly reported adverse events were falls (24%), urinary tract infections (15%), headache (13%), syncope (13%), and dizziness (10%). Post marketing studies are necessary to define the rate of adverse responses in clinical practice.
All available drugs that raise BP in the standing position also raise BP in the supine position, therefore increasing the risk of supine hypertension. Although there are no specific data on cardio- and cerebrovascular events induced by supine hypertension in patients with nOH, treating physicians should be aware of this potential side effect. Before beginning treatment with droxidopa, careful review of the patient’s medication is required as administering droxidopa in combination with other agents that increase BP (e.g., fludrocortisone, ephedrine, midodrine and triptans) would be expected to increase the risk of supine hypertension. Patients should be instructed to avoid the supine position during the day, to sleep with the head of the bed raised 30-degrees, and ensure that their final dose of droxidopa is taken at least 4-h before bed. Droxidopa should be reduced and, if necessary, discontinued if supine hypertension persists. Blood pressure should be rechecked supine at a 30-degree angle if increased doses are required. Safety in patients with blood pressure higher than 180 mmHg at a 30-degree angle has not been established as these patients were excluded from the trials.
The safety and efficacy of droxidopa in pediatric patients has not been established, although there was one study in Japan involving children (aged 11–15) with orthostatic intolerance that showed no side effects [74]. Patients with mild or moderate renal impairment (GFR greater than 30 mL/min) were included in clinical trials and did not have a higher frequency of adverse reactions.
No prolongation of the QTc interval was observed at single oral droxidopa doses up to 2,000 mg.
6. REGULATORY AFFAIRS
Based on the results of the phase III clinical trials, the Food and Drug Administration’s Cardiovascular and Renal Drugs Advisory Committee approved droxidopa (Northera®) under the accelerated approval program. Questions remain regarding the long-term durability of the clinical benefit, which was not shown beyond 2-weeks. The approval was granted under the condition that additional trials are performed to verify the drug’s long-term clinical benefit. Similarly to midodrine, the drug carries a “black box” warning, cautioning against the risks of supine hypertension and advising patients that they must sleep with the head and upper body elevated. Droxidopa is available in the US, Japan, China, South Korea, and Taiwan.
7. CONCLUSION
nOH is a disabling condition that affects patients with autonomic failure. It is the result of defective norepinephrine release from sympathetic vasomotor neurons upon standing. nOH is an independent risk factor for falls and overall mortality. Despite its importance, there is a paucity of treatment options for this condition. Midodrine was approved for the treatment of nOH in 1996. It took nearly two decades for a second drug, droxidopa, to be approved for the same indication. Droxidopa is a synthetic amino acid that is converted into norepinephrine by the enzyme aromatic aminoacid decarboxylase (AAAD or dopa-decarboxylase), the same enzyme that converts L-DOPA to dopamine. Droxidopa increases BP and thus can prevent cerebral perfusion pressure falling below the critical limit and inducing symptoms of cerebral hypoperfusion. In multicenter clinical trials, droxidopa has been shown to significantly improve specific symptoms of nOH including dizziness, vision disturbance, weakness, and fatigue. Clinical trials showed that droxidopa also improved patients’ abilities to perform activities of daily living that involved standing or walking. Efficacy was only shown for a 2-week period. The US Food and Drug Administration’s approval for clinical use in patients with nOH was granted in early 2014 with the understanding that further research be completed to show persistence of its beneficial effects during chronic use.
8. EXPERT COMMENTARY
The role of droxidopa for the treatment of nOH becomes immediately apparent when comparing it with L-DOPA. After almost six decades, L-DOPA is still the best treatment for overcoming dopaminergic deficits in PD [75]. But droxidopa, as L-DOPA, does have limitations: its therapeutic efficacy depends on a careful titration, and only patients with an appropriate diagnosis respond adequately. As some patients with parkinsonism do not respond to L-DOPA, some patients with nOH do not respond to droxidopa. The identification of the factors that predict response to droxidopa will certainly be a forthcoming area of research and should help us improve the therapeutic approach to patients with nOH.
Also, although in clinical trials droxidopa was taken orally three times daily, clinical experience (rather than clinical trials) indicate that the dosage should be tailored to each patient’s needs considering the periods of time when he/she is going to be active or inactive. For instance, a patient with MSA with reduced mobility that remains active for a few hours in the morning (when going to the restroom, cooking breakfast and going to physical therapy), it is reasonable to administer a single morning dose of droxidopa and skip the afternoon and evening dosages. In other patients it may be needed to administer droxidopa twice daily only, or to give a higher dose in the morning and then lower doses in the afternoon and evening, depending on the patient’s needs.
9. FIVE YEAR REVIEW
The FDA approved droxidopa under the accelerated approval program based on clinical data showing that the drug has a significant effect on an intermediate clinical measure (in this case, short-term relief of dizziness/lightheadedness) that is reasonably likely to predict the outcome of ultimate interest (relief of dizziness/lightheadedness during chronic treatment). The approval was contingent upon the company that commercializes droxidopa conducting post-approval clinical trials to verify the drug’s clinical benefit. Therefore, additional clinical trials to assess the long-term efficacy of droxidopa in patients with nOH will be conducted.
Droxidopa use may be expanded to include the treatment of nOH due to other causes of autonomic failure, such as diabetes mellitus [76], spinal cord injury [77,78], or hemodialysis [53,79] as well as in postprandial hypotension [80]. It will be interesting to compare droxidopa with midodrine, the only other FDA–approved drug for the treatment of nOH.
10. KEY ISSUES.
Neurogenic OH is a severely disabling disorder suffered by patients with autonomic failure due to defective norepinephrine release from sympathetic postganglionic terminals upon standing.
The FDA approved Droxidopa in 2014 for the treatment of orthostatic dizziness, lightheadedness or “feeling about to faint” in adult patients with symptomatic neurogenic OH associated with PD, MSA, PAF, dopamine β-hydroxylase deficiency and non diabetic autonomic neuropathy.
Droxidopa is an orally active synthetic amino acid that is converted to norepinephrine by the aromatic L-amino acid decarboxylase (dopa-decarboxylase), the same enzyme that converts L-DOPA into dopamine in the treatment of PD.
Phase III clinical trials showed that droxidopa treatment led to significant improvement in symptoms of neurogenic OH (dizziness, lightheadedness or feeling about to faint) with an associated increase in standing systolic BP; the effectiveness of droxidopa beyond 2 weeks has not been proven.
The most significant concern when using droxidopa is supine hypertension. To avoid it, droxidopa should be carefully titrated and patients should sleep in a semi-sitting position (30-degree elevation of head and torso).
Acknowledgments
Financial disclosures
Dr. Kaufmann serves as Editor in Chief for Clinical Autonomic Research; receives research support from the National Institutes of Health (U54NS065736 and 1U01NS078025-01) and the Dysautonomia Foundation, Inc. He has received compensation as a consultant/advisory board member for Lundbeck, Eli Lilly, Pfizer, and Astra Zeneca.
Dr. Norcliffe-Kaufmann receives research support from the National Institutes for Health (U54NS065736), the Dysautonomia Foundation, Inc and the MSA Coalition.
Dr. Palma receives research support from the Dysautonomia Foundation, Inc.; and has received compensation as a consultant/advisory board member for Lundbeck.
REFERENCES
* Of interest ** Of considerable interest
- 1.FDA. Definition of orphan disease and orphan products. 2012 (Ed.^(Eds)
- 2.Freeman R, Wieling W, Axelrod FB, et al. Consensus statement on the definition of orthostatic hypotension, neurally mediated syncope and the postural tachycardia syndrome. Clin. Auton. Res. 2011;21(2):69–72. doi: 10.1007/s10286-011-0119-5. [DOI] [PubMed] [Google Scholar]
- 3.Goldstein DS, Holmes CS, Dendi R, Bruce SR, Li ST. Orthostatic hypotension from sympathetic denervation in Parkinson's disease. Neurology. 2002;58(8):1247–1255. doi: 10.1212/wnl.58.8.1247. [DOI] [PubMed] [Google Scholar]
- 4.Kaufmann H, Biaggioni I. Autonomic failure in neurodegenerative disorders. Semin. Neurol. 2003;23(4):351–363. doi: 10.1055/s-2004-817719. [DOI] [PubMed] [Google Scholar]
- 5.Freeman R. Clinical practice. Neurogenic orthostatic hypotension. N. Engl. J. Med. 2008;358(6):615–624. doi: 10.1056/NEJMcp074189. [DOI] [PubMed] [Google Scholar]
- 6.Masaki KH, Schatz IJ, Burchfiel CM, et al. Orthostatic hypotension predicts mortality in elderly men: the Honolulu Heart Program. Circulation. 1998;98(21):2290–2295. doi: 10.1161/01.cir.98.21.2290. [DOI] [PubMed] [Google Scholar]
- 7.Berganzo K, Diez-Arrola B, Tijero B, et al. Nocturnal hypertension and dysautonomia in patients with Parkinson's disease: are they related? J. Neurol. 2013;260(7):1752–1756. doi: 10.1007/s00415-013-6859-5. [DOI] [PubMed] [Google Scholar]
- 8.Palma JA, Gomez-Esteban JC, Norcliffe-Kaufmann L, et al. Orthostatic Hypotension in Parkinson Disease: How Much You Fall or How Low You Go? Mov. Disord. 2015 doi: 10.1002/mds.26079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaufmann H. Consensus statement on the definition of orthostatic hypotension, pure autonomic failure and multiple system atrophy. Clin. Auton. Res. 1996;6(2):125–126. doi: 10.1007/BF02291236. [DOI] [PubMed] [Google Scholar]
- 10.Horowitz DR, Kaufmann H. Autoregulatory cerebral vasodilation occurs during orthostatic hypotension in patients with primary autonomic failure. Clin. Auton. Res. 2001;11(6):363–367. doi: 10.1007/BF02292768. [DOI] [PubMed] [Google Scholar]
- 11.Arnold AC, Biaggioni I. Management approaches to hypertension in autonomic failure. Curr. Opin. Nephrol. Hypertens. 2012;21(5):481–485. doi: 10.1097/MNH.0b013e328356c52f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Robertson D, Garland EM. Dopamine Beta-Hydroxylase Deficiency. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews(R) Seattle (WA): 1993. [Google Scholar]
- 13.Norcliffe-Kaufmann L, Axelrod F, Kaufmann H. Afferent baroreflex failure in familial dysautonomia. Neurology. 2010;75(21):1904–1911. doi: 10.1212/WNL.0b013e3181feb283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palma JA, Carmona-Abellan MM, Barriobero N, et al. Is cardiac function impaired in premotor Parkinson's disease? A retrospective cohort study. Mov. Disord. 2013;28(5):591–596. doi: 10.1002/mds.25431. [DOI] [PubMed] [Google Scholar]
- 15.Norcliffe-Kaufmann L, Kaufmann H. Is ambulatory blood pressure monitoring useful in patients with chronic autonomic failure? Clin. Auton. Res. 2014;24(4):189–192. doi: 10.1007/s10286-014-0229-y. [DOI] [PubMed] [Google Scholar]
- 16.Ejaz AA, Kazory A, Heinig ME. 24-hour blood pressure monitoring in the evaluation of supine hypertension and orthostatic hypotension. J. Clin. Hypertens. (Greenwich) 2007;9(12):952–955. doi: 10.1111/j.1524-6175.2007.07298.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shibao C, Lipsitz LA, Biaggioni I. ASH position paper: evaluation and treatment of orthostatic hypotension. J. Clin. Hypertens. (Greenwich) 2013;15(3):147–153. doi: 10.1111/jch.12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schroeder C, Jordan J, Kaufmann H. Management of neurogenic orthostatic hypotension in patients with autonomic failure. Drugs. 2013;73(12):1267–1279. doi: 10.1007/s40265-013-0097-0. [DOI] [PubMed] [Google Scholar]
- 19.Perera R, Isola L, Kaufmann H. Effect of recombinant erythropoietin on anemia and orthostatic hypotension in primary autonomic failure. Clin. Auton. Res. 1995;5(4):211–213. doi: 10.1007/BF01824009. [DOI] [PubMed] [Google Scholar]
- 20.Weinberg AD, Minaker KL. Dehydration. Evaluation and management in older adults. Council on Scientific Affairs, American Medical Association. JAMA. 1995;274(19):1552–1556. doi: 10.1001/jama.274.19.1552. [DOI] [PubMed] [Google Scholar]
- 21.Krediet CT, van Lieshout JJ, Bogert LW, Immink RV, Kim YS, Wieling W. Leg crossing improves orthostatic tolerance in healthy subjects: a placebo-controlled crossover study. Am. J. Physiol. Heart Circ. Physiol. 2006;291(4):H1768–H1772. doi: 10.1152/ajpheart.00287.2006. [DOI] [PubMed] [Google Scholar]
- 22.Wieling W, van Lieshout JJ, van Leeuwen AM. Physical manoeuvres that reduce postural hypotension in autonomic failure. In: Wieling W, editor. 2014. (Ed.^(Eds) [DOI] [PubMed] [Google Scholar]
- 23.May M, Jordan J. The osmopressor response to water drinking. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011;300(1):R40–R46. doi: 10.1152/ajpregu.00544.2010. [DOI] [PubMed] [Google Scholar]
- 24.McHugh J, Keller NR, Appalsamy M, et al. Portal osmopressor mechanism linked to transient receptor potential vanilloid 4 and blood pressure control. Hypertension. 2010;55(6):1438–1443. doi: 10.1161/HYPERTENSIONAHA.110.151860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MacLean AR, Allen EV. Orthostatic hypotension and orthostatic tachycardia - Treatment with the "head-up" bed. J. Am. Med. Assoc. 1940;115:2162–2167. [Google Scholar]
- 26.Diedrich A, Biaggioni I. Segmental orthostatic fluid shifts. Clin. Auton. Res. 2004;14(3):146–147. doi: 10.1007/s10286-004-0188-9. [DOI] [PubMed] [Google Scholar]
- 27.Smit AA, Wieling W, Fujimura J, et al. Use of lower abdominal compression to combat orthostatic hypotension in patients with autonomic dysfunction. Clin. Auton. Res. 2004;14(3):167–175. doi: 10.1007/s10286-004-0187-x. [DOI] [PubMed] [Google Scholar]
- 28.Kooner JS, Raimbach S, Watson L, Bannister R, Peart S, Mathias CJ. Relationship between splanchnic vasodilation and postprandial hypotension in patients with primary autonomic failure. J. Hypertens. Suppl. 1989;7(6):S40–S41. doi: 10.1097/00004872-198900076-00017. [DOI] [PubMed] [Google Scholar]
- 29.Berne C, Fagius J, Niklasson F. Sympathetic response to oral carbohydrate administration. Evidence from microelectrode nerve recordings. J. Clin. Invest. 1989;84(5):1403–1409. doi: 10.1172/JCI114313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jansen RW, Lipsitz LA. Postprandial hypotension: epidemiology, pathophysiology, and clinical management. Ann. Intern. Med. 1995;122(4):286–295. doi: 10.7326/0003-4819-122-4-199502150-00009. [DOI] [PubMed] [Google Scholar]
- 31.Chobanian AV, Volicer L, Tifft CP, Gavras H, Liang CS, Faxon D. Mineralocorticoid-induced hypertension in patients with orthostatic hypotension. N. Engl. J. Med. 1979;301(2):68–73. doi: 10.1056/NEJM197907123010202. [DOI] [PubMed] [Google Scholar]
- 32.Norcliffe-Kaufmann L, Axelrod FB, Kaufmann H. Developmental abnormalities, blood pressure variability and renal disease in Riley Day syndrome. J. Hum. Hypertens. 2013;27(1):51–55. doi: 10.1038/jhh.2011.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaufmann H, Brannan T, Krakoff L, Yahr MD, Mandeli J. Treatment of orthostatic hypotension due to autonomic failure with a peripheral alpha-adrenergic agonist (midodrine) Neurology. 1988;38(6):951–956. doi: 10.1212/wnl.38.6.951. [DOI] [PubMed] [Google Scholar]
- 34.Wright RA, Kaufmann HC, Perera R, et al. A double-blind, dose-response study of midodrine in neurogenic orthostatic hypotension. Neurology. 1998;51(1):120–124. doi: 10.1212/wnl.51.1.120. [DOI] [PubMed] [Google Scholar]
- 35.Ramirez CE, Okamoto LE, Arnold AC, et al. Efficacy of atomoxetine versus midodrine for the treatment of orthostatic hypotension in autonomic failure. Hypertension. 2014;64(6):1235–1240. doi: 10.1161/HYPERTENSIONAHA.114.04225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singer W, Sandroni P, Opfer-Gehrking TL, et al. Pyridostigmine treatment trial in neurogenic orthostatic hypotension. Arch. Neurol. 2006;63(4):513–518. doi: 10.1001/archneur.63.4.noc50340. [DOI] [PubMed] [Google Scholar]
- 37.Jordan J, Shannon JR, Biaggioni I, Norman R, Black BK, Robertson D. Contrasting actions of pressor agents in severe autonomic failure. Am. J. Med. 1998;105(2):116–124. doi: 10.1016/s0002-9343(98)00193-4. [DOI] [PubMed] [Google Scholar]
- 38.Hoeldtke RD, Israel BC. Treatment of orthostatic hypotension with octreotide. J. Clin. Endocrinol. Metab. 1989;68(6):1051–1059. doi: 10.1210/jcem-68-6-1051. [DOI] [PubMed] [Google Scholar]
- 39.Ooi WL, Hossain M, Lipsitz LA. The association between orthostatic hypotension and recurrent falls in nursing home residents. Am. J. Med. 2000;108(2):106–111. doi: 10.1016/s0002-9343(99)00425-8. [DOI] [PubMed] [Google Scholar]
- 40.Rosenmund KW, Dornsaft H. Über Oxy- und Hioxyphenylserin und die Muttersubstanz des Adrenalins. Ber. Dtsch. Chem. Ges. 1919;52:1734–1749. [Google Scholar]
- 41.Blaschko H, Holton P, Stanley GH. The decarboxylation of -3: 4-dihydroxyphenylserine (noradrenaline carboxylic acid) Br. J. Pharmacol. Chemother. 1948;3(4):315–319. doi: 10.1111/j.1476-5381.1948.tb00393.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beyer KH, Blaschko H, Burn JH, Langemann H. Enzymic formation of noradrenaline in mammalian tissue extracts. Nature. 1950;165(4206):926. doi: 10.1038/165926a0. [DOI] [PubMed] [Google Scholar]
- 43.Blaschko H, Burn JH, Langemann H. The formation of noradrenaline from dihydroxyphenylserine. Br. J. Pharmacol. Chemother. 1950;5(3):431–437. doi: 10.1111/j.1476-5381.1950.tb00593.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmiterlow CG. The formation in vivo of noradrenaline from 3:4-dihydroxyphenylserine (noradrenaline carboxylic acid) Br. J. Pharmacol. Chemother. 1951;6(1):127–134. doi: 10.1111/j.1476-5381.1951.tb00628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Araki H, Tanaka C, Fujiwara H, Nakamura M, Ohmura I. Pressor effect of L-threo-3,4-dihydroxyphenylserine in rats. J. Pharm. Pharmacol. 1981;33(12):772–777. doi: 10.1111/j.2042-7158.1981.tb13929.x. [DOI] [PubMed] [Google Scholar]
- 46.Suzuki T, Higa S, Sakoda S, et al. Orthostatic hypotension in familial amyloid polyneuropathy: treatment with DL-threo-3,4-dihydroxyphenylserine. Neurology. 1981;31(10):1323–1326. doi: 10.1212/wnl.31.10.1323. [DOI] [PubMed] [Google Scholar]
- 47.Biaggioni I, Robertson D. Endogenous restoration of noradrenaline by precursor therapy in dopamine-beta-hydroxylase deficiency. Lancet. 1987;2(8569):1170–1172. doi: 10.1016/s0140-6736(87)91317-1. [DOI] [PubMed] [Google Scholar]
- 48.Man in 't Veld AJ, Boomsma F, van den Meiracker AH, Schalekamp MA. Effect of unnatural noradrenaline precursor on sympathetic control and orthostatic hypotension in dopamine-beta-hydroxylase deficiency. Lancet. 1987;2(8569):1172–1175. doi: 10.1016/s0140-6736(87)91318-3. [DOI] [PubMed] [Google Scholar]
- 49.Birkmayer W, Birkmayer G, Lechner H, Riederer P. DL-3,4-threo-DOPS in Parkinson's disease: effects on orthostatic hypotension and dizziness. J. Neural Transm. 1983;58(3–4):305–313. doi: 10.1007/BF01252816. [DOI] [PubMed] [Google Scholar]
- 50.Sakoda S, Suzuki T, Higa S, et al. Treatment of orthostatic hypotension in Shy-Drager syndrome with DL-threo-3,4-dihydroxyphenylserine: a case report. Eur. Neurol. 1985;24(5):330–334. doi: 10.1159/000115820. [DOI] [PubMed] [Google Scholar]
- 51.Senda Y, Muto T, Matsuoka Y, Takahashi A, Sobue I. [Clinical effects of oral L-threo-3,4-dihydroxyphenylserine on orthostatic hypotension in patients with Shy-Drager syndrome] Rinsho Shinkeigaku. 1987;27(3):300–304. [PubMed] [Google Scholar]
- 52.Kachi T, Iwase S, Mano T, Saito M, Kunimoto M, Sobue I. Effect of L-threo-3,4-dihydroxyphenylserine on muscle sympathetic nerve activities in Shy-Drager syndrome. Neurology. 1988;38(7):1091–1094. doi: 10.1212/wnl.38.7.1091. [DOI] [PubMed] [Google Scholar]
- 53.Iida N, Tsubakihara Y, Shirai D, Imada A, Suzuki M. Treatment of dialysis-induced hypotension with L-threo-3,4-dihydroxyphenylserine. Nephrol. Dial. Transplant. 1994;9(8):1130–1135. doi: 10.1093/ndt/9.8.1130. [DOI] [PubMed] [Google Scholar]
- 54.Bartholini J, Constantinidis J, Puig M, Tissot R, Pletscher A. The stereoisomers of 3,4-dihydroxyphenylserine as precursors of norepinephrine. J. Pharmacol. Exp. Ther. 1975;193(2):523–532. [PubMed] [Google Scholar]
- 55.Inagaki C, Fujiwara H, Tanaka C. Inhibitory effect of (+)threo-3,4-dihydroxy-phenylserine (DOPS) on decarboxylation of (−)threo-dops. Jpn. J. Pharmacol. 1976;26(3):380–382. doi: 10.1254/jjp.26.380. [DOI] [PubMed] [Google Scholar]
