

SUMMARY
Immune checkpoint blockade therapies fail to induce responses in majority of cancer patients; so how to increase the objective response rate becomes an urgent challenge. Here we demonstrate that sufficient T cell infiltration in tumor tissues is a prerequisite for response to PD-L1 blockade. Targeting tumors with tumor necrosis factor superfamily member LIGHT activates lymphotoxin beta receptor signaling, leading to the production of chemokines that recruit massive numbers of T cells. Furthermore, targeting non-T cell-inflamed tumor tissues by antibody-guided LIGHT creates a T cell-inflamed microenvironment and overcomes tumor resistance to checkpoint blockade. Our data indicates that targeting LIGHT might be a potent strategy to increase the responses to checkpoint blockades and other immunotherapies in non-T cell-inflamed tumors.
Graphical Abstract
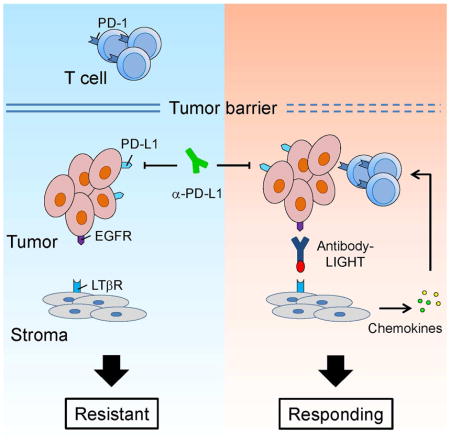
INTRODUCTION
The Programmed Cell Death Protein 1 (also known as CD279 and PD-1) and its ligand PD-1 Ligand (PD-L1) signaling pathway is a critical immune checkpoint that functions normally to protect against autoimmunity (Keir et al., 2008; Nishimura et al., 2001). Increasing evidence has suggested that PD-1 signaling is also an important mechanism utilized by tumors to escape antitumor immune responses (Dong et al., 2002; Iwai et al., 2002; Shin and Ribas, 2015). Recent clinical trials with anti-PD-1 and PD-L1 monoclonal antibodies have shown unprecedented durable responses in some patients with a variety of cancers (Brahmer et al., 2012; Topalian et al., 2012). Unfortunately, only a minority of total treated patients respond to the current immunotherapy treatment. Thus, it has become a top priority to identify the factors that determine the responsiveness to checkpoint blockade, and to develop strategies that could potentially increase the patient response rates (Sznol and Chen, 2013).
Some recent retrospective clinical studies have shown correlations between tumor PD-L1 expression and response to PD-1/PD-L1 checkpoint blockade therapy (Herbst et al., 2014; Topalian et al., 2012). In contrast, other studies have also suggested that the presence of tumor-infiltrating lymphocytes (TILs) is an important biomarker for predicting responses to PD-L1 blockade therapy (Tumeh et al., 2014). Interestingly, the presence of TILs has been previously shown to correlate with better patient outcomes during various antitumor therapies in multitude of cancers (Galon et al., 2006; Hwang et al., 2012; Mahmoud et al., 2011). However, it is commonly known that the tumor microenvironment often inhibits activated T cells from entering tumor tissues or prevents effective T cell priming for tumor control through various pathways (Gajewski et al., 2013). By using only clinical samples and data, it is hard to dissect the relative contribution of PD-L1 and TILs for responsiveness to PD-L1 blockade; thus, proper mouse tumor models are needed for conclusive mechanism studies.
Our lab has previously shown that upregulation of LIGHT (stands for “homologous to lymphotoxin, exhibits inducible expression and competes with HSV glycoprotein D for binding to herpesvirus entry mediator, a receptor expressed on T lymphocytes”) in peripheral tissues results in T cell activation and migration into non-lymphoid tissues and the formation of lymphoid-like structures, which can lead to rapid T cell-mediated tissue destruction (Lee et al., 2006). LIGHