

SUMMARY
Defective neutrophils in patients with chronic granulomatous disease (CGD) cause susceptibility to extracellular and intracellular infections. As microbes must first be ejected from intracellular niches to expose them to neutrophil attack, we hypothesized that inflammasomes detect certain CGD pathogens upstream of neutrophil killing. Here, we identified one such ubiquitous environmental bacterium, Chromobacterium violaceum, whose extreme virulence was fully counteracted by the NLRC4 inflammasome. Caspase-1 protected via two parallel pathways that eliminated intracellular replication niches. Pyroptosis was the primary bacterial clearance mechanism in the spleen, but both pyroptosis and interleukin-18 (IL-18)-driven natural killer (NK) cell responses were required for liver defense. NK cells cleared hepatocyte replication niches via perforin-dependent cytotoxicity, whereas interferon-γ was not required. These insights suggested a therapeutic approach; exogenous IL-18 restored perforin-dependent cytotoxicity during infection by the inflammasome-evasive bacterium Listeria monocytogenes. Therefore, inflammasomes can trigger complementary programmed cell death mechanisms, directing sterilizing immunity against intracellular bacterial pathogens.
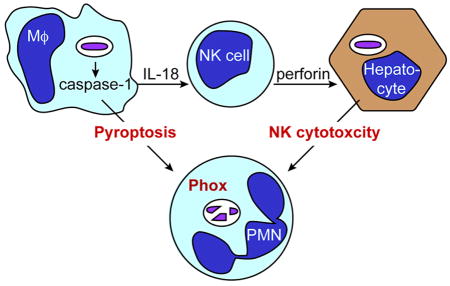
Graphical abstract
INTRODUCTION
Inflammasomes are innate immune sensors that detect cytosolic contaminations or perturbations. While canonical inflammasomes activate caspase-1, the noncanonical inflammasome pathway activates caspase-11. Caspase-1 processes interleukin-1β (IL-1β) and IL-18, leading to their secretion, and also triggers a form of programmed lytic cell death known as pyroptosis. The NLRC4 inflammasome responds to bacterial type III and IV secretion systems (T3SS and T4SS) by specifically detecting three conserved bacterial proteins when they are aberrantly translocated into the cytosol of host cells: flagellin, T3SS rod, and T3SS needle (Moltke et al. 2013). Like most inflammasomes, NLRC4 must signal though the adaptor protein ASC to promote IL-1β and IL-18 secretion. However, unlike the PYD-containing inflammasomes (including NLRP3, AIM2, and pyrin) it can also mediate pyroptosis directly via CARD-CARD domain interactions with caspase-1 (Moltke et al. 2013).
Inflammasome-deficient mice are more susceptible to a variety of pathogens, yet the defense conferred is typically only incremental. This was perhaps first and most thoroughly described for Salmonella typhimurium, which replicates somewhat faster in inflammasome-deficient mice. The overall effect on the infection is that inflammasome-competent mice die two days later than inflammasome-deficient mice (Lara-Tejero et al., 2006; Raupach et al., 2006). Normalization of death kinetics can be accomplished by only a 4-fold change in the infectious dose (Aachoui et al., 2013a; Moltke et al., 2013). Similar or lesser phenotypes are observed with Listeria monocytogenes, Yersinia pseudotuberculosis, Y. pestis, Francisella novicida, Mycobacterium tuberculosis, Klebsiella pneumoniae, Influenza, and Candida albicans (Allen et al., 2009; Fernandes-Alnemri et al., 2010; Gross et al., 2009; Jones et al., 2012; LaRock and Cookson, 2012; Lightfield et al., 2011; Mayer-Barber et al., 2010; Moltke et al., 2013; Sauer et al., 2011; Thomas et al., 2009; Tsuji et al., 2004; Willingham et al., 2009). This has led to the conclusion that inflammasomes merely slow the infectious process, allowing the host to survive until an adaptive immune response clears the infection.
Indeed, inflammasomes are ineffective in defense against systemic S. typhimurium and L. monocytogenes because these bacteria evade NLRC4, in part by repressing flagellin expression (Lara-Tejero et al., 2006; Moltke et al., 2013; Raupach et al., 2006). We previously engineered S. typhimurium to express flagellin during systemic infection and found that it is completely attenuated in wild-type (WT) mice but remains fully virulent in inflammasome-deficient mice. We showed that pyroptosis ejects bacteria from intracellular replication niches, exposing them to attack by neutrophils, which kill the bacteria via reactive oxygen species (ROS) (Miao et al., 2010a). The extreme efficiency by which the innate immune system eradicates evasion-deficient bacteria starkly contrasts with the typical role for inflammasomes in vivo as mechanisms to slow, but not halt, WT bacterial replication. We therefore hypothesized that there were naturally occurring environmental microbes that were analogous to engineered evasion-deficient bacteria. Such microbes would almost never cause disease in immune-competent individuals because inflammasome detection results in completely penetrant innate immunity.
Although caspase-1-deficient patients have not yet been identified, ROS-deficient patients are well studied. Chronic granulomatous disease (CGD) is caused by a variety of germline mutations in the genes for NADPH oxidase 2 (NOX2, encoded by Phox) that greatly reduce or eliminate ROS production in phagocytes (Holland, 2013). Patients with CGD are susceptible to infection by a characteristic array of not only extracellular, but also intracellular pathogens (Marciano et al., 2014). In order for neutrophils to attack intracellular bacteria, these microbes must first be ejected from their intracellular niche. Thus, we hypothesized that a subset of CGD-associated pathogens are detected by inflammasomes and released from intracellular niches via pyroptosis. Herein we identified one such bacterium, C. violaceum, and used it to reveal unappreciated inflammasome-driven tissue-specific clearance mechanisms.
RESULTS
Inflammasomes prevent lethal infection by specific CGD pathogens
We performed a survey of five CGD-specific bacteria in inflammasome-deficient mice. These were selected based on their inability to infect immunocompetent humans, and the bacterial species are further described in Extended Experimental Procedures.
Burkholderia thailandensis is a ubiquitous soil bacterium that uses a T3SS to invade the cytosol, where it is detected by caspase-11 (Aachoui et al., 2013a). Consistent with the hypothesis that caspase-11-dependent pyroptosis exposes intracellular bacteria to killing by neutrophils, mice with the most common form of autosomal recessive CGD (Ncf1−/−, which encodes p47phox) were also susceptible to B. thailandensis infection, surviving slightly longer than Casp1−/−Casp11−/−mice while WT mice were fully resistant (Figure 1A–B). In contrast, species within the related Burkholderia cepacia complex (B. cepacia, B. cenocepacia, B. multivorans) that do not encode the T3SS were efficiently cleared by WT and Casp1−/−Casp11−/−mice, but were highly virulent in Ncf1−/−mice (Figure 1C and S1A–D). A similar result was observed for the CGD-tropic pathogens Francisella philomiragia (Friis-Moller et al., 2004; Mailman and Schmidt, 2005) and Serratia marcescens (Lee and Lau, 2013; Marciano et al., 2014) (Figure 1D and S1C–F), illustrating that inflammasomes were not strictly required for defense against all CGD-tropic pathogens.