- 56.Kaufmann H, Freeman R, Biaggioni I, et al. Droxidopa for neurogenic orthostatic hypotension: a randomized, placebo-controlled, phase 3 trial. Neurology. 2014;83(4):328–335. doi: 10.1212/WNL.0000000000000615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kaufmann H, Saadia D, Voustianiouk A, et al. Norepinephrine precursor therapy in neurogenic orthostatic hypotension. Circulation. 2003;108(6):724–728. doi: 10.1161/01.CIR.0000083721.49847.D7. [DOI] [PubMed] [Google Scholar]
- 58.Goldstein DS, Holmes C, Kaufmann H, Freeman R. Clinical pharmacokinetics of the norepinephrine precursor L-threo-DOPS in primary chronic autonomic failure. Clin. Auton. Res. 2004;14(6):363–368. doi: 10.1007/s10286-004-0221-z. [DOI] [PubMed] [Google Scholar]
- 59.Goldstein DS. L-Dihydroxyphenylserine (L-DOPS): a norepinephrine prodrug. Cardiovasc. Drug Rev. 2006;24(3–4):189–203. doi: 10.1111/j.1527-3466.2006.00189.x. [DOI] [PubMed] [Google Scholar]
- 60.Ishikawa Y, Kato Y, Murakami Y, Inoue T, Koshiyama H, Imura H. Effect of L-threo-3,4-dihydroxyphenylserine (L-DOPS) on catecholamine levels in plasma and cerebrospinal fluid (CSF) in anesthetized rats. Proc. Soc. Exp. Biol. Med. 1987;184(2):197–200. doi: 10.3181/00379727-184-42467. [DOI] [PubMed] [Google Scholar]
- 61.Tohgi H, Abe T, Takahashi S. The effects of L-threo-3,4-dihydroxyphenylserine on the total norepinephrine and dopamine concentrations in the cerebrospinal fluid and freezing gait in parkinsonian patients. J. Neural Transm. Park. Dis. Dement. Sect. 1993;5(1):27–34. doi: 10.1007/BF02260912. [DOI] [PubMed] [Google Scholar]
- 62.Tohgi H, Abe T, Takahashi S, Takahashi J, Ueno M, Nozaki Y. Effect of a synthetic norepinephrine precursor, L-threo-3,4- dihydroxyphenylserine on the total norepinephrine concentration in the cerebrospinal fluid of parkinsonian patients. Neurosci. Lett. 1990;116(1–2):194–197. doi: 10.1016/0304-3940(90)90409-3. [DOI] [PubMed] [Google Scholar]
- 63.Hoeldtke RD, Cilmi KM, Mattis-Graves K. DL-Threo-3,4-dihydroxyphenylserine does not exert a pressor effect in orthostatic hypotension. Clin. Pharmacol. Ther. 1984;36(3):302–306. doi: 10.1038/clpt.1984.179. [DOI] [PubMed] [Google Scholar]
- 64.Freeman R, Landsberg L, Young J. The treatment of neurogenic orthostatic hypotension with 3,4-DL-threo-dihydroxyphenylserine: a randomized, placebo-controlled, crossover trial. Neurology. 1999;53(9):2151–2157. doi: 10.1212/wnl.53.9.2151. [DOI] [PubMed] [Google Scholar]
- 65.Kaufmann H, Oribe E, Yahr MD. Differential effect of L-threo-3,4-dihydroxyphenylserine in pure autonomic failure and multiple system atrophy with autonomic failure. J. Neural Transm. Park. Dis. Dement. Sect. 1991;3(2):143–148. doi: 10.1007/BF02260889. [DOI] [PubMed] [Google Scholar]
- 66.Mathias CJ, Senard JM, Braune S, et al. L-threo-dihydroxyphenylserine (L-threo-DOPS; droxidopa) in the management of neurogenic orthostatic hypotension: a multi-national, multi-center, dose-ranging study in multiple system atrophy and pure autonomic failure. Clin. Auton. Res. 2001;11(4):235–242. doi: 10.1007/BF02298955. [DOI] [PubMed] [Google Scholar]
- 67.Kaufmann H, Malamut R, Norcliffe-Kaufmann L, Rosa K, Freeman R. The Orthostatic Hypotension Questionnaire (OHQ): validation of a novel symptom assessment scale. Clin. Auton. Res. 2012;22(2):79–90. doi: 10.1007/s10286-011-0146-2. [DOI] [PubMed] [Google Scholar]
- 68.Biaggioni I, Freeman R, Mathias CJ, et al. Randomized withdrawal study of patients with symptomatic neurogenic orthostatic hypotension responsive to droxidopa. Hypertension. 2015;65(1):101–107. doi: 10.1161/HYPERTENSIONAHA.114.04035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hauser RA, Isaacson S, Lisk JP, Hewitt LA, Rowse G. Droxidopa for the Short-Term Treatment of Symptomatic Neurogenic Orthostatic Hypotension in Parkinson's Disease (nOH306B) Mov. Disord. 2014 doi: 10.1002/mds.26086. [DOI] [PubMed] [Google Scholar]
- 70.Hauser RA, Hewitt LA, Isaacson S. Droxidopa in patients with neurogenic orthostatic hypotension associated with Parkinson's disease (NOH306A) J. Parkinsons Dis. 2014;4(1):57–65. doi: 10.3233/JPD-130259. [DOI] [PubMed] [Google Scholar]
- 71.Yamamoto K, Morita S, Ikeda S, Yanagisawa N. Hyperthermia in a Shy-Drager syndrome patient--pathophysiological effects of body temperature and L-DOPS on orthostatic hypotension. Rinsho Shinkeigaku. 1993;33(1):68–73. [PubMed] [Google Scholar]
- 72.Ueda M, Hamamoto M, Otsubo K, Miyazaki T, Terashi A. Monoamine imbalance of the central nervous system in a case of Shy-Drager syndrome with recurrent attacks of a neuroleptic malignant syndrome. Rinsho Shinkeigaku. 1996;36(5):696–698. [PubMed] [Google Scholar]
- 73.Yoshikawa H, Oda Y, Sakajiri K, et al. Pure akinesia manifested neuroleptic malignant syndrome: a clinical variant of progressive supranuclear palsy. Acta Neuropathol. 1997;93(3):306–309. doi: 10.1007/s004010050619. [DOI] [PubMed] [Google Scholar]
- 74.Tanaka H, Yamaguchi H, Mino M. The effects of the noradrenaline precursor, L-threo-3,4-dihydroxyphenylserine, in children with orthostatic intolerance. Clin. Auton. Res. 1996;6(4):189–193. doi: 10.1007/BF02291133. [DOI] [PubMed] [Google Scholar]
- 75.Kaufmann H. Could treatment with DOPS do for autonomic failure what DOPA did for Parkinson's disease? Neurology. 1996;47(6):1370–1371. doi: 10.1212/wnl.47.6.1370. [DOI] [PubMed] [Google Scholar]
- 76.Yoshikawa H, Kawai K, Inoue S, et al. A case of diabetic orthostatic hypotension effectively managed with L-dops (L-threo-3,4-dihydroxyphenylserine) Nihon Naika Gakkai Zasshi. 1987;76(11):1695–1700. doi: 10.2169/naika.76.1695. [DOI] [PubMed] [Google Scholar]
- 77.Wecht JM, Rosado-Rivera D, Weir JP, Ivan A, Yen C, Bauman WA. Hemodynamic effects of L-threo-3,4-dihydroxyphenylserine (Droxidopa) in hypotensive individuals with spinal cord injury. Arch. Phys. Med. Rehabil. 2013;94(10):2006–2012. doi: 10.1016/j.apmr.2013.03.028. [DOI] [PubMed] [Google Scholar]
- 78.Muneta S, Iwata T, Hiwada K, Murakami E, Sato Y, Imamura Y. Effect of L-threo-3, 4-dihydroxyphenylserine on orthostatic hypotension in a patient with spinal cord injury. Jpn. Circ. J. 1992;56(3):243–247. doi: 10.1253/jcj.56.243. [DOI] [PubMed] [Google Scholar]
- 79.Akizawa T, Koshikawa S, Iida N, et al. Clinical effects of L-threo-3,4-dihydroxyphenylserine on orthostatic hypotension in hemodialysis patients. Nephron. 2002;90(4):384–390. doi: 10.1159/000054725. [DOI] [PubMed] [Google Scholar]
- 80.Freeman R, Young J, Landsberg L, Lipsitz L. The treatment of postprandial hypotension in autonomic failure with 3,4-DL-threo-dihydroxyphenylserine. Neurology. 1996;47(6):1414–1420. doi: 10.1212/wnl.47.6.1414. [DOI] [PubMed] [Google Scholar]