

Abstract
Interleukin 17–producing helper T cells (TH17 cells) have a major role in protection against infections and in mediating autoimmune diseases, yet the mechanisms involved are incompletely understood. We found that interleukin 26 (IL-26), a human TH17 cell–derived cytokine, is a cationic amphipathic protein that kills extracellular bacteria via membrane-pore formation. Furthermore, TH17 cell–derived IL-26 formed complexes with bacterial DNA and self-DNA released by dying bacteria and host cells. The resulting IL-26–DNA complexes triggered the production of type I interferon by plasmacytoid dendritic cells via activation of Toll-like receptor 9, but independently of the IL-26 receptor. These findings provide insights into the potent antimicrobial and proinflammatory function of TH17 cells by showing that IL-26 is a natural human antimicrobial that promotes immune sensing of bacterial and host cell death.
Human interleukin 17–producing helper T cells 7 (TH17 cells) are a subset of T cells that drive inflammatory responses by producing interleukin 17A (IL-17A), IL-17F, IL-21, IL-22 and IL-26 (refs. 1–3). Defective TH17 cell responses in patients deficient in the transcription factor STAT3 have been associated with increased susceptibility to infection by Staphylococcus aureus and Streptococcus pyogenes4,5, which indicates that this T cell subset has a major role in the defense against extracellular bacterial infections, particularly in skin and mucosal surfaces. However, excessive TH17 cell responses can drive chronic inflammation and the development of autoimmunity in predisposed individuals. TH17 cell–associated cytokines are indeed associated with disease activity and were found to be present in increased amounts in the skin of subjects with psoriasis, in the intestine of individuals with Crohn’s disease, in the brain of people with multiple sclerosis and in the synovium of people with rheumatoid arthritis and ankylosing spondylitis compared with healthy individuals6. Furthermore, variants of genes involved in IL-23–induced TH17 cell differentiation have been identified as susceptibility factors in psoriasis, multiple sclerosis, rheumatoid arthritis, ankylosing spondylitis and Crohn’s disease, which suggests a genetic basis for the enhanced TH17 cell responses in these diseases6.
The mechanisms by which TH17 cell cytokines drive antimicrobial and inflammatory responses are incompletely understood. IL-17A and IL-17F induce the expression of various CXC chemokines in epithelial cells, which drives the recruitment of neutrophils and monocytes7. IL-22 induces the proliferation of epithelial cells and is involved in epidermal remodeling8. IL-22 also cooperates with IL-17 to activate epithelial cells and induce the production of antimicrobial peptides8. IL-21 does not have a direct inflammatory role but amplifies and stabilizes the development of TH17 cells9. The functions of IL-26, a 19-kDa α-helical protein that belongs to the IL-20 cytokine family, are less well characterized. IL-26 was first identified in herpesvirus saimiri–transformed T cells10 and was subsequently found to be conserved in most vertebrate species but absent in mice11. Like other TH17 cell cytokines, IL-26 shows high expression in psoriatic skin lesions1, colonic lesions from individuals with inflammatory bowel disease12 and synovia of individuals with rheumatoid arthritis13, and it is strongly associated with inflammatory activity. A risk locus containing IL26 and single-nucleotide polymorphisms within the IL26 gene region have been associated with multiple sclerosis14, rheumatoid arthritis15 and inflammatory bowel disease16, which suggests a particularly important role for IL-26 in TH17 cell–mediated inflammatory disease.
IL-26 signals through the IL-10R2–IL-20R1 heterodimeric receptor, which is expressed exclusively by epithelial cells17,18. Via its receptor, IL-26 inhibits the proliferation of intestinal epithelial cells and, in parallel, induces expression of immunosuppressive IL-10, but also of the proinflammatory cytokines tumor necrosis factor (TNF) and IL-8 (ref. 12). How these functions fit with the proinflammatory role of IL-26 in the context of TH17 cell responses remains unclear. Here we identified a distinctive cationic, amphipathic and multimeric structure of IL-26 that enabled TH17 cells to engage in direct antimicrobial activity. This function was mediated by the ability of IL-26 to directly kill extracellular bacteria through pore formation. Furthermore, IL-26 was found to form complexes with extracellular DNA released by dying bacteria and host cells and to promote Toll-like receptor 9 (TLR9) activation of plasmacytoid dendritic cells (pDCs), providing evidence for a potent proinflammatory function of TH17 cells.
RESULTS
IL-26 is a cationic and amphipathic multimeric protein
Our sequence analysis of IL-26 showed unusual cationicity of this cytokine (calculated charge of +18.1 at pH 7 and isoelectric point of 10.4), as previously described17. The majority of the cationic charges were found to be contained in, or adjacent to, two of the six predicted helices of the protein, helices B and E, which contain three arginines and seven lysines (Supplementary Fig. 1a). Three-dimensional modeling of the protein showed that helices B and E were close to each other (Fig. 1a), which led to cluster formation and surface exposure of the cationic residues (Fig. 1b). On the opposite side of this cluster, we observed a hydrophobic patch (helix A) composed of several hydrophobic side chains (alanine 23, isoleucine 26, alanine 29, tryptophan 30 and alanine 33) (Fig. 1b). The predominance of polar (cationic) residues on one side of the molecule and hydrophobic amino acids on the opposite side indicated that IL-26 is a cationic amphipathic protein. In contrast, IL-22, a close homolog of IL-26 with 27% amino acid identity, has a net charge of +0.2 and an even distribution of cationic, anionic and hydrophobic residues across the surface of the molecule19 (Fig. 1b).
Figure 1.

IL-26 is a cationic amphipathic protein that forms oligomers. (a) Protein ribbon of IL-26 obtained by homology modeling. Six predicted α-helices are indicated as αA–αF and are represented in different colors. (b) Color-coded electrostatic potentials mapped onto the surfaces of IL-26 (top row) and IL-22 (bottom row). Regions of positive charges are in blue, negative charges are in red and hydrophobic charges are in white. (c) Small-angle X-ray scattering analysis and rigid-body modeling (via Sasref) of IL-26 suggested the formation of elongated tetramers. The four IL-26 molecules (secondary-structure representation) are shown in different colors.
To gain further insights into the structure of IL-26, we carried out small-angle X-ray scattering analysis of recombinant human IL-26 (rhIL-26). IL-26 not only formed dimers but also was able to form higher-degree multimers (Fig. 1c and Supplementary Fig. 1b,c). This represented an atypical structure compared with close homologs IL-10 and IL-22, which can only dimerize. IL-26 multimers were found to adopt a beads-on-string shape (Fig. 1c and Supplementary Fig. 1b,c), which gave rise to elongated structures. This structure is distinct from the dimeric structure of IL-22, which is compact and results from extensive interactions among helices A, B and F of two monomers19. The structure of IL-26 is also distinct from the compact arm-exchange structure of IL-10 dimers20, which results from the exchange of helices E and F between two monomers. The absence of arm-exchange dimers in IL-26 is in agreement with the fact that proline 113 of IL-10, a key residue for the arm exchange in IL-10, is not conserved in IL-26. Moreover, the positions of IL-26 cysteines 11 and 100 and of cysteines 58 and 103 in the 3D homology model suggest that these residues form two disulfide bonds to stabilize the helices of IL-26, rather than allowing an arm-exchanged conformation (Supplementary Fig. 1d). Thus, IL-26 is a highly cationic and amphipathic protein that forms elongated multimers, representing a highly atypical structure compared with the structures of other cytokines from the same family. Because IL-26 monomers or dimers would be sufficient for binding to the dimeric IL-26 receptor, the formation of multimeric structures suggests a functional adaptation of IL-26 to a specialized task.
IL-26 kills extracellular bacteria by pore formation
Because an amphipathic structure, the clustering of cationic charges and the formation of multimers are hallmarks of naturally occurring antimicrobial peptides21, we asked whether IL-26 has antimicrobial properties. Using microbroth dilution assays, we found that 5–10 μM rhIL-26 inhibited the growth of several gram-negative bacterial strains, including Pseudomonas aeruginosa (ATCC 27853, PA14), Escherichia coli (O1:K1:H7, O18:K1:H7, O111:B4, O111:K58:H2) and Klebsiella pneumoniae (O1:K2), as well as gram-positive S. aureus (ATCC 6538) (Fig. 2a and Supplementary Fig. 2a). No inhibition of colony formation was observed with Enterococcus faecalis or Candida albicans at IL-26 concentrations up to 50 μM (Table 1). rhIL-26, but not rhIL-17 or rhIL-22, efficiently inhibited P. aeruginosa colony formation at a concentration of 10 μM (Fig. 2b). The observed inhibition of colony formation was due to bacterial killing, as the number of colonies decreased over time (>log 2 over 24 h) (Fig. 2c). A loss of membrane integrity was confirmed by decreased bacterial staining with a fluorescent membrane-potential indicator dye (Fig. 2d,e) and increased bacterial staining with an otherwise impermeable DNA dye (Supplementary Fig. 2b,c). IL-26 concentrations between 1 and 10 μM were required for bacterial killing, similar to the concentrations described for other cationic antimicrobial peptides such as LL-37 and hBD3 (Supplementary Fig. 2d,e). To determine whether IL-26, like other antimicrobial peptides, kills bacteria by pore formation and membrane disruption, we used scanning electron microscopy to analyze the ultrastructure of IL-26–treated bacteria. As early as 30 min after treatment with 10 μM IL-26, we observed the formation of membrane blebs on the surface of the bacteria (Fig. 2f). In some instances we observed disruption of the blebs with leakage of the cytosol into the extracellular environment (Fig. 2f). These findings indicated that, similar to other cationic antimicrobial peptides, IL-26 disrupts bacterial membranes via pore formation. Although the molecular basis for the interaction of IL-26 with gram-negative and gram-positive bacteria needs to be elucidated, we found that IL-26 had the ability to bind lipopolysaccharide (LPS) from gram- negative bacteria and lipoteichoic acid (LTA) from gram-positive bacteria (Fig. 2g and Table 2). Finally, we tested the antimicrobial activity of rhIL-26 in vivo using a mouse model of K. pneumoniae sepsis. This model uses intranasal administration of K. pneumoniae (O1:K2) to induce lung infection followed by rapid spreading of the bacteria into the circulation and the spleen22. Treatment of the mice with IL-26 resulted in significantly reduced bacterial titers in lungs, spleen and blood compared with amounts in control mice (Fig. 2h). The in vivo antibacterial activity of IL-26 was similar to that observed with the antimicrobial peptide LL-37. A strong reduction in bacterial titers was also observed when bacteria were pretreated in vitro with IL-26, before intranasal administration (Fig. 2h). Together, these data suggest that IL-26 is an antimicrobial protein that efficiently kills extracellular bacteria in vitro and in vivo.