Fig. 1. Inflammasomes and the NADPH oxidase are required to clear specific CGD pathogens.

(A–D) Mice were infected IP with (A) 2×107 or (B) 1×104 cfu of B. thailandensis, (C) 1×106 cfu of B. cepcacia, (D) or 1×106 cfu of F. philomiragia and monitored for survival. Casp1−/−Casp11−/−mice referred to as Casp1-11DKO. Mice: (A) WT=9, Casp1-11DKO=10, Ncf1−/−=10; (B) WT=4, Casp1-11DKO=6, Ncf1−/−=9; (C) n=4 for all; (D) n=5 for all.
(E) Mice were infected IP with C. violaceum (Cv) at the indicated doses and monitored for survival. Mice: WT=4, Casp1-11DKO=7, Ncf1−/−=4.
(F–I) Mice were infected IP with 100 cfu of Cv and bacterial burdens were measured in the liver and spleen (F–G) 72 hours later or (H–I) 17 hours later.
(J) Mice were infected IP with 100 cfu of Cv and 72 hours later livers and spleens were embedded in paraffin and H&E stained.
Data and statistical analyses are representative of at least two experiments (A–D, H–I), three experiments (F–G), or pooled from two experiments (E). Dashed line: limit of detection. * p ≤ 0.05; survival curve analysis (A–E), two-tailed unpaired t-test (F–I). See also Supp Figure 1.
Chromobacterium violaceum is a Gram-negative bacterium that derives its name from the purple pigment that it produces, called violacin (Durán et al., 2010). C. violaceum is a ubiquitous environmental organism that inhabits aquatic environments in the tropics and subtropics (Lima-Bittencourt et al., 2010). It encodes two T3SSs, Cpi-1 and Cpi-2, with similarity to the S. typhimurium SPI-1 and SPI-2 T3SS, respectively (Brito et al., 2004; Miki et al., 2010). Although the natural host that C. violaceum infects is unknown, the Cpi-1 needle and rod proteins can be detected by the mammalian NLRC4 inflammasome in vitro (Yang et al., 2013; Zhao et al., 2011). Most instances of C. violaceum infection in humans occur in immune compromised individuals, with 8.5% of cases occurring in confirmed CGD patients (Yang and Li, 2011). C. violaceum infection in CGD patients is typified by rapid bacteremia often without a nidus of infection, and a 53% mortality rate (Yang and Li, 2011). This is experimentally demonstrated in mice, where C. violaceum has minimal virulence for WT C57BL/6 mice, while it is highly virulent in Ncf1−/−mice (Segal et al., 2003) (Figure 1E). We found that like Ncf1−/−mice, Casp1−/−Casp11−/−mice were acutely susceptible to C. violaceum infection. As few as 100 CFUs were lethal to Casp1−/−Casp11−/−mice, while WT mice survived a challenge of 1,000,000 CFUs (Figure 1E). This susceptibility was mirrored by high bacterial burdens in both the liver and spleen in comparison to WT mice, which cleared the infection (Figure 1F–I and S1G–J). The exquisite sensitivity of Casp1−/−Casp11−/−mice to C. violaceum represents the strongest effect of inflammasomes in defense against pathogens in the published literature, equaled only by their susceptibility to B. thailandensis (Aachoui et al., 2015).
C. violaceum is liver-tropic in CGD patients (Yang and Li, 2011). Upon examination of mice 3 days post infection with 100 CFU of C. violaceum, we observed numerous macroscopic lesions on the livers of Casp1−/−Casp11−/−mice (Figure S1K), associated with extensive neutrophil infiltration and a complete loss of structure around lesion foci in the liver (Figure 1J). These results revealed that certain environmental microbes, such as C. violaceum and B. thailandensis, had significant virulence potential that could not be contained in the absence of a coordinated action between inflammasomes and phagocyte-specific NADPH oxidase.
NLRC4 is required for protection against infection by C. violaceum in vivo
The T3SS of C. violaceum can be detected by NLRC4 in vitro (Zhao et al., 2011), but the role of NLRC4 during in vivo infection of C. violaceum has not been examined. Therefore, we next set out to determine whether the canonical inflammasome pathway (activating caspase-1) or the noncanonical inflammasome pathway (caspase-11) was more important for defense against C. violaceum. We found that caspase-11 was dispensable for defense, whereas Nlrc4−/−Asc−/−mice (which lack all canonical inflammasomes except NLRP1a/1b) remained highly susceptible to infection (Figure 2A). ASC is completely required for signaling by the PYD-containing inflammasomes, and for the cytokine outputs from NLRC4 (Wen et al., 2013). However, ASC is dispensable for NLRC4-driven pyroptosis (Miao et al., 2010a). While ASC-deficient mice had an intermediate susceptibility, NLRC4 was absolutely required for resistance to infection (Figure 2B–F and S2A). These results suggest a role for pyroptosis and IL-1β and/or IL-18 in defense against C. violaceum.
Fig. 2. Canonical inflammasome signaling via NLRC4 prevents lethal infection by C. violaceum in vivo.

(A–B) Mice were infected IP with 100 cfu of Cv and monitored for survival. Nlrc4−/−Asc−/−mice referred to as Nlrc4-AscDKO. Mice: (A) Casp1-11DKO=4, Nlrc4-AscDKO =4, Casp11−/−=5; (B) n=4 for all.
(C–F) Mice were infected IP with 100 cfu of Cv, and bacterial burdens were measured in livers and spleens 72 hours later.
All data and statistical analyses are representative of at least three experiments. * p ≤ 0.05; survival curve analysis (A–B) two-tailed unpaired t-test (C–F). See also Supp Figure 2.
Differential role for pyroptosis against C. violaceum in the spleen and liver
We next compared caspase-deficient mice to Il1b−/−Il18−/−mice to better understand the contributions of pyroptosis and cytokine secretion. Similar to the Asc−/−mice, Il1b−/−Il18−/−mice had an intermediate phenotype (Figure 3A–B), again consistent with roles for both pyroptosis and cytokines. Interestingly, we observed a different phenotype in the spleen, where Il1b−/−Il18−/−mice had few or no bacteria, like WT mice, while Casp1−/−Casp11−/−mice carried significant burdens (Figure 3C). This suggested that pyroptosis was the primary effector mechanism in the spleen, but that a more complex defense mechanism existed in the liver.
Fig. 3. Differential role for pyroptosis in the liver and spleen.

(A and D) Mice were infected with Cv at the indicated doses and monitored for survival. Il1b−/−Il18−/−mice referred to as Il1b-Il18DKO. Mice: (A) Casp1-11DKO=5, Il1b-Il18DKO =7, Il1b−/−=5; (D) Il1b-Il18DKO =5, Il1b−/−=5, Il18−/−=4, Asc−/−=5.
(B–C and E–F) Mice were infected IP with 100 cfu of Cv and bacterial burdens were measured in livers and spleens 72 hours later.
Data and statistical analyses are representative of at least two experiment (A and D–F) or three experiments (B–C), or pooled from two experiments (E–F). * p ≤ 0.05; survival curve analysis (A and D), two-tailed unpaired t-test (B–C and E–F). * over a line indicates that all data sets under the line have p ≤ 0.05 for each pair wise comparison possible. See also Supp Figure 3.
IL-18 is protective in the liver whereas IL-1β is dispensable
IL-1β and IL-18 are pleiotropic cytokines that promote innate and adaptive immune responses. IL-1β is well known to induce fever, and to potentiate local inflammatory responses, resulting in neutrophil influx (Dinarello, 2010). IL-18 was first described as an interferon-γ (IFN-γ) stimulating cytokine, and IFN-γ responses are well established to promote defense against intracellular pathogens (Souza-Fonseca-Guimaraes et al., 2012). In order to study the role of these cytokines in a more penetrant survival challenge, we increased the infectious dose to 104 CFUs, resulting in uniform lethality in Il1b−/−Il18−/−mice. At this dose, Il18−/−and Asc−/−mice were also highly susceptible to infection, while Il1b−/−mice were fully resistant (Figure 3D). In support of this, Asc−/−, Il18−/−, and Il1b−/−Il18−/−mice all had elevated bacterial burdens in the liver, but not spleen, in comparison to WT and Il1b−/−mice (Figure 3E–F and S3A–B). These results further supported our hypothesis that pyroptosis was the primary defense mechanism against C. violaceum in the spleen, and revealed that IL-18, but not IL-1β, was protective in the liver.
Natural killer cell cytotoxicity is required to clear intracellular replication niches
In considering IL-18 responsive cell types, we found that NK cells have higher IL-18R expression than any other cell type or organ (Figure S4A). Accordingly, depletion of NK and NKT cells from WT mice resulted in elevated bacterial burden in the liver, but not the spleen (Figure 4A–B). As IL-18 secretion drives IFN-γ production in natural killer (NK) cells and certain T cells (Chaix et al., 2008), and because IFN-γ is well established as a potent cytokine in defense against intracellular pathogens (Adib-Conquy et al., 2013; Le-Barillec et al., 2005; Souza-Fonseca-Guimaraes et al., 2012), we hypothesized that IFN-γ would be the primary effector mechanism downstream of IL-18. Surprisingly, this was not the case, as Ifng−/−mice had bacterial burdens comparable to those seen in WT mice (Figure 4C–D).
Fig. 4. Cytotoxic activity of NK cells is required for clearance of C. violaceum.

(A–D and K–L) Mice were infected IP with Cv at the indicated doses and bacterial burdens were measured in livers and spleens 72 hours later. (A–B) Mice were injected IP with 75 μg of isotype or anti-NK1.1 antibody at 1 and 3 days prior to infection.
(E) Primary hepatocytes and splenocytes from naïve WT mice were treated with LPS, lysed, and examined for caspase-1 via Western blot.
(F) Mice were infected IP with 100 cfu of Cv and bacterial burdens were measured in livers 72 hours later. Equal weight liver sections were removed and the hepatocyte fraction was graphed as a percentage of the total burden by weight.
(G) Primary hepatocytes were infected with Cv at an MOI of 25 and percentage of invasive bacteria was determined via gentamycin protection assay
(H) Primary hepatocytes, liver immune cells, and splenocytes were infected with Cv at an MOI of 10, treated with gentamycin, and LDH release was determined (note, different splenocytes have a distinct LDH content per cell, thus in this case LDH release does not directly translate to percent lysis).
(I–J) Mice were infected IP with 103 cfu of L. monocytogenes (Lm) and bacterial burdens were measured in livers and spleens 72 hours later.
Data and statistical analyses are representative of two experiments (C–D and I–J) or pooled from two (K–L) or three experiments (A–B). * p ≤ 0.05; two-tailed unpaired t-test (A–D and I–L). See also Supp Figure 4.
We then considered whether the predominance of IL-18 for liver but not splenic defense was in part due to the differences in cell populations between the liver and spleen. The spleen is primarily composed of macrophages, T cells, B cells, and dendritic cells. In contrast, 80–90% of the liver is composed of hepatocytes, with the remainder composed of Kupffer cells (liver resident macrophages), NK cells, and other immune cells (Grégoire et al., 2007). Thus, when considering target cells that C. violaceum may infect, the liver is dramatically different from the spleen. Interestingly, primary hepatocytes did not express detectable amounts of caspase-1 after LPS stimulation (Figure 4E). In contrast, splenocytes expressed abundant caspase-1 (Figure 4E), as do liver mononuclear cells (Petrasek et al., 2012). This may explain why pyroptosis appears to be sufficient to defend the spleen, but not the liver. Indeed, hepatocytes contained 90% of the C. violaceum bacterial burden in the liver (Figure 4F), and C. violaceum also efficiently infected primary hepatocytes in vitro (Figure 4G). However, consistent with the lack of caspase-1 expression, we did not observe appreciable lytic cell death in infected primary hepatocytes (Figure 4H and S4D). In contrast, rapid and robust caspase-1/11-dependent lytic cell death was observed in liver immune cells and splenocytes (Figure 4H). Consequently, we hypothesized that a cell death pathway parallel to pyroptosis was required to eject the bacteria from their replicative niche in hepatocytes, and expose them to neutrophil killing.