Figure 2.

IL-26 has direct bactericidal properties. (a) Growth of Pseudomonas aeruginosa ATCC 27853 (P. aerug.), Escherichia coli O1:K1:H7 (E. coli), Staphylococcus aureus ATCC 6538 (S. aureus) and Klebsiella pneumoniae O1:K2 (K. pneumo.) in culture with increasing concentrations of rhIL-26. (b) Growth of P. aeruginosa in culture with increasing concentrations of rhIL-26, rhIL-17 and rhIL-22. (c) Growth kinetics of P. aeruginosa cultured with no IL-26, with 10 μM rhIL-26, and with rhIL-26 in the presence of either blocking anti–IL-26 or isotype control antibodies (ctl IgG). (d) Membrane potential of P. aeruginosa cultured for 24 h as in c and stained with the green fluorescent dye DiOC2(3), which forms red fluorescent aggregates in polarized membranes. A loss of red fluorescence indicates membrane disruption. The proton ionophore CCCP was used as a positive control. (e) Kinetics of membrane potential of P. aeruginosa cultured as in d. (f) P. aeruginosa cultured for 30 min with or without 10 μM rhIL-26 and visualized by scanning electron microscopy. Arrows in the top right image show the formation of membrane blebs; arrow in the lower right image shows extracellular leakage of bacterial content. (g) Capacity of IL-26 to bind LPS or LTA as assessed by enzyme-linked immunosorbent assay (ELISA). (h) Bacterial loads measured in mouse lungs, spleen and blood collected 3 d after intranasal infection with K. pneumoniae (100 CFU) followed by intranasal treatment with 20 μg rhIL-26 or 50 μg LL-37. As a control, K. pneumoniae was pretreated with rhIL-26 ex vivo. Each data point represents a separate mouse; ††† indicates a mouse that died from overwhelming infection within the 3-d period between initial infection and organ collection. Data are representative of three (a–c,e), two (g) or four (h) independent experiments and were obtained from eight mice per group; error bars represent ±s.d. of duplicate (a–c,e) or triplicate (g) wells. Data were statistically analyzed via unpaired non-parametric Mann-Whitney U-test. *P < 0.001. CFU, colony-forming units.
Table 1.
MIC50 determination of IL-26 antimicrobial activity against several microorganisms
| Organism | MIC50 of IL-26 (μM) |
|---|---|
| Pseudomonas aeruginosa (ATCC 27853) | 8.6 |
| Escherichia coli (ATCC 11775) | 18.6 |
| Staphylococcus aureus (ATCC 6538) | 8.8 |
| Enterococcus faecalis (ATCC 29212) | >50 |
| Candida albicans (ATCC 24433) | >50 |
MIC50 of IL-26 against P. aeruginosa, E. coli, S. aureus, E. faecalis and C. albicans determined by microbroth dilution assay.
Table 2.
IL-26 binds to the bacterial cell wall components LPS and LTA
| Protein | IL-26 | IL-26 | LL-37 | IL-22 | IL-22 |
|---|---|---|---|---|---|
| Ligand | LPS | LTA | LPS | LPS | LTA |
|
KD (mean ± s.d.) |
40.5 ± 4.95 nM | 20.91 ± 2.84 nM | 3.45 ± 0.8 nM | NI | NI |
Dissociation constants (KD) of IL-26, IL-22 and LL-37 binding to LPS or LTA, measured by ELISA. NI, no interaction.
TH17 cells induce antimicrobial effects via the production of IL-26
Microarray analysis of IL-26 expression on peripheral blood cell subsets showed that IL-26 was expressed only on CD4+ T cells, and not on CD8+ T cells, NK cells, B cells, CD11c+ dendritic cells, pDCs, monocyte subsets or granulocytes (Supplementary Fig. 3a). In the CD4+ T cell subset, IL-26 seemed to be expressed mainly by TH17 cells, moderately by type 1 helper T cells (TH1 cells) and not at all by type 2 helper T cells (TH2 cells) (Supplementary Fig. 3a). To confirm these results, we carried out RT-PCR in in vitro–differentiated TH17, type 0 helper T (TH0), TH1 and TH2 cells under well-established polarizing conditions23. We found that IL26 mRNA expression was indeed high in TH17 cells and moderate in TH1 cells, but no expression was observed in TH0 or TH2 cells (Fig. 3a). We then tested the ability of these supernatants to kill extracellular bacteria with a microbroth dilution assay. TH17 cell supernatants but not TH0 supernatants efficiently killed P. aeruginosa (Fig. 3b). Bacterial growth was completely restored by the addition of a blocking antibody to IL-26, which was shown to inhibit killing of bacteria by recombinant IL-26 (Fig. 2c–e). We also performed experiments with three different human TH17 cell clones (1G3, 7H1 and 72G6) and found that supernatants derived from activated clones killed bacteria efficiently (Fig. 3c). This activity was entirely dependent on IL-26, as small interfering RNA (siRNA)-mediated knockdown of IL-26 completely abrogated the ability of the TH17 cell clones to kill bacteria (Fig. 3c and Supplementary Fig. 3b). We noted that the concentration of IL-26 in the supernatants of both activated primary TH17 cells and activated TH17 cell clones ranged from 0.10 to 0.15 μM (Fig. 3d) and exerted roughly the same antimicrobial activity as 10 μM rhIL-26. This suggested that the natural IL-26 released by TH17 cell clones has an antimicrobial potency 80-fold to 120-fold greater than that of rhIL-26 (median minimum inhibitory concentration (MIC50) for rhIL-26 of 8.6 μM, versus 0.068–0.1 μM for natural IL-26) (Supplementary Fig. 4a–c). Collectively these data demonstrate the direct antimicrobial capacity of TH17 cells enacted via their production of IL-26.
Figure 3.

TH17 cells exert direct antimicrobial activity via the production of IL-26. (a) Real-time PCR analysis of IL26, IL17A, IL17F, IL22, IL13 and IFNG mRNA expression by primary TH17, TH1, TH2 and TH0 cells generated in vitro from purified naive T cells and re-stimulated by plate-bound anti-CD3 and soluble anti-CD28. Cytokine mRNA expression was normalized to GAPDH mRNA expression. Each data point represents a separate donor (n = 10–11); horizontal bars denote mean values. (b) Growth of P. aeruginosa cultured with supernatants of primary TH0 and TH17 cells and measured by microbroth dilution assay. Assays were performed with and without blocking anti–IL-26 and control antibodies (Ctl IgG). (c) Growth of P. aeruginosa cultured with supernatants of three TH17 cell clones (1G3, 7H1 and 72G6) transfected with siRNA against IL-26 (siIL26, white bars) or a control siRNA (siCtl, black bars). n.s., not significant. (d) IL-26 production by re-stimulated primary TH0 and TH17 cells and transfected TH17 cell clones as measured by ELISA. Data are representative of at least three (b,c) or two (d) independent experiments. Error bars represent the s.d. of triplicate wells (b,c). Data were statistically analyzed via unpaired two-tailed Student’s t-test; *P < 0.05, **P < 0.01.
IL-26 promotes sensing of DNA released during bacterial killing
As clustered cationic charges in antimicrobial peptides have been shown to form complexes with extracellular DNA24, we asked whether IL-26 binds DNA during bacterial killing. Confocal microscopy of P. aeruginosa treated with rhIL-26 showed that dying bacteria released DNA structures that formed complexes with IL-26 (Fig. 4a). The ability of IL-26 to bind bacterial DNA was also confirmed when we mixed fragments of bacterial DNA with increasing concentrations of IL-26. IL-26, but not IL-17 or IL-22, efficiently bound to DNA, as shown by decreased staining of the DNA and retarded DNA migration in an agarose gel assay (Fig. 4b,c and Supplementary Fig. 5a). Furthermore, we found that bacterial DNA mixed with IL-26 formed insoluble particles, but DNA mixed with IL-17 or IL-22 did not (Fig. 4d and Supplementary Fig. 5b). This suggested that IL-26 is able to package and condense bacterial DNA. These IL-26–bacterial DNA particles were found to be highly protected from extracellular DNAse degradation (not shown), a process that has been shown to confer immunogenicity to DNA24–27.
Figure 4.

IL-26 promotes bacterial killing and concomitant sensing of DNA released by pDCs. (a) Confocal microscopy images of DNA–IL-26 complexes (yellow) in cultures of P. aeruginosa treated with rhIL-26 and stained with DAPI (green) and anti–IL-26 (red). Images shown are representative of three independent experiments. (b,c) Fluorimetric quantification of DNA staining by picogreen dye after mixing of bacterial DNA with increasing concentrations of IL-26 (b) or 1 μM IL-26, IL-17 or IL-22 (c). A.U., arbitrary units. (d) Number of particles formed after mixing of bacterial DNA with 1 μM IL-17, IL-22 or IL-26, counted as insoluble precipitates by microscopy. (e) Production of IL-1β, IL-6, TNF and IFN-α by PBMCs stimulated overnight with 1 μM IL-26 alone or in complex with bacterial DNA (bactDNA, 3 μg ml−1). Bars show mean ± s.d. of pooled data. (f) IFN-α produced by human PBMCs, monocytes, macrophages, NK cells, neutrophils, pDCs and pDC-depleted PBMCs stimulated overnight with 1 μM IL-26 alone or in complex with bactDNA. Bars show mean ± s.d. of pooled data. (g–i) IFN-α produced by pDCs stimulated overnight with increasing concentrations of live P. aeruginosa (titrated according to CFU) (g) or of P. aeruginosa lysate (titrated according to DNA content) (h) in the presence or not of IL-26 (10 or 1 μM, respectively). (i) IFN-α produced by pDCs stimulated overnight with P. aeruginosa lysate (1 μg ml−1 DNA) or purified bactDNA (1 μg ml−1) alone or in the presence of IL-26, with or without DNase pretreatment. Data are representative of four independent experiments (b–d,g–i) or four independent donors (e,f); error bars represent the s.d. of triplicate wells (b–d,g–i). Data were statistically analyzed via unpaired two-tailed Student’s t-test; *P < 0.05, **P < 0.01.
To test whether IL-26–DNA complexes are immunogenic, we tested their ability to trigger the production of inflammatory cytokines by human peripheral blood mononuclear cells (PBMCs). IL-26–DNA complexes induced the production of IL-1β, IL-6 and interferon-α (IFN-α by PBMCs), but only the production of IFN-α was significantly enhanced compared with production after stimulation with bacterial DNA alone or IL-26 alone (Fig. 4e). To determine which cell type was responsible for the production of IFN-α, we stimulated monocytes, NK cells, neutrophils, pDCs and macrophages with IL-26–DNA complexes. Only pDCs produced high amounts of IFN-α upon stimulation with complexes (Fig. 4f), and depletion of pDCs from PBMCs completely abrogated their ability to produce IFN-α, which indicated that pDCs are a key target of bacterial DNA–IL-26 complexes. Purified bacterial DNA alone induced weak or no IFN-α production by pDCs (Fig. 4f), and IL-26 was unable to induce any IFN-α production in pDCs in the absence of DNA (Fig. 4f and Supplementary Fig. 5c). Thus these data demonstrate that IL-26 can form complexes with bacterial DNA and thereby promote the immunogenicity of the DNA, leading to IFN-α production by pDCs. Next we tested whether IL-26–DNA complexes generated in the context of bacterial killing also promote pDC activation. Whereas live P. aeruginosa was unable to activate pDCs, P. aeruginosa killed by IL-26 induced strong IFN-α production (Fig. 4g). Supernatant of P. aeruginosa killed by freeze-thaw cycles was also unable to activate pDCs, but it acquired this ability when mixed with IL-26 (Fig. 4h). In these cultures, IFN-α production paralleled the DNA content (Fig. 4h and Supplementary Fig. 5d) and was completely abrogated when DNA was depleted by DNase treatment (Fig. 4i and Supplementary Fig. 5e). These findings indicate that IL-26 has the ability to kill bacteria, bind bacterial DNA and promote immunogenicity by triggering pDC activation.
IL-26 promotes sensing of human DNA released by dying cells
Because antimicrobial peptides can also promote the immunogenicity of human DNA24–27, we next asked whether IL-26 also kills human cells and promotes sensing of released self-DNA. We tested the ability of rhIL-26 to kill a range of human cells, including human embryonic kidney (HEK) cells and primary immune cells, but we did not observe any cytotoxic activity at 10 μM, the concentration required for efficient microbial killing (Fig. 5a). Similarly, rhIL-26 mixed with live human cells did not induce pDC activation (Fig. 5b). However, when rhIL-26 was mixed with irradiated human cells to trigger apoptosis and secondary necrosis, pDC activation was observed (Fig. 5b). As expected, pDC activation was dependent on extracellular DNA released by dying human cells, as it was largely abrogated by DNase treatment of the cultures (not shown). At higher rhIL-26 concentrations, we detected cytotoxic activity on human cells similar to that described for other antimicrobial peptides such as LL-37 (Supplementary Fig. 6a,b). This in vitro cytotoxicity was observed at 4.9–6.6-fold the effective antimicrobial concentration for IL-26, compared to 4.0–7.6-fold that for LL-37 (Supplementary Fig. 6a,b).
Figure 5.

IL-26 promotes pDC sensing of human DNA released in the context of cell death. (a) Cell viability of monocytes, macrophages, NK cells, neutrophils, pDCs and HEK cells as determined by staining with 7-AAD after overnight culture in medium alone or in the presence of either 10 μM rhIL-26 or undiluted supernatants (SN) of primary TH17 cells. (b) IFN-α produced by pDCs stimulated overnight with live or UV-irradiated HEK293 cells in the presence or not of 10 μM rhIL-26. (c) Fluorimetric quantification of DNA staining by picogreen dye upon mixing of human DNA fragments (huDNA) with increasing concentrations of rhIL-26. (d) Visualization of DNA–IL-26 complexes (yellow) formed after mixing of human DNA with rhIL-26 and appearing as insoluble precipitates that stained with DAPI (green) and anti–IL-26 (red). (e) IFN-α produced by human PBMCs, monocytes, macrophages, NK cells, neutrophils, pDCs and pDC-depleted PBMCs stimulated overnight with IL-26 alone, human DNA alone or IL-26–human DNA complexes. Each data point represents an individual donor (n = 4 for each condition). Bars show mean ± s.d. of pooled data. Data were statistically analyzed via unpaired two-tailed Student’s t-test; *P < 0.05, **P < 0.0001. (f) IFN-α produced by pDCs stimulated with increasing concentrationsof human DNA either alone or in complex with 1 μM IL-26. In panels a–c and f, data are representative of three independent experiments, and error bars represent ±s.d. of triplicate wells.
We confirmed the ability of IL-26 to bind extracellular human DNA fragments and to form particulate complexes that resembled those described for bacterial DNA (Fig. 5c,d). Like IL-26–bacterial DNA complexes, IL-26–human DNA complexes induced preferential IFN-α production by PBMCs that was largely pDC dependent (Fig. 5e). Human DNA induced IFN-α production in a dose-dependent manner and only in the presence of IL-26 (Fig. 5e,f), which confirmed that IL-26 converts otherwise non-stimulatory human DNA into a potent activator of pDCs. IL-26–human DNA complexes were also found to induce some activation of monocytes, leading to the production of small amounts of IFN-α (Fig. 5e); this suggested broader action of IL-26 in DNA-mediated innate immune activation, potentially via cytosolic DNA sensors, as monocytes do not express TLR9. Furthermore, IL-26 alone was found to trigger the production of some IL-6 and IL-1β by purified monocytes, although this activity was intrinsic to IL-26 and was not enhanced by the presence of DNA (not shown). Taken together, these data indicate that effective antibacterial concentrations of IL-26 are unable to kill human cells but do bind human DNA released in the context of cell death, leading to pDC activation.
IL-26–DNA complexes activate TLR9 in pDCs
Human pDCs specifically express TLR9, an endosomal receptor for DNA. To investigate whether IL-26 promotes immunogenicity of DNA by facilitating entry into TLR9-containing endosomal compartments, we first used fluorochrome-labeled DNA to track IL-26–DNA complexes in pDCs. We observed that IL-26–DNA complexes, but not DNA alone, rapidly associated with pDCs (Fig. 6a) and accessed intracellular vesicular structures in those cells (Fig. 6b). We concluded that internalization of the complexes occurred via endocytosis, as the vesicular structures expressed the early endosomal marker CD71 (Fig. 6b) and complex internalization was inhibited by culturing of pDCs either at 4 °C or in the presence of the endocytosis inhibitor Cytochalasin D (Fig. 6c,d).
Figure 6.
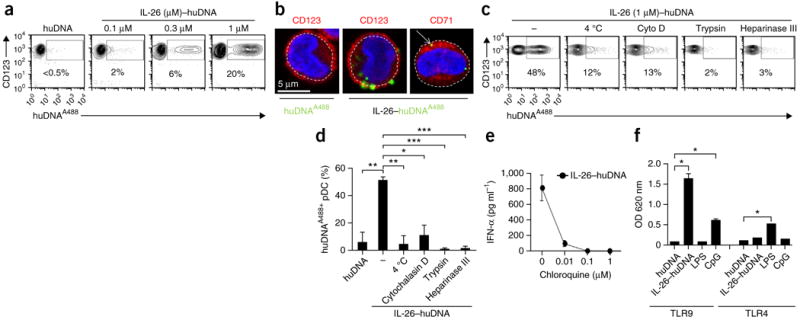
IL-26–DNA complexes are endocytosed via heparan-sulfate proteoglycans and activate TLR9 in pDCs. (a) Flow cytometry of CD123+ pDCs stimulated for 4 h with Alexa Fluor 488–conjugated human DNA (huDNAA488) alone or complexed with increasing concentrations of IL-26. (b) Confocal microscopy images of pDCs stimulated with huDNAA488 alone or with IL-26–huDNAA488 complexes. We used DAPI to stain the nucleus, and we used phycoerythrin-conjugated anti-CD123 to visualize the contour of pDCs (left and middle panels) or biotin-conjugated anti-CD71 plus phycoerythrin-conjugated streptavidin to visualize early endocytic compartments (right panel). (c) Flow cytometry of human DNA–positive CD123+ pDCs stimulated for 4 h with huDNAA488 complexed with IL-26 after pretreatment with 1 μg ml−1 cytochalasin D (Cyto D) to block endocytosis, 0.5 μg ml−1 trypsin to cleave membrane proteins and 100 mU Heparinase III to cleave surface heparan-sulfate proteoglycans. Cells were also cultured at 4 °C to inhibit active internalization processes such as endocytosis. (d) Quantification of human DNA–positive CD123+ pDCs stimulated as in c. *P < 0.01, **P < 0.001, ***P < 0.0001. (e) IFN-α produced by pDCs stimulated overnight with IL-26–human DNA complexes in the presence of increasing concentrations of chloroquine to inhibit endosomal TLR activation. (f) Fluorimetric quantification of embryonic alkaline phosphatase secreted by HEK293 cells transfected with either TLR9 or TLR4 along with an NF-κB–inducible secreted embryonic alkaline phosphatase reporter gene and stimulated under the indicated conditions. CpG, synthetic phosphothioate TLR9 agonist CpG-2006. *P < 0.0001. Data are representative of three independent experiments. Error bars in d–f represent the s.d. of triplicate wells. Data were statistically analyzed via unpaired two-tailed Student’s t-test.
Next we investigated how IL-26–DNA complexes are endocytosed by pDCs. These cells do not express the IL-20R1 chain28, which means that the IL-26 receptor cannot serve as a scavenger receptor. IL-26–DNA complexes were disrupted by the addition of increasing concentrations of the anionic molecule heparin (Supplementary Fig. 7a), which indicated that complex formation with the DNA is driven by the cationic residues of IL-26. Because polycation–nucleic acid complexes enter cells through heparan-sulfate proteoglycan (HSPG)-mediated endocytosis29, we investigated whether endocytosis of IL-26–DNA complexes is mediated by a similar mechanism. We found that internalization of the complexes was blocked both by pretreatment of pDCs with trypsin and by heparinase III (Fig. 6c,d), which suggested that HSPGs are required for membrane attachment and endocytosis by pDCs.
To determine whether, upon endosomal entry, IL-26–DNA complexes trigger activation of TLR9, we first measured IFN-α production by pDCs pretreated with chloroquine, an inhibitor of endosomal TLR signaling. IFN-α production was completely inhibited by chloroquine (Fig. 6e), which suggested the involvement of an endosomal TLR. IFN-α production was not inhibited when pDCs were treated with chloroquine and activated by cGAMP, which triggers the cytoplasmic sensor STING (Supplementary Fig. 8a). Furthermore, IFN-α production was not inhibited by a neutralizing antibody to IL-10R2 that blocks activation of the IL-26 receptor18 (Supplementary Fig. 8b) or by a STAT3 inhibitor that blocks IL-26–receptor signaling (not shown). Gain-of-function experiments using TLR9- and TLR4-transfected HEK293 cells confirmed that IL-26–DNA complexes induced NF-κB activation in TLR9-transfected HEK293 cells but not in TLR4-transfected cells (Fig. 6f). Thus, we concluded that IL-26–DNA complexes are endocytosed by pDCs upon attachment to membrane HSPGs and trigger IFN-α production via activation of TLR9.
TH17 cells promote activation of pDCs by producing IL-26
TH17 cell supernatants containing concentrations of natural IL-26 sufficient to exert antimicrobial activity did not exert any cytotoxic activity on human cells (Fig. 5a). However, we observed that supernatants of re-stimulated TH17 cells contained insoluble particles that stained for DNA and IL-26 (Fig. 7a), which suggested that TH17 cell–derived IL-26 forms complexes with endogenous DNA released in the context of cell turnover during cell culture. Consistent with the production of high amounts of IL-26 by TH17 cells, these particles were numerous in TH17 cell supernatants compared with their amounts in supernatants of other helper T cells, including TH0 cells (Fig. 7b). To determine whether these natural IL-26–DNA complexes present in TH17 cell supernatants trigger IFN-α production by pDCs, we stimulated purified pDCs with the TH17 cell supernatants. Half of the TH17 cell supernatants induced IFN-α production by pDCs (Fig. 7c,d), and this production was inhibited by the addition of an antibody to IL-26 that blocks binding to DNA (Supplementary Fig. 7b) and inhibits pDC activation (Supplementary Fig. 7c), as well as by DNase pretreatment of the supernatants (Fig. 7e). The other half of the TH17 cell supernatants, which did not activate pDCs, acquired the ability to induce IFN-α production when exogenous DNA was added to the culture (Fig. 7f), which indicated that the presence of endogenous DNA in TH17 cell supernatants was a limiting factor for pDC activation. Supernatants of TH17 cell clones expressing IL-26 were also able to induce IFN-α production by pDCs, and this was inhibited by siRNA-mediated knockdown of IL-26 (Fig. 7g). The addition of exogenous DNA to the cultures enhanced interferon production by pDCs and confirmed that activation was driven by IL-26, as interferon production by pDCs was completely blocked in knockdown experiments (Fig. 7g). These findings suggest that TH17 cell–derived IL-26 drives innate immune activation of pDCs through the formation of complexes with human DNA released into the extra-cellular environment during cell turnover. We did not observe any differences between the concentration of natural IL-26 contained in TH17 cell supernatants and the concentration of rhIL-26 required to induce IFN-α production in pDCs (Supplementary Fig. 4c). Both recombinant and natural IL-26 induced IFN-α production by pDCs at 0.1 μM, a concentration that seems to be relevant in vivo, as tissue concentrations of up to 0.15 μM have been measured in psoriatic skin lesions known to contain large numbers of activated pDCs producing IFN-α (ref. 30) (Fig. 7h).
Figure 7.

TH17 cells activate pDCs via IL-26 production. (a) Confocal microscopy images of insoluble DNA–IL-26 particles in supernatants of re-stimulated TH17 cells stained with DAPI and unlabeled anti–IL-26 plus Alexa Fluor 546–conjugated mouse anti-IgG. Images shown are representative of three independent experiments. (b) DNA–IL-26 particle count in supernatants of re-stimulated TH17 and TH0 cells. Each data point represents an independent donor (n = 4–6). Horizontal bars represent means. Data were statistically analyzed via unpaired two-tailed Student’s t-test; *P < 0.05. (c–g) IFN-α produced by pDCs stimulated overnight with (c) various dilutions of TH17 cell–derived supernatants in culture medium; (d) supernatants of re-stimulated TH17 or TH0 cells derived from multiple donors (n = 9); (e) supernatant derived from TH17 cells re-stimulated in the presence of anti–IL-26, anti–IL-17 or DNase I treatment or untreated; (f) TH17 cell supernatants derived from multiple donors (n = 3–7), either alone or supplemented by exogenous human DNA (3 μg ml−1), with or without blocking anti–IL-26; and (g) supernatants of TH17 cell clones (n = 3) transfected with siRNA against IL-26 (siIL26) or a control siRNA (siCtl) supplemented or not by human DNA. In a–g, data are representative of at least three independent experiments and were statistically analyzed via unpaired two-tailed Student’s t-test; *P < 0.05, **P < 0.01. Horizontal bars in d, f and g represent means. (h) IL-26 concentrations in healthy skin and psoriatic skin lesions measured by ELISA of total skin extracts derived from healthy donors (n = 15) or psoriasis patients (n = 15). Each data point represents an individual donor. Data were statistically analyzed via unpaired non-parametric Mann-Whitney U-test; *P < 0.001.
DISCUSSION
Our findings demonstrate an unexpected function of the human cytokine IL-26 in TH17 cell–mediated antimicrobial and inflammatory responses. As predicted on the basis of its cationicity, amphipathicity and ability to form multimers, IL-26 exerted direct antimicrobial activity against extracellular bacteria. Like classical cationic antimicrobial peptides, IL-26 forms pores in bacterial membranes, which leads to microbial lysis. Via the expression of IL-26, TH17 cells exert direct antimicrobial activity, and this provides new explanations for the known role of TH17 cells in the host defense. TH17 cells indeed mediate protection against extracellular bacteria: patients with defective TH17 cells are highly susceptible to S. aureus infections4, and mucosal TH17 cell depletion in the context of simian immunodeficiency virus infection leads to dissemination of gut bacteria31.
As a consequence of bacterial killing, TH17 cell–derived IL-26 also triggers potent immune activation. IL-26 binds to bacterial DNA released during the antimicrobial response and promotes internalization of the DNA into endosomal TLR9 compartments of pDCs, leading to the production of IFN-α. The mechanism underlying this process seems to be similar to the one described for the cationic antimicrobial peptide LL-37. First, IL-26 aggregates and condenses DNA fragments to form insoluble particles that are protected from extra-cellular degradation. Second, exposed cationic residues enable attachment of the DNA complexes to heparan proteoglycans on the cell membrane, which leads to receptor-independent endocytosis. Finally, the DNA complexes gain access to endosomal TLR9, which leads to pDC activation and the production of high amounts of type I interferons.
During microbial infections, type I interferons (IFN-α and IFN-β) can have several functions. In the context of infections caused by intracellular bacteria, type I interferons can be detrimental because of their proapoptotic activity and their ability to interfere with anti-bacterial mechanisms32–34. However, type I interferons can be beneficial in the context of infections by extracellular bacteria. Induction of type I interferon was shown to be associated with the resolution of P. aeruginosa lung infections35 and to be required for the control of infection by Group B streptococcus, Streptococcus pneumoniae and E. coli36. These effects might be driven by proinflammatory functions of type I interferons37 such as induction of dendritic cell maturation38,39, promotion of CD8+ T cell activation40,41 and TH1 cell differentiation41, activation of NK cells, and differentiation of B cells into antibody-secreting plasma cells42. Alternatively, type I interferon can also contribute to a beneficial disease outcome by downregulating certain inflammatory responses to bacterial infections. In fact, IFN-α production by pDCs seems to be responsible for controlling inflammation in a model of bacterial sepsis43. Given our findings, we speculate that IL-26 might have evolved from a pure antimicrobial protein to become a potent trigger of IFN-α production that both amplifies and regulates innate and adaptive immune responses to extracellular bacteria.
IL-26 was also found to bind fragments of extracellular human (self-) DNA and trigger IFN-α production via TLR9 activation in pDCs. This finding suggests that in addition to its antimicrobial role or as part of bacterial killing followed by binding of DNA, IL-26 also represents a danger signal that promotes IFN-α expression in injured tissue.
IL-26 is expressed in high amounts in TH17 cell–mediated autoimmune diseases such as psoriasis1, inflammatory bowel disease12 and rheumatoid arthritis13. These diseases are characterized by high numbers of pDCs and high IFN-α expression in tissues30,44,45, which suggests that IL-26 overexpression drives uncontrolled inflammatory responses via exaggerated IFN-α induction. As a consequence of this process, IL-26 may further enhance tissue injury by releasing self-DNA, leading to a self-sustaining inflammatory cycle that unleashes autoimmunity. Genetic evidence for a potential pathogenic role of IL-26 in these diseases is provided by the identification of a risk locus containing the gene IL26 and single-nucleotide polymorphisms within the IL26 region in subjects with rheumatoid arthritis15 and inflammatory bowel disease16.
As is true of other antimicrobial peptides, high concentrations of recombinant IL-26 can kill normal human cells directly in vitro. In psoriasis, tissue concentrations of IL-26 were found to be relevant for antimicrobial and interferon-inducing activity, but not for cytotoxic activity against normal human cells. Although we cannot exclude the possibility that higher microconcentrations of IL-26 can occur in tissues, these data suggest that interferon induction in vivo might be limited to situations in which host cells are destroyed by infection or inflammation and self-DNA is already present in the extracellular environment.
The regulation of IL-26 expression in TH17 cells is currently unknown. Analysis of in vitro–generated helper T cell subsets suggests that IL-26 expression directly parallels the expression not of IL-17, but rather of IL-22. Distinct regulation of IL-26 expression is also suggested by the fact that bacteria and bacterial products that trigger IL-17 expression do not lead to increased expression of IL-26 in TH17 cells (data not shown).
Although pDCs seem to be the main target of IL-26–DNA complexes, we observed some activation of monocytes that led to the production of small amounts of IFN-α; this suggested broader implication of IL-26 in DNA-mediated innate immune activation, potentially via cytosolic STING-dependent DNA sensors as described for LL-37 (ref. 25). Future studies will be needed to determine whether, like LL-37, IL-26 can bind extracellular RNA fragments and trigger activation of conventional DCs and monocytes via TLR8 (ref. 46).
Our finding that the natural IL-26 in TH17 cell supernatants is more potent than recombinant IL-26 is consistent with previously documented differences in post-transcriptional modifications between natural and recombinant proteins, including glycosylation47. These differences were not observed for the IFN-inducing function of IL-26, which suggests that they are selectively required to promote the antimicrobial process, as suggested by others48. Another possibility is that IL-26 acts in synergy with other factors in TH17 cells to mediate antimicrobial activity.
In conclusion, this study showed that TH17 cells, via the production of IL-26, have direct antimicrobial activity and the ability to trigger potent immune responses via the expression of type I interferon. Future strategies could be developed to modulate IL-26 function, either to enhance host antimicrobial activity or to control inflammation in response to tissue injury and in the context of autoimmune diseases.
METHODS
Methods and any associated references are available in the online version of the paper.
ONLINE METHODS
Human samples
Studies were approved by the institutional review board of the University of Texas MD Anderson Cancer Center, the Heinrich-Heine University of Düsseldorf, and the University of Lausanne and were performed in accordance with the guidelines of the Declaration of Helsinki. Skin-punch biopsies were taken with informed consent from subjects with plaque psoriasis, defined according to standard clinical and histopathological criteria, and from healthy donors. Samples were snap-frozen and stored at −80 °C. Blood buffy coats from healthy donors were obtained from the Gulf Coast Regional Blood Center, Houston, Texas, or from the Service Vaudois de Transfusion, Epalinges, Switzerland.
Reagents
Recombinant human IL-26 (rhIL-26)12,13,18, rhIL-17 and rhIL-22 were from R&D Systems. Human genomic DNA isolated from fetal skin, lung or leukocytes was from BioChain. Synthetic LL-37 and hBD3 were from Innovagen. To generate antibodies to IL-26, we immunized 6–8- week-old BALB/c mice with rhIL-26 protein. Hybridomas secreting antibodies specific for IL-26 were established. Two clones were used in these studies: clone 142-84-1, with the ability to inhibit binding of IL-26 to DNA, and clone 142-69-1, with the ability to stain IL-26 for confocal microscopy. Both clones were used at 10 μg/ml. Mouse IgG1 isotype-matched antibodies were used as controls. Synthetic phosphothioate TLR9 agonist CpG-2006 (1 μM) (5′-tcgtcgttttgtcgttttgtcgtt-3′) was from Trilink. The TLR1/2 agonist Pam3CSK4, the TLR2 agonist LTA and the TLR4 agonist LPS were from Invivogen. Blocking goat anti-human IL-10R2 was from R&D Systems (AF874). Chloroquine was from Sigma.
Protein modeling
Human IL-26 models (UniProtKP sequence Q9NPH9) were generated using the software i-Tasser49. Further modeling of IL-26 was based on published atomic structures of IL-19 (PDB entry 1N1F) and IL-22 (1M4R), and a putative arm-exchanged IL-26 model was based on the atomic structure of IL-10 (2ILK). The sequences of IL-19, IL-22 and IL-10 are about 25% identical and about 55% similar to that of IL-26. The best i-Tasser model (a compact monomer) had a high-significance C-score of 0.36, with an expected r.m.s. deviation of 4.0 ± 2.7 Å.
Small-angle X-ray scattering (SAXS)
IL-26 was suspended in 16 mM sodium phosphate buffer, pH 6.5, 560 mM NaCl, 8% glycerol, 2 mM β-mercaptoethanol and then concentrated to 1.1 mg/ml. Data were recorded at the ALS (Berkeley, CA) beamline 12.3.1. (SIBYLS) at 10 °C using a wavelength of 1 Å, for values of the momentum transfer vector q = (4πsinθ)/λ between 0.01 and 0.32 Å−1. The same sample was sequentially exposed to X-rays for 0.5 s, 1 s and 5 s. Samples containing buffer only were measured before and after protein samples. The buffer contributions were subtracted from protein scattering data with the Ogre software program available at SIBYLS. Data were not affected by radiation damage, as indicated by a constant Rg of the Guinier region for subsequent sample exposures. We obtained a combined SAXS pattern by scaling and merging selected regions of buffer-subtracted scattering patterns for the 0.5-s, 1-s and 5-s exposures. Data with q values between 0.01 and 0.13 Å−1 were analyzed with PRIMUS, SASREF and DAMMIF of the ATSAS software package50.