Previous studies show that exposure to IL-18 can prime the cytotoxic effects of NK and NKT cells in addition to promoting IFN-γ production (Dao et al., 1998; Son et al., 2001). These cytotoxic effects require perforin, a pore-forming protein that NK/NKT and cytotoxic T lymphocytes (CTLs) use to kill target cells (Kägi et al., 1994; Lopez et al., 2013). However, while NK and/or NKT cell cytotoxic activity is a well-established antiviral effector mechanism (Shabani et al., 2014), it has not been established as an important defense mechanism against intracellular bacteria. For example, WT and Prf1−/−mice had similar bacterial burdens after infection with the model intracellular pathogens L. monocytogenes or S. typhimurium (Figure 4I–J and S4B–C). However, interpretation of these results were confounded by the fact that both L. monocytogenes and S. typhimurium largely evade NLRC4 in vivo (Miao et al., 2010a; Sauer et al., 2011), thereby minimizing IL-18 secretion. Nevertheless, we examined whether perforin has a role in defense against C. violaceum, which was readily detected by NLRC4 in vivo. Both Prf1−/− and Casp1−/−Casp11−/−mice had significantly higher bacterial burdens than WT and Ifng−/−mice in the liver, but again, only Casp1−/−Casp11−/−mice had substantial bacterial burdens in the spleen (Figure 4K–L and S4E–F). These results indicated that NK and/or NKT cells were protective in the liver but were dispensable for splenic clearance (where pyroptosis was sufficient), and suggested that their cytotoxic activity as the primary protective mechanism.
The adaptive immune system is dispensable for defense against C. violaceum
Several cell types express perforin, including NK cells, NKT cells, and CTLs (Kägi et al., 1994; Lopez et al., 2013). To determine whether NKT cells and/or CTLs, were required for resistance to C. violaceum, we examined Rag1−/−mice, which lack all B and T cells. Interestingly, Rag1−/−and WT mice had similar bacterial burdens and clearance kinetics (Figure 5A–B), demonstrating that neither NKT cells nor CTLs were the primary cytotoxic effector cells against C. violaceum infection. Additionally, we concluded that inflammasomes provided innate immunity against this ubiquitous environmental microbe that did not require an adaptive immune response.
Fig. 5. Adaptive immune responses are not required for sterilizing immunity against C. violaceum.

(A–B) Mice were infected IP with 103 cfu of Cv and bacterial burdens were measured in livers and spleens at the indicated times.
Data and statistical analyses are representative of two experiments. * p ≤0.05; two-tailed unpaired t-test.
IL-18 administration improves NK cell recruitment and cytotoxicity in vivo
We hypothesized that a lack of IL-18 in inflammasome-deficient mice prevented appropriate recruitment and/or stimulation of NK cells, so we examined whether administration of exogenous IL-18 could restore NK cell function. IL-18 therapy resulted in significantly reduced bacterial burdens in Casp1−/−Casp11−/−and Il1b−/−Il18−/−mice (Figure 6A–B). However, because exogenous IL-18 did not fully normalize bacterial burdens in Il1b−/−Il18−/−mice, it was likely that IL-18 therapy could be further optimized from a pharmacologic standpoint. To examine the response of NK cells to IL-18, we performed flow cytometry on liver immune cells isolated from untreated and IL-18-treated Nlrc4−/−Asc−/−mice. IL-18 therapy promoted a three-fold increase in conventional NK cell numbers, but no increase of liver resident NK cells and NK1.1+CD3+ cell (likely NKT cells) numbers (Figure 6C–D and S5A). Granzyme B is a serine protease that is secreted from NK cells and CTLs along with perforin to mediate target cell death, and is an established marker for cyotolytic activation of these cell types. Notably, we saw increased granzyme B mean fluorescence intensity (MFI) in conventional NK cells, but no significant increase in the MFI of liver resident NK or NKT cells (Figure 6E and S5B). These results were consistent with the lack of phenotype in Rag1−/−mice (Figure 5), and suggested that IL-18 was driving both NK cell recruitment to infected target organs and increased activation.
Fig. 6. Exogenous IL-18 recruits NK cells and primes their cytotoxic effects.

(A–G) Mice were infected IP with 100 cfu of Cv and (A–B and F–G) bacterial burdens were measured in livers and spleens 72 hours later. (A–B and F) Mice were injected IP with PBS or recombinant mouse IL-18 (0.2 μg/dose at 0, 24, and 48 hours). (F) Mice were injected IP with 75 μg of isotype or anti-NK1.1 antibody at 1 and 3 days prior to infection. (C–E) Liver immune cells were collected and prepared for flow cytometry 1 day post infection. Markers for conventional NK (NK1.1hiDX5+), liver resident NK (NK1.1hi, DX5 ), and (likely) NKT (NK1.1+DX5 CD3+) cell populations were determined in addition to granzyme B staining.
(H) Hepatocytes were harvested from Ncf1−/−mice infected with 100 Cv 17 hpi. Liver immune cells were harvested from WT mice infected with 104 Cv under 3 conditions: isotype antibody treatment, anti-NK1.1 antibody treatment, or IL-18 therapy. Cells were coincubated in a 96 well plate and hepatocyte killing was determined by LDH assay 6 hours later. A model of the experimental setup is provided.
Data and statistical analyses are representative of at least two experiments (A–E, F), or pooled from two experiments (F).
* p ≤ 0.05; two-tailed unpaired t-test (A–B and D–H). See also Supp Figure 5.
Our model thus far suggested that pyroptosis was the primary defense mechanism in the spleen, while IL-18 drove defense in the liver. Thus, the observation that IL-18 treatment was therapeutic in the spleen of Casp1−/−Casp11−/−mice was unexpected (Figure 6B). However, Casp1−/−Casp11−/−mice are deficient for both pyroptosis and IL-18 in the spleen, therefore, pyroptosis and NK cytotoxicity could be independently sufficient to prevent bacterial colonization in the spleen. Unfortunately, pyroptosis-deficient mice have not been developed, so we cannot directly assess whether endogenous IL-18 would be sufficient in the absence of pyroptosis.
We next sought to provide evidence that IL-18 therapy acts through NK cells. Consistent with our findings, Nlrc4−/−Asc−/−mice were highly susceptible to C. violaceum infection, and bacterial clearance could be rescued by IL-18 therapy (Figure 6F). However, when simultaneously depleted of NK cells, IL-18 therapy failed. Further, IL-18 therapy also required cytotoxic activity, as Prf1−/−mice failed to respond (Figure 6G). Therefore, IL-18 acted upstream of NK cell cytotoxic activity against intracellular bacterial infection in vivo.
In order to demonstrate that C. violaceum infected hepatocytes can be killed by NK cells, we combined ex vivo and in vitro techniques. Hepatocytes were isolated from C. violaceum-infected Ncf1−/−mice, which carried huge bacterial burdens in their liver. These were used as target cells for lysis by liver immune cells harvested from infected WT mice (Figure 6H). We observed hepatocyte killing that was ablated by NK cell depletion. Furthermore, hepatocyte killing increased with cells from IL-18 treated mice (Figure 6H). In summation, NK cells killed hepatocytes in vitro, and this killing was enhanced with IL-18 therapy, consistent with our in vivo results (Figure 6).
Exogenous IL-18 is therapeutic against L. monocytogenes
Our observations revealed a unique link between inflammasomes and NK cell cytotoxicity in defense against a ubiquitous intracellular environmental bacterium, so we sought to discover if this mechanism was applicable to a bona fide human pathogen. Like C. violaceum, L. monocytogenes is well established to infect hepatocytes (Cousens and Wing, 2000). However, unlike C. violaceum, L. monocytogenes evades inflammasome detection. Therefore, we examined the efficacy of IL-18 treatment during L. monocytogenes infection. Bacterial burdens were significantly decreased in the liver and spleen of IL-18 treated WT mice (Figure 7A–B), but there was no difference between untreated and treated Prf1−/−mice in the liver (Figure 7A). Splenic burdens were decreased in Prf1−/−mice upon IL-18 therapy (Figure 7B), suggesting that there were also perforin-independent mechanisms of clearing the spleen, perhaps through IFN-γ. Importantly, despite the increased susceptibility of Ifng−/−mice to L. monocytogenes infection, IL-18 therapy was still beneficial to these mice (Figure 7C–D). Moreover, IL-18 therapy was still effective in Rag−/−mice, indicating that IL-18 acted independently of the adaptive immune system, as it did during C. violaceum infection (Figure 7E–F). Thus, perforin-dependent cytotoxicity against intracellular bacteria was a bona fide defense mechanism downstream of IL-18 secretion that was underappreciated due to the ability of many bacteria, such as L. monocytogenes, to evade inflammasome detection in vivo.
Fig. 7. L. monocytogenes burdens are reduced upon IL-18 administration.

(A–F) Mice were infected IP with 103 cfu of Lm and injected IP with PBS or recombinant mouse IL-18 (24, 48 and 72 hours), and bacterial burdens were measured in livers and spleens 4 days later. Data and statistical analyses are pooled from two experiments (A–F). * p ≤ 0.05; two-tailed unpaired t-test (A–F).
DISCUSSION
While inflammasomes are appreciated to be an innate immune mechanism that slows the course of infection, prevailing published evidence indicates that inflammasomes ultimately fail to eradicate pathogens. However, studies using bacteria engineered to remove inflammasome evasion hint at the converse hypothesis: that inflammasomes have the unrealized capacity of directing sterilizing immunity (Miao et al., 2010a; 2010b; Sauer et al., 2011). We propose that the importance of inflammasomes during infection is underappreciated because phenotypes have been interpreted in the face of confounding pathogenic evasion strategies. We further propose that animals are constantly exposed to a milieu of highly virulent environmental microbes that have remained uncharacterized because inflammasomes completely prevent disease in immunocompetent individuals. It is important to note that under our model, inflammasomes would be unimportant against environmental microbes that do not encode such potent virulence traits.
Concordantly, C. violaceum and B. thailandensis are ubiquitous water and soil bacteria that rarely cause infection in humans, despite the fact that they encode potent virulence traits, including T3SSs that enable intracellular replication or cytosolic invasion (Wiersinga et al., 2006; Yang and Li, 2011). Our results herein show that inflammasomes detect these virulence traits, and initiate a specific innate immune defense program that provides sterilizing innate immunity. To our knowledge, the role of inflammasomes in defense against C. violaceum (shown here) and B. thailandensis (Aachoui et al., 2015) are the most penetrant inflammasome phenotype against any infection, be it bacterial, viral, fungal, or parasitic. We hypothesize that this is one example of a wide array of potentially lethal environmental microbes. Exposure to such microbes might exert a strong evolutionary pressure that could explain the maintenance and expansion of inflammasome genes. We propose that if people lacked NLRC4, swimming in freshwater lakes or streams might carry an incredible risk of death subsequent to infection by C. violaceum.
Why should we be interested in studying bacteria to which almost all people are innately immune? The answer lies in the fact that these bacteria offer us the opportunity to study the innate immune system in its fully functional state, unobscured by pathogenic evasion strategies. Our studies of C. violaceum reveal the importance of inflammasomes in driving parallel mechanisms to clear intracellular replication niches. Pyroptosis appears to be fully competent to protect the spleen, where inflammasome-expressing macrophages are abundant. This likely occurs in liver macrophages as well. It should be noted that we have not provided definitive evidence that pyroptosis is the mechanism of clearance in the spleen; our data address 4 of 7 criteria that we have previously described (Aachoui et al., 2013b). A rapid pyroptotic response in the early hours of infection is thus predicted to eliminate the bacteria before they can replicate to high numbers. In addition to pyroptosis, inflammasome-driven IL-18 primes NK cells to kill infected hepatocytes in the liver, providing an example where NK cell cytotoxicity is critical to bacterial clearance in vivo.
The two primary effector mechanisms used by NK cells are production of IFN-γ and cytotoxicity. NK cell cytotoxicity, a well-established mechanism against viral pathogens, is not established in defense against bacterial pathogens in vivo (Souza-Fonseca-Guimaraes et al., 2012). There are numerous studies that demonstrate NK cell cytotoxicity in vitro against bacterially infected cells (Katz et al., 1990; Souza-Fonseca-Guimaraes et al., 2012), yet these responses were later found to be ineffective during infection in vivo (Junqueira-Kipnis et al., 2003; Souza-Fonseca-Guimaraes et al., 2012), likely because of pathogenic evasion strategies. We identified cytotoxicity, instead of IFN-γ, as the NK effector mechanism employed against C. violaceum infected hepatocytes in vivo. L. monocytogenes is also highly tropic for hepatocytes, yet NK cell cytotoxicity fails to clear this infection (Cousens and Wing, 2000). We propose that this is due to inflammasome evasion strategies employed by L. monocytogenes in vivo, whereby IL-18 secretion is limited (Sauer et al., 2011). Here we used IL-18 therapy as an innate immune booster to harness NK cytotoxicity and specifically eliminate L. monocytogenes-infected cells in the innate phase of infection.
Therefore, when we study bona fide pathogens, it is difficult to distinguish between innate immune mechanisms that are simply not useful from those that are actively evaded. The latter could be harnessed therapeutically to combat the infection, and environmental bacteria like C. violaceum and B. thailandensis may be the key to uncovering such insights. We used the well-studied susceptibility of CGD patients to identify extremely virulent bacteria that are normally cleared by the innate immune system with exquisite efficiency. It is likely that other environmental microbes, in a similar fashion, will inform our understanding of previously underappreciated innate immune clearance mechanisms. These microbes provide a lens with which to understand the capabilities of the innate immune system when it is correctly activated, acting as powerful tools to help develop therapeutics that bypass the evasion strategies of bona fide pathogens.
Experimental Procedures
Bacterial strains and growth conditions
The following strains were used in this work: C. violaceum (ATCC 12472), S. typhimurium (CS401), L. monocytogenes (10403s; kind gift from D.A.P.), and B. thailandensis (passaged through Casp1−/−Casp11−/−mouse strain). Additionally, the following strains were isolated from CGD patients (by S.M.H.): B. cepacia, B. multivorans, B. cenocepacia, F. philomiragia, and S. marcescens.