Antimicrobial assays
The following bacteria strains were used: Pseudomonas aeruginosa (ATCC 27853 and PA14), Escherichia coli (O1:K1:H7, O18:K1:H7, O111:K58:H2 and J5 O111:B4), Staphylococcus aureus (ATCC 6538), Klebsiella pneumoniae (O1:K2), and Enterococcus faecalis (ATCC 29212). Bacteria were cultured at 37 °C overnight in trypticase soy broth with 10 mM NaCl and then subcultured for an additional 3 h to achieve mid–logarithmic phase growth. Bacterial concentrations were measured by spectrophotometry at 620 nm and diluted to a final concentration of 105 CFU/ml, after which they were incubated at 37 °C for 24 h under low-ionic-strength conditions (10 mM NaCl). IL-26, LL-37 and hBD3 were added to these cultures to test their antimicrobial activity. After 24 h, serial dilutions of bacterial cultures were plated onto lysogeny broth (LB) agar plates. The number of colonies formed after overnight incubation was counted by two independent investigators. The MIC50 of IL-26 was defined as the lowest concentration that inhibited >50% of the growth of bacteria after overnight incubation. In some experiments, bacteria were incubated in antibiotic-free RPMI medium or TH17 cell supernatants supplemented or not with the blocking anti–IL-26 clone 142-84-1 at 10 μg/ml. For analysis of the membrane potential loss, bacteria were incubated with 10 μM IL-26 and harvested at different time points to be stained with the fluorescent membrane-potential indicator dye DiOC2(3) (BacLight bacterial membrane potential kit, Molecular Probes) according to the manufacturer’s protocol. As a positive control, we used 5 μM CCCP, a proton ionophore. For further analysis of bacterial death, we stained bacterial nucleic acids with 5 μM cell-permeant SYTO 13 Green dye and 5 μM cell-impermeant SYTOX Orange dye (Life Technologies). Viable bacteria will be stained by SYTO 13 Green but not SYTOX Orange, whereas dead bacteria will be stained with both dyes.
Scanning electron microscopy of P. aeruginosa
We incubated 5 × 105 CFU of P. aeruginosa with IL-26 or vehicle (sodium phosphate, 1.0 M NaCl, 10% glycerol, pH 6.5) for 30–180 min in trypticase soy broth. Samples were placed on poly-L-lysine–treated glass coverslips and allowed to dry overnight. Samples on coverslips were mounted onto double-stick carbon tabs (Ted Pella) that had been previously mounted on aluminum specimen mounts (Electron Microscopy Sciences). The samples were then coated under vacuum with a Balzer MED 010 evaporator (Technotrade International) with platinum alloy to a thickness of 25 nm, after which they were immediately flash–carbon coated under vacuum. The samples were transferred to a desiccator for examination at a later date. Samples were examined in a JSM-5910 scanning electron microscope (JEOL) at an accelerating voltage of 5 kV.
ELISA binding assay
Nunc MaxiSorp 96-well plates were coated with PBS 1% BSA, 1 μg/ml LPS or 1 μg/ml LTA for 24 h at 4 °C. Plates were then washed, and increasing concentrations of IL-26, LL-37 or IL-22 diluted in PBS 1% BSA were added for 1 h at room temperature. Plates were then washed, and 0.5 μg/ml of mouse anti–IL-26 (clone 142-69-1), anti–LL-37 (sc-166770, Santa Cruz Biotechnology) or anti–IL-22 (clone 142906; MAB7822, R&D Systems) diluted in PBS 1% BSA was added for 1 h at room temperature. Plates were then washed, and horseradish peroxidase–conjugated goat anti-mouse IgG (Thermo Scientific) diluted 1/5,000 in PBS 1% BSA was added to all wells for 30 min at room temperature. Plates were then washed, and TMB substrate was added. Plates were then read at 450 nm with a spectrometer.
In vivo lung infection model
Animal procedures were approved by the Office Vétérinaire du Canton de Vaud, Lausanne, Switzerland, and performed according to the Swiss federal guidelines for animal experimentation. K. pneumoniae strain Caroli (O1:K2)22 was cultured at 37 °C overnight in 10 ml brain-heart infusion medium, subcultured for 2 h, and further diluted to obtain 104 CFU/ml. Eight-week-old female BALB/c mice (Harlan Lab) were used. We administered 102 CFU of K. pneumoniae intranasally (i.n.) in a volume of 10 μl by holding the mice in an upright position under anesthesia. After infection, one of the following in a 20-μl volume was administered i.n.: 20 μg of IL-26, 50 μg LL-37 (positive control) or PBS (negative control). Another control condition was intranasal infection of mice with IL-26–pretreated bacteria (102 CFU + 20 μg IL-26 mixed for 1 h before administration). Seventy-two hours post-challenge, mice were euthanized, and blood, lungs and spleen were collected. Lungs and spleens were weighed and homogenized with a Potter-Elvehjem Pestle. Diluted blood, lung and spleen homogenates in NaCl were cultured overnight on blood agar plates before bacterial colonies were counted by two independent investigators.
Isolation of peripheral blood cells
Peripheral blood mononuclear cells (PBMCs) were obtained by Ficoll centrifugation of blood buffy coats. Monocytes, NK cells, and pDCs were then isolated by magnetic separation with the following commercially available kits: Monocyte Isolation Kit II (Miltenyi), EasySep Human NK Cell Enrichment Kit (StemCell), and Diamond pDC Isolation Kit II (Miltenyi). We differentiated macrophages by culturing monocytes with 50 ng/nl of the cell-signaling molecule GM-CSF for 5 d. After Ficoll centrifugation, we used the high-density granulocyte fraction to isolate neutrophils with CD15 Microbeads (Miltenyi). Purity was routinely >95%, as assessed by flow cytometry of CD14+ monocytes, CD56+ NK cells, BDCA2+CD123+ pDCs, CD14+CD68+FSChi macrophages and CD15+SSChi neutrophils.
Microarray analysis
Total RNA from sorted cells was immediately isolated with the RNeasy kit from Qiagen and used to generate cDNA according to the Expression Analysis Technical Manual (Affymetrix). cRNA samples were generated with the Bioarray High-Yield RNA Transcript Labeling kit (Enzo) and the Human Genome U133 Plus 2.0 array according to the manufacturer’s protocol (Affymetrix). The scanned images were aligned and analyzed with GeneChip Microarray Suite 5.0 (Affymetrix) according to the manufacturer’s instructions. The signal intensities were normalized to the mean intensity of all the genes represented on the array, and global scaling (to all probe sets) was done before comparison analysis. Genes with variable expression levels were selected on the basis of the following criteria: gene expression (i.e., presence calls) in at least one of the three samples, and a σi/μi ratio of >0.65, where σi and μi are the s.d. and mean of the hybridization intensity values of each particular gene across all samples, respectively.
In vitro generation and characterization of IL-26–DNA complexes
We generated IL-26–DNA complexes by mixing 600 ng of bacterial DNA (Life Technologies) or human DNA (BioChain) with different concentrations of recombinant IL-26 in 40 μl of nuclease-free water (Ambion) and then diluted them into 200 μl of complete medium for cell stimulation (final concentrations: 3 μg/ml DNA and 0.1–1 μM IL-26). IL-26–DNA complexes were visualized by 1.5% agarose gel migration or by confocal microscopy after DNA had been stained with DAPI (0.1 ng/ml; Sigma-Aldrich) or labeled with Alexa Fluor 488. DNA labeling with Alexa Fluor 488 was done with the Ulysis Nucleic Acid Labeling Kit (Molecular Probes), which uses a platinum dye complex to form a stable adduct with the N7 position of a guanine and, with less efficiency, with adenine bases in DNA. For detection of IL-26, suspensions containing precipitating complexes were stained overnight with mouse anti–IL-26 (clone 142-69-1), carefully washed, and incubated with Alexa Fluor 546–labeled anti-mouse secondary antibody for 1 h.
Generation of TH17 cells and TH17 cell clones
CD4+ naive T cells were enriched from buffy coats of healthy volunteers with the CD4+ Naive T Cell Isolation Kit II (Miltenyi Biotec). Afterward, the cells were sorted on a FACSAria (Becton Dickinson) as CD45RA+, CD4+, CD8−, CD14−, CD16−, CD20−, CD56−, γδTCR−, BDCA2−, CD11c−, CD25−, or CD45RO− to reach a purity of >95% naive CD4+CD45RA+ T cells. These cells were then plated in flat-bottom plates at 5 × 104 cells/well and cultured for 5 d in the presence of plate-bound anti-CD3 (10 μg ml−1) and soluble anti-CD28 (1 μg ml−1) + IL-1β (10 ng/ml) + IL-6 (20 ng/ml) + TGF-β (1 ng/ml) + TNF-α (10 ng/ml) + IL-23 (100 ng/ml) in Yssel’s medium (Gemini)23. As a control, we also stimulated naive T cells (i) without cytokines; (ii) with IL-12 (5 ng/ml), IFN-γ (20 ng/ml) and anti–IL-4 (10 μg/ml); and (iii) with IL-4 (25 ng/ml) and anti–IFN-γ (10 μg/ml) to generate TH0, TH1 and TH2 cells, respectively. All cytokines used for polarization were purchased from R&D Systems. For the TH17 cell clones, 1G3, 7H1 and 72G6 cells were generated as described previously and cultured in RPMI 1640 medium (Invitrogen) supplemented with 2 mmol/L L-glutamine, 0.05 mmol/L β-mercaptoethanol, 10% human male AB serum (GemCell), 300 IU/mL IL-2, 5 ng/mL IL-1β and 10 ng/mL IL-23 in the presence of the irradiated feeder EBV B cell line LCL111 at 0.3 × 106/mL. To generate supernatants, we re-stimulated primary TH17 cells or TH17 cell clones with anti-CD3 or anti-CD28 in antibiotic-free medium and harvested cell-free supernatants 24 h later. IL-26 protein levels in TH17 cell supernatants were measured by ELISA (USCN Life Science). In some experiments, TH17 cell clones were transfected at 37 °C for 6 h with 80 pmol of control siRNA-A (scrambled sequence) or siRNA targeting IL-26 complexed with 6 μl of siRNA Transfection Reagent according to the manufacturer’s protocol (Santa Cruz Biotechnology). Cells were then washed and re-stimulated with anti-CD3 or anti-CD28 in antibiotic-free medium, and cell-free supernatants were harvested 24 h later. The IL-26 concentration in the different supernatants was measured by ELISA (Cusabio Biotech).