S. typhimurium and all Burkholderia species were grown in Luria-Bertani medium (LB). C. violaceum, S. marcescens, and L. monocytogenes were grown in Brain heart infusion (BHI). F. philomiragia was grown in BHI + isovitalex. All strains were grown overnight at 37°C and back-diluted (1:40) for 2 hours in their respective media for use in both in vitro and in vivo experiments.
Mouse infections
Mouse strain information and housing conditions are described in the Extended Experimental Procedures. For all mouse infections, 8–12 week old mice were infected with the designated colony forming units (Cfu) of log phase bacteria in PBS by intraperitoneal (IP) injection. Livers and spleens were harvested at the indicated time point (3, 4, 10, or 21 days post infection), homogenized and dilutions plated on LB or BHI as described above. For NK cell depletion, mice were injected IP with 75 μg anti-NK1.1 PK136 or isotype control C1.18.4 (BioXCell) at -3 and -1 dpi. Depletion of NK1.1-positive cells was confirmed by flow cytometry. For IL-18 therapies, mice were injected IP with 0.2 μg recombinant mouse IL-18 (rmIL-18; MBL) at the time of infection, 24, and 48 hpi (C. violaceum infections), or 24, 48, and 72 hpi (L. monocytogenes infections).
Histology
Livers and spleens were fixed in formalin and embedded in paraffin to generate section of 5 μm thickness. Paraffin sections were counterstained with hemotoxylin and eosin.
Cell isolation
A detailed protocol is in the Extended Experimental Procedures. In brief, livers were extracted, perfused with Collagenase D, mashed through a cell strainer (Falcon), and washed with DMEM. Cells were spun and the supernatant removed. Pellets were resuspended in 44% Percoll (GE Healthcare) in DMEM and under laid with 66% Percoll. Cells were spun at 2,000 x g and the hepatocytes were harvested from the upper layer. Lymphocytes were treated with RBC lysis buffer, spun, the supernatant removed, and the pellet resuspended in DMEM or 1X PBS depending on usage. Splenocytes were harvested in the same manner but without the Percoll gradient. Average hepatocyte purity as assessed by CD45 staining was determined to be 99.5%. Average liver immune cell purity as assessed visually by hemacytometer counting was determined to be >99.5%.
Western blots
For Western blots, total protein from lysates of 5 × 104 hepatocytes or splenocytes was analyzed. Caspase-1 expression was determined using an anti-caspase-1 antibody (clone 4B4, Genentech). Blots were stripped and equivalent loading of protein was ensured by Western blot using anti-β-actin HRP antibody (Cat. # 20272, AbCam) diluted 1:20,000.
Bacterial count enumeration
For percentages of intracellular C. violaceum in vivo, equal weights of livers were harvested. Total cfu were obtained from one section, cfu specific to the hepatocyte fraction from the second section via the cell isolation technique, and the hepatocyte cfu graphed as a percentage of the total. For percentages of intracellular C. violaceum in vitro, hepatocytes were isolated, seeded in a 96 well plate, and allowed to rest for 2 hours. C. violaceum were grown to log phase and hepatocytes were infected at an MOI of 25, with Gentamicin (5 ng/ml) added 30 min p.i. At 1, 2, 3, and 4 hours post infection, intracellular bacterial burden was determined by plating cell lysates on BHI.
Flow cytometry
For flow preparation, cells were Fc-blocked with anti-CD16/CD32 and stained with anti-CD45-PercPCy5.5 (104), NK1.1-FITC (PK136), CD3, Granzyme B, and the cell viability marker Aqua dead live-AmCyan, all from BD. Cells were fixed in 2% PFA and samples were analyzed on a Becton Dickinson LRSIII (HTS) (UNC Flow Cytometry Core Facility).
Assessment of NK killing in vitro
Hepatocytes were harvested from Ncf1−/−mice 17 hpi with 100 Cv. Liver immune cells were harvested from WT mice under 3 conditions 48 hpi. (1–2) WT mice were treated with isotype or anti-NK1.1 antibody days 1 and 3 prior to infection with 104 Cv. (3) WT mice were infected with 104 Cv and treated with 0.2 μg rm IL-18 at the time of infection and 24 hours later. Hepatocytes and immune cells were seeded into 96 well plates and allowed to rest for 2 hours. Non-adherent immune cells were used to enrich for NK cells. The hepatocytes and immune cells were then coincubated at an effector: target ratio (E:T) of 50:1 and cell death was evaluated 6 hours later. LDH release was defined as (LDHEXPERIMENTAL – LDHEFFECTORS – LDHSPONTANEOUS)/(LDHMAXIMAL – LDHSPONTANEOUS), where LDHEFFECTOR refers to the liver immune cells, LDHSPONTANEOUS refers to hepatocytes, and LDHMAXIMAL refers to total lysis of hepatocytes by Triton X-100.
Statistics
Error bars represent the standard deviation of technical replicates. A standard two-tailed unpaired t-test (Prism 5; GraphPad) was used for statistical analysis, wherein P values of ≤ 0.05 were considered significant.
Supplementary Material
Acknowledgments
We wish to thank Vishva Dixit, Richard Flavell, Shizuo Akira, and David Chaplin for sharing mice. We also thank Dat Mao, Davis Trihn, and Taylor Atherton for upkeep of our mouse colony. This work was supported by the following NIH grants: AI097518, AI057141, and AI119073 (E.A.M), AI097518-02S1 (V.I.M), R56AI110682 and R01AI074862 (J.K.W.), and T32 AI7273-27 (K.D.C.). The UNC Flow Cytometry Core Facility is supported in part by an NCI Center Core Support Grant (P30CA016086) to the UNC Lineberger Comprehensive Cancer Center.
Footnotes
AUTHOR CONTRIBUTIONS
V.I.M., A.L.T. and E.A.M. conceived the work. V.I.M., A.L.T., K.D.C., Y.A., J.K.W. and E.A.M. planned the experiments and analyzed the data. V.I.M., A.L.T., K.D.C. and Y.A. performed experiments. E.L.F. and S.M.H. provided bacterial strains and clinically relevant expertise. V.I.M. and E.A.M. wrote the paper. All authors helped to edit the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aachoui Y, Kajiwara Y, Leaf IA, Mao D, Ting JPY, Coers J, Aderem A, Buxbaum JD, Miao EA. Canonical Inflammasomes Drive IFN-γ to Prime Caspase-11 in Defense against a Cytosol-Invasive Bacterium. Cell Host Microbe. 2015 doi: 10.1016/j.chom.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, Tan MH, Cotter PA, Vance RE, Aderem A, et al. Caspase-11 protects against bacteria that escape the vacuole. Science. 2013a;339:975–978. doi: 10.1126/science.1230751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aachoui Y, Sagulenko V, Miao EA, Stacey KJ. Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Current Opinion in Microbiology. 2013b:1–8. doi: 10.1016/j.mib.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adib-Conquy M, Scott-Algara D, Cavaillon JM, Souza-Fonseca-Guimaraes F. TLR-mediated activation of NK cells and their role in bacterial/viral immune responses in mammals. Immunol Cell Biol. 2013;92:256–262. doi: 10.1038/icb.2013.99. [DOI] [PubMed] [Google Scholar]
- Allen IC, Scull MA, Moore CB, Holl EK, McElvania-TeKippe E, Taxman DJ, Guthrie EH, Pickles RJ, Ting JPY. The NLRP3 Inflammasome MediatesIn Vivo Innate Immunity to Influenza A Virus through Recognition of Viral RNA. Immunity. 2009;30:556–565. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito CFA, de Carvalho CB, Santos F, Gazzinelli RT, Oliveira SC, Azevedo V, Teixeira SMR. Chromobacterium violaceum genome: molecular mechanisms associated with pathogenicity. Genet Mol Res. 2004;3:148–161. [PubMed] [Google Scholar]
- Chaix J, Tessmer MS, Hoebe K, Fuseri N, Ryffel B, Dalod M, Alexopoulou L, Beutler B, Brossay L, Vivier E, et al. Cutting Edge: Priming of NK Cells by IL-18. The Journal of Immunology. 2008;181:1627–1631. doi: 10.4049/jimmunol.181.3.1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousens LP, Wing EJ. Innate defenses in the liver during Listeria infection. Immunol Rev. 2000;174:150–159. doi: 10.1034/j.1600-0528.2002.017407.x. [DOI] [PubMed] [Google Scholar]
- Dao T, Mehal WZ, Crispe IN. IL-18 augments perforin-dependent cytotoxicity of liver NK-T cells. J Immunol. 1998;161:2217–2222. [PubMed] [Google Scholar]
- Dinarello CA. IL-1: Discoveries, controversies and future directions. Eur J Immunol. 2010;40:599–606. doi: 10.1002/eji.201040319. [DOI] [PubMed] [Google Scholar]
- Durán M, Faljoni-Alario A, Durán N. Chromobacterium violaceum and its important metabolites--review. Folia Microbiol (Praha) 2010;55:535–547. doi: 10.1007/s12223-010-0088-4. [DOI] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Yu J-W, Juliana C, Solorzano L, Kang S, Wu J, Datta P, McCormick M, Huang L, Mcdermott E, et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol. 2010;11:385–393. doi: 10.1038/ni.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friis-Moller A, Lemming LE, Valerius NH, Bruun B. Problems in Identification of Francisella philomiragia Associated with Fatal Bacteremia in a Patient with Chronic Granulomatous Disease. Journal of Clinical Microbiology. 2004;42:1840–1842. doi: 10.1128/JCM.42.4.1840-1842.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grégoire C, Chasson L, Luci C, Tomasello E, Geissmann F, Vivier E, Walzer T. The trafficking of natural killer cells. Immunol Rev. 2007;220:169–182. doi: 10.1111/j.1600-065X.2007.00563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross O, Poeck H, Bscheider M, Dostert C, Hannesschläger N, Endres S, Hartmann G, Tardivel A, Schweighoffer E, Tybulewicz V, et al. Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature. 2009;459:433–436. doi: 10.1038/nature07965. [DOI] [PubMed] [Google Scholar]
- Holland SM. Chronic Granulomatous Disease. Hematology/Oncology Clinics of North America. 2013;27:89–99. doi: 10.1016/j.hoc.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CL, Napier BA, Sampson TR, Llewellyn AC, Schroeder MR, Weiss DS. Subversion of Host Recognition and Defense Systems by Francisella spp. Microbiology and Molecular Biology Reviews. 2012;76:383–404. doi: 10.1128/MMBR.05027-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junqueira-Kipnis AP, Kipnis A, Jamieson A, Juarrero MG, Diefenbach A, Raulet DH, Turner J, Orme IM. NK Cells Respond to Pulmonary Infection with Mycobacterium tuberculosis, but Play a Minimal Role in Protection. The Journal of Immunology. 2003;171:6039–6045. doi: 10.4049/jimmunol.171.11.6039. [DOI] [PubMed] [Google Scholar]
- Katz P, Yeager H, Whalen G, Evans M, Swartz RP, Roecklein J. Natural killer cell-mediated lysis of Mycobacterium-avium complex-infected monocytes. J Clin Immunol. 1990;10:71–77. doi: 10.1007/BF00917500. [DOI] [PubMed] [Google Scholar]
- Kägi D, Ledermann B, Bürki K, Seiler P, Odermatt B, Olsen KJ, Podack ER, Zinkernagel RM, Hengartner H. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature. 1994;369:31–37. doi: 10.1038/369031a0. [DOI] [PubMed] [Google Scholar]
- Lara-Tejero M, Sutterwala FS, Ogura Y, Grant EP, Bertin J, Coyle AJ, Flavell RA, Galán JE. Role of the caspase-1 inflammasome in Salmonella typhimurium pathogenesis. J Exp Med. 2006;203:1407–1412. doi: 10.1084/jem.20060206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRock CN, Cookson BT. The Yersinia Virulence Effector YopM Binds Caspase-1 to Arrest Inflammasome Assembly and Processing. Cell Host Microbe. 2012;12:799–805. doi: 10.1016/j.chom.2012.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le-Barillec K, Magalhaes JG, Corcuff E, Thuizat A, Sansonetti PJ, Phalipon A, Di Santo JP. Roles for T and NK cells in the innate immune response to Shigella flexneri. J Immunol. 2005;175:1735–1740. doi: 10.4049/jimmunol.175.3.1735. [DOI] [PubMed] [Google Scholar]
- Lee PP-W, Lau Y-L. Endemic infections in Southeast Asia provide new insights to the phenotypic spectrum of primary immunodeficiency disorders. Asian Pac J Allergy Immunol. 2013;31:217–226. [PubMed] [Google Scholar]