Real-time PCR analysis
Polarized TH0, TH1, TH2, and TH17 cells were harvested, lysed with TRI Reagent (Ambion), and stored at −20 °C. Total RNA was extracted with the RiboPure kit (Ambion) and cleaned with the RNAqueous kit (Ambion). One microgram of RNA was then reverse-transcribed into cDNA (High Capacity cDNA, Applied Biosystems). For each individual gene, 20 ng of cDNA was amplified for expression analysis with TaqMan on an ABI 7500 Fast system. Human GAPDH mRNA levels were quantified and used for normalization as described30. Human TaqMan probes used were IL26 (Hs00218189_m1), IL17A (Hs00174383_m1), IL17F (Hs00369400_m1), IL22 (Hs01574154_m1), IFNG (Hs00989291_m1) and IL13 (Hs00174379_m1), all from Life Technologies.
Imaging of IL-26–DNA complexes in TH17 cell supernatants
We visualized DNA–IL-26 complexes in TH17 cell supernatants by staining precipitating complexes with 10 μg/ml mouse anti–IL-26 (clone 142-69-1) for 2 h and then incubating them with Alexa Fluor 546–labeled anti-mouse secondary antibody for 1 h. After washing, DAPI (0.1 ng/ml) was added to stain DNA and visualized by confocal microscopy.
Stimulation of immune cells by IL-26–DNA complexes or TH17 cell supernatants
Purified immune cells were cultured in 96-well round-bottom plates at 5 × 104 cells/well in 200 μl RPMI 1640 medium (Gibco) supplemented with 10% FCS (Atlanta Biologicals) and penicillin-streptomycin in the presence of 1 μM IL-26, bacterial DNA or human DNA, or with IL-26–DNA complexes. The viability of cells after culture was assessed by flow cytometry with 7-AAD staining according to the manufacturer’s protocol (BioLegend). In some experiments, pDCs were stimulated with different amounts of live bacteria (P. aeruginosa), different concentrations of DNA content from chemically lysed bacteria (50 mM Tris, pH 8.0, 0.1% Triton X-100) or different amounts of live or UV-treated dying HEK293T cells (1 × 106 cells/ml) in the presence of 10 μM IL-26. The HEK293T cell line was from ATCC (CRL-11268). Cell cultures were routinely tested for mycoplasma contamination. pDCs were also stimulated with various amounts of supernatants derived from TH0 or TH17 cells alone or supplemented with 3 μg/ml human DNA. In some experiments the supernatants or in vitro–generated complexes were pretreated with DNase I (1,000 U/ml) or heparin (up to 10 U/ml).The different immune-cell supernatants were collected after overnight culture, and the levels of cytokine production (IL-1β, IL-6, TNF-α (R&D Systems) and IFN-α (PBL Biomedical Laboratories)) were measured by ELISA.
Uptake and internalization of IL-26–DNA complexes by pDCs
Purified pDCs were incubated at 37 °C for 3 h with 3 μg/ml Alexa Fluor 488–labeled DNA mixed with 1 μM IL-26 at 5 × 104 cells/well in 200 μl RPMI 1640 medium (Gibco) supplemented with 10% FCS (Atlanta Biologicals). Cells were then fixed with 4% paraformaldehyde, stained for the pDC-specific marker CD123 (anti-CD123–allophycocyanin, BD Biosciences) and analyzed by flow cytometry (FACS Calibur, BD Biosciences). Alternatively, cells were allowed to attach to poly-L-lysine–coated coverslips for 3 h, fixed with 4% paraformaldehyde and stained for the pDC-specific marker CD123 (anti-CD123–phycoerythrin, clone 7G3, BD Biosciences) or the early endosome marker CD71 (anti-CD71–biotin, clone M-A712, BD Biosciences, plus streptavidin-phycoerythrin) and mounted in ProLong with DAPI (Life Technologies).
TLR9 reporter gene assay
HEK293 cells that stably co-express a human TLR4 or TLR9 gene and an NF-κB–inducible secreted embryonic alkaline phosphatase (SEAP) reporter gene were purchased from Invivogen. After TLR4 or TLR9 activation, secreted SEAP levels were detected in the cell-free supernatant with QUANTI-Blue medium (InvivoGen) and quantified on the basis of optical density at 620 nm.
Tissue homogenization for IL-26 ELISA
The skin biopsies were transferred into polypropylene tubes containing 1 ml buffer consisting of PBS supplemented with 1 M NaCl and protease inhibitor (Protease Inhibitor Cocktail Set III, Merck Millipore) before homogenization was performed with a Polytron PT 2500 E (Kinematica, Switzerland). The homogenates were then transferred into Protein Low Bind Tubes (Eppendorf) and centrifuged at 5,000g for 5 min at 4 °C. The resulting supernatant was stored at −20 °C. To determine the concentration of total protein, we used the Pierce BCA Protein Assay Kit (Thermo Scientific) according to the manufacturer’s protocol. We adjusted the samples’ total protein concentration to 100 μg/ml before we measured IL-26 concentrations by ELISA (Cusabio Biotech). We then estimated the concentration of IL-26 in the original skin-biopsy sample by dividing the total amount of IL-26 measured in each sample by the skin-biopsy volume. The skin-biopsy volume in a cylindrical 3-mm punch-biopsy specimen was estimated as follows: h × (πr2) = 3 × (π × 1.52) = 21.2 mm3 = 21.2 μl.
Statistical analysis
For statistical analyses, we used the two-tailed unpaired Student’s t-test for in vitro experiments and the unpaired non-parametric Mann-Whitney U-test for in vivo experiments and patient-sample analyses. P < 0.05 was considered significant. Group sizes, reproducibility and P values for each experiment are given in figure legends.
Supplementary Material
Acknowledgments
We thank K. Dunner for scanning electron microscopy at the HREM Facility, MD Anderson Cancer Center. We thank L. Bover and the Monoclonal Antibody Core Facility staff at the MD Anderson Cancer Center for assistance in generating monoclonal antibodies to IL-26. We thank the Berkeley Laboratory Advanced Light Source and the SIBYLS beamline staff at 12.3.1 for assistance with the collection of small-angle X-ray scattering data, and we thank K. Dyer for the mail-in service provided by SIBYLS. This work was funded by the Swiss National Science Foundation (grant 144072), the National Cancer Institute (PO1 grant CA128913) and the DANA Foundation (all to M.G.). S.M. was partly supported by the German Research Foundation and the research commission of the Medical Faculty of the University of Düsseldorf. Research by S.T.A. was supported by the King Abdullah University of Science and Technology (KAUST). T.R. is supported by the Swiss National Science Foundation (grant 149511).