- Lightfield KL, Persson J, Trinidad NJ, Brubaker SW, Kofoed EM, Sauer JD, Dunipace EA, Warren SE, Miao EA, Vance RE. Differential Requirements for NAIP5 in Activation of the NLRC4 Inflammasome. Infect Immun. 2011;79:1606–1614. doi: 10.1128/IAI.01187-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima-Bittencourt CI, Costa PS, Hollatz C, Raposeiras R, Santos FR, Chartone-Souza E, Nascimento AMA. Comparative biogeography of Chromobacterium from the neotropics. Antonie Van Leeuwenhoek. 2010;99:355–370. doi: 10.1007/s10482-010-9501-x. [DOI] [PubMed] [Google Scholar]
- Lopez JA, Susanto O, Jenkins MR, Lukoyanova N, Sutton VR, Law RHP, Johnston A, Bird CH, Bird PI, Whisstock JC, et al. Perforin forms transient pores on the target cell plasma membrane to facilitate rapid access of granzymes during killer cell attack. Blood. 2013;121:2659–2668. doi: 10.1182/blood-2012-07-446146. [DOI] [PubMed] [Google Scholar]
- Mailman TL, Schmidt MH. Francisella philomiragia adenitis and pulmonary nodules in a child with chronic granulomatous disease. Can J Infect Dis Med Microbiol. 2005;16:245–248. doi: 10.1155/2005/486417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciano BE, Spalding C, Fitzgerald A, Mann D, Brown T, Osgood S, Yockey L, Darnell DN, Barnhart L, Daub J, et al. Common Severe Infections in Chronic Granulomatous Disease. Clinical Infectious Diseases. 2014 doi: 10.1093/cid/ciu1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer-Barber KD, Barber DL, Shenderov K, White SD, Wilson MS, Cheever A, Kugler D, Hieny S, Caspar P, Nunez G, et al. Cutting Edge: Caspase-1 Independent IL-1 Production Is Critical for Host Resistance to Mycobacterium tuberculosis and Does Not Require TLR Signaling In Vivo. The Journal of Immunology. 2010;184:3326–3330. doi: 10.4049/jimmunol.0904189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010a;11:1136–1142. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, Leaf IA, Aderem A. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proceedings of the National Academy of Sciences. 2010b;107:3076–3080. doi: 10.1073/pnas.0913087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki T, Iguchi M, Akiba K, Hosono M, Sobue T, Danbara H, Okada N. Chromobacterium pathogenicity island 1 type III secretion system is a major virulence determinant for Chromobacterium violaceum-induced cell death in hepatocytes. Molecular Microbiology no no. 2010 doi: 10.1111/j.1365-2958.2010.07248.x. [DOI] [PubMed] [Google Scholar]
- von Moltke J, Ayres JS, Kofoed EM, Chavarría-Smith J, Vance RE. Recognition of Bacteria by Inflammasomes. Annu Rev Immunol. 2013;31:73–106. doi: 10.1146/annurev-immunol-032712-095944. [DOI] [PubMed] [Google Scholar]
- Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, Barrieau M, Min S-Y, Kurt-Jones EA, Szabo G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest. 2012;122:3476–3489. doi: 10.1172/JCI60777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raupach B, Peuschel SK, Monack DM, Zychlinsky A. Caspase-1-Mediated Activation of Interleukin-1 (IL-1 ) and IL-18 Contributes to Innate Immune Defenses against Salmonella enterica Serovar Typhimurium Infection. Infect Immun. 2006;74:4922–4926. doi: 10.1128/IAI.00417-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer JD, Pereyre S, Archer KA, Burke TP, Hanson B, Lauer P, Portnoy DA. Listeria monocytogenes engineered to activate the Nlrc4 inflammasome are severely attenuated and are poor inducers of protective immunity. Proceedings of the National Academy of Sciences. 2011;108:12419–12424. doi: 10.1073/pnas.1019041108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal BH, Ding L, Holland SM. Phagocyte NADPH Oxidase, but Not Inducible Nitric Oxide Synthase, Is Essential for Early Control of Burkholderia cepacia and Chromobacterium violaceum Infection in Mice. Infect Immun. 2003;71:205–210. doi: 10.1128/IAI.71.1.205-210.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabani Z, Bagheri M, Zare-Bidaki M, Hassanshahi G, Arababadi MK, Mohammadi Nejad M, Kennedy D. NK cells in hepatitis B virus infection: a potent target for immunotherapy. Arch Virol. 2014 doi: 10.1007/s00705-013-1965-3. [DOI] [PubMed] [Google Scholar]
- Son YI, Dallal RM, Mailliard RB, Egawa S, Jonak ZL, Lotze MT. Interleukin-18 (IL-18) synergizes with IL-2 to enhance cytotoxicity, interferon-gamma production, and expansion of natural killer cells. Cancer Res. 2001;61:884–888. [PubMed] [Google Scholar]
- Souza-Fonseca-Guimaraes F, Adib-Conquy M, Cavaillon J-M. Natural killer (NK) cells in antibacterial innate immunity: angels or devils? Mol Med. 2012;18:270–285. doi: 10.2119/molmed.2011.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PG, Dash P, Aldridge JR, Jr, Ellebedy AH, Reynolds C, Funk AJ, Martin WJ, Lamkanfi M, Webby RJ, Boyd KL, et al. The Intracellular Sensor NLRP3 MediatesKey Innate and Healing Responses to Influenza A Virus via the Regulation of Caspase-1. Immunity. 2009;30:566–575. doi: 10.1016/j.immuni.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji NM, Tsutsui H, Seki E, Kuida K, Okamura H, Nakanishi K, Flavell RA. Roles of caspase-1 in Listeria infection in mice. International Immunology. 2004;16:335–343. doi: 10.1093/intimm/dxh041. [DOI] [PubMed] [Google Scholar]
- Wen H, Miao EA, Ting JPY. Mechanisms of NOD-like receptor-associated inflammasome activation. Immunity. 2013;39:432–441. doi: 10.1016/j.immuni.2013.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiersinga WJ, van der Poll T, White NJ, Day NP, Peacock SJ. Melioidosis: insights into the pathogenicity of Burkholderia pseudomallei. Nat Rev Micro. 2006;4:272–282. doi: 10.1038/nrmicro1385. [DOI] [PubMed] [Google Scholar]
- Willingham SB, Allen IC, Bergstralh DT, Brickey WJ, Huang MTH, Taxman DJ, Duncan JA, Ting JPY. NLRP3 (NALP3, Cryopyrin) Facilitates In Vivo Caspase-1 Activation, Necrosis, and HMGB1 Release via Inflammasome-Dependent and -Independent Pathways. The Journal of Immunology. 2009;183:2008–2015. doi: 10.4049/jimmunol.0900138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CH, Li YH. Chromobacterium violaceum infection: A clinical review of an important but neglected infection. Journal of the Chinese Medical Association. 2011;74:435–441. doi: 10.1016/j.jcma.2011.08.013. [DOI] [PubMed] [Google Scholar]
- Yang J, Zhao Y, Shi J, Shao F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proceedings of the National Academy of Sciences. 2013;110:14408–14413. doi: 10.1073/pnas.1306376110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature. 2011;477:596–600. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.