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
S.M. and J.D.D. performed and analyzed most of the experiments and participated in their design. G.C. and J.G. helped establish, perform and analyze antimicrobial assays. D.G. performed TLR9 reporter assays and participated in the imaging studies. C.C. and K.S.V. helped establish the T cell culture experiments. H.C.F. performed ELISA analysis of the skin biopsies. J.E.L. and S.T.A. designed and performed the protein-modeling and small-angle X-ray scattering experiments. D.L.R. and T.R. provided virulent strains of bacteria and helped with the in vivo infectious model. B.H., D.P.K., Y.-J.L. and R.L.M. provided suggestions and discussions throughout the study. S.W. provided initial modeling data for IL-26. M.G. conceived and supervised the study, was involved in the design and evaluation of all experiments, and wrote the manuscript along with S.M. and J.D.D., with comments from other co-authors.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Wilson NJ, et al. Development, cytokine profile and function of human interleukin 17–producing helper T cells. Nat Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 2.Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor–β and induction of the nuclear receptor RORγt. Nat Immunol. 2008;9:641–649. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of T(H)17 cells. Nature. 2008;453:1051–1057. doi: 10.1038/nature07036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma CS, et al. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med. 2008;205:1551–1557. doi: 10.1084/jem.20080218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Beaucoudrey L, et al. Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J Exp Med. 2008;205:1543–1550. doi: 10.1084/jem.20080321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23–IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. 2014;14:585–600. doi: 10.1038/nri3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ye P, et al. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolk K, Witte E, Witte K, Warszawska K, Sabat R. Biology of interleukin-22. Semin Immunopathol. 2010;32:17–31. doi: 10.1007/s00281-009-0188-x. [DOI] [PubMed] [Google Scholar]
- 9.Nurieva R, et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- 10.Knappe A, Hor S, Wittmann S, Fickenscher H. Induction of a novel cellular homolog of interleukin-10, AK155, by transformation of T lymphocytes with herpesvirus saimiri. J Virol. 2000;74:3881–3887. doi: 10.1128/jvi.74.8.3881-3887.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Donnelly RP, et al. Interleukin-26: an IL-10-related cytokine produced by Th17 cells. Cytokine Growth Factor Rev. 2010;21:393–401. doi: 10.1016/j.cytogfr.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dambacher J, et al. The role of the novel Th17 cytokine IL-26 in intestinal inflammation. Gut. 2009;58:1207–1217. doi: 10.1136/gut.2007.130112. [DOI] [PubMed] [Google Scholar]
- 13.Corvaisier M, et al. IL-26 is overexpressed in rheumatoid arthritis and induces proinflammatory cytokine production and Th17 cell generation. PLoS Biol. 2012;10:e1001395. doi: 10.1371/journal.pbio.1001395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goris A, Marrosu MG, Vandenbroeck K. Novel polymorphisms in the IL-10 related AK155 gene (chromosome 12q15) Genes Immun. 2001;2:284–286. doi: 10.1038/sj.gene.6363772. [DOI] [PubMed] [Google Scholar]
- 15.Vandenbroeck K, et al. Polymorphisms in the interferon-gamma/interleukin-26 gene region contribute to sex bias in susceptibility to rheumatoid arthritis. Arthritis Rheum. 2003;48:2773–2778. doi: 10.1002/art.11236. [DOI] [PubMed] [Google Scholar]
- 16.Silverberg MS, et al. Ulcerative colitis-risk loci on chromosomes 1p36 and 12q15 found by genome-wide association study. Nat Genet. 2009;41:216–220. doi: 10.1038/ng.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hör S, et al. The T-cell lymphokine interleukin-26 targets epithelial cells through the interleukin-20 receptor 1 and interleukin-10 receptor 2 chains. J Biol Chem. 2004;279:33343–33351. doi: 10.1074/jbc.M405000200. [DOI] [PubMed] [Google Scholar]
- 18.Sheikh F, et al. Cutting edge: IL-26 signals through a novel receptor complex composed of IL-20 receptor 1 and IL-10 receptor 2. J Immunol. 2004;172:2006–2010. doi: 10.4049/jimmunol.172.4.2006. [DOI] [PubMed] [Google Scholar]
- 19.Nagem RA, et al. Crystal structure of recombinant human interleukin-22. Structure. 2002;10:1051–1062. doi: 10.1016/s0969-2126(02)00797-9. [DOI] [PubMed] [Google Scholar]
- 20.Zdanov A, et al. Crystal structure of interleukin-10 reveals the functional dimer with an unexpected topological similarity to interferon gamma. Structure. 1995;3:591–601. doi: 10.1016/s0969-2126(01)00193-9. [DOI] [PubMed] [Google Scholar]
- 21.Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- 22.Roger T, et al. Macrophage migration inhibitory factor deficiency is associated with impaired killing of gram-negative bacteria by macrophages and increased susceptibility to Klebsiella pneumoniae sepsis. J Infect Dis. 2013;207:331–339. doi: 10.1093/infdis/jis673. [DOI] [PubMed] [Google Scholar]
- 23.Volpe E, et al. A critical function for transforming growth factor–β, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol. 2008;9:650–657. doi: 10.1038/ni.1613. [DOI] [PubMed] [Google Scholar]
- 24.Lande R, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449:564–569. doi: 10.1038/nature06116. [DOI] [PubMed] [Google Scholar]
- 25.Chamilos G, et al. Cytosolic sensing of extracellular self-DNA transported into monocytes by the antimicrobial peptide LL37. Blood. 2012;120:3699–3707. doi: 10.1182/blood-2012-01-401364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lande R, et al. Cationic antimicrobial peptides in psoriatic skin cooperate to break innate tolerance to self-DNA. Eur J Immunol. 2015;45:203–213. doi: 10.1002/eji.201344277. [DOI] [PubMed] [Google Scholar]
- 27.Lande R, et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA–peptide complexes in systemic lupus erythematosus. Sci Transl Med. 2011;3:73ra19. doi: 10.1126/scitranslmed.3001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghirelli C, Zollinger R, Soumelis V. Systematic cytokine receptor profiling reveals GM-CSF as a novel TLR-independent activator of human plasmacytoid predendritic cells. Blood. 2010;115:5037–5040. doi: 10.1182/blood-2010-01-266932. [DOI] [PubMed] [Google Scholar]
- 29.Sandgren S, et al. The human antimicrobial peptide LL-37 transfers extracellular DNA plasmid to the nuclear compartment of mammalian cells via lipid rafts and proteoglycan-dependent endocytosis. J Biol Chem. 2004;279:17951–17956. doi: 10.1074/jbc.M311440200. [DOI] [PubMed] [Google Scholar]
- 30.Nestle FO, et al. Plasmacytoid predendritic cells initiate psoriasis through interferon-α production. J Exp Med. 2005;202:135–143. doi: 10.1084/jem.20050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raffatellu M, et al. Simian immunodeficiency virus–induced mucosal interleukin-17 deficiency promotes Salmonella dissemination from the gut. Nat Med. 2008;14:421–428. doi: 10.1038/nm1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Connell RM, et al. Type I interferon production enhances susceptibility to Listeria monocytogenes infection. J Exp Med. 2004;200:437–445. doi: 10.1084/jem.20040712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Teles RM, et al. Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science. 2013;339:1448–1453. doi: 10.1126/science.1233665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qiu H, et al. Type I IFNs enhance susceptibility to Chlamydia muridarum lung infection by enhancing apoptosis of local macrophages. J Immunol. 2008;181:2092–2102. doi: 10.4049/jimmunol.181.3.2092. [DOI] [PubMed] [Google Scholar]
- 35.Parker D, et al. Induction of type I interferon signaling by Pseudomonas aeruginosa is diminished in cystic fibrosis epithelial cells. Am J Respir Cell Mol Biol. 2012;46:6–13. doi: 10.1165/rcmb.2011-0080OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mancuso G, et al. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol. 2007;178:3126–3133. doi: 10.4049/jimmunol.178.5.3126. [DOI] [PubMed] [Google Scholar]
- 37.Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (α/β) in immunity and autoimmunity. Annu Rev Immunol. 2005;23:307–336. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 38.Santini SM, et al. Type I interferon as a powerful adjuvant for monocyte-derived dendritic cell development and activity in vitro and in Hu-PBL-SCID mice. J Exp Med. 2000;191:1777–1788. doi: 10.1084/jem.191.10.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Luft T, et al. Type I IFNs enhance the terminal differentiation of dendritic cells. J Immunol. 1998;161:1947–1953. [PubMed] [Google Scholar]
- 40.Le Bon A, et al. Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat Immunol. 2003;4:1009–1015. doi: 10.1038/ni978. [DOI] [PubMed] [Google Scholar]
- 41.Hibbert L, Pflanz S, De Waal Malefyt R, Kastelein RA. IL-27 and IFN-α signal via Stat1 and Stat3 and induce T-Bet and IL-12Rβ2 in naive T cells. J Interferon Cytokine Res. 2003;23:513–522. doi: 10.1089/10799900360708632. [DOI] [PubMed] [Google Scholar]
- 42.Jego G, et al. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity. 2003;19:225–234. doi: 10.1016/s1074-7613(03)00208-5. [DOI] [PubMed] [Google Scholar]
- 43.Venet F, Huang X, Chung CS, Chen Y, Ayala A. Plasmacytoid dendritic cells control lung inflammation and monocyte recruitment in indirect acute lung injury in mice. Am J Pathol. 2010;176:764–773. doi: 10.2353/ajpath.2010.090765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lande R, et al. Characterization and recruitment of plasmacytoid dendritic cells in synovial fluid and tissue of patients with chronic inflammatory arthritis. J Immunol. 2004;173:2815–2824. doi: 10.4049/jimmunol.173.4.2815. [DOI] [PubMed] [Google Scholar]
- 45.Baumgart DC, et al. Aberrant plasmacytoid dendritic cell distribution and function in patients with Crohn’s disease and ulcerative colitis. Clin Exp Immunol. 2011;166:46–54. doi: 10.1111/j.1365-2249.2011.04439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ganguly D, et al. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med. 2009;206:1983–1994. doi: 10.1084/jem.20090480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Opdenakker G, Rudd PM, Wormald M, Dwek RA, Van Damme J. Cells regulate the activities of cytokines by glycosylation. FASEB J. 1995;9:453–457. doi: 10.1096/fasebj.9.5.7896019. [DOI] [PubMed] [Google Scholar]
- 48.Wang G. Post-translational modifications of natural antimicrobial peptides and strategies for peptide engineering. Curr Biotechnol. 2012;1:72–79. doi: 10.2174/2211550111201010072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Konarev PV, Petoukhov MV, Volkov VV, Svergun DI. ATSAS 2.1, a program package for small-angle scattering data analysis. J Appl Crystallogr. 2006;39:277–286. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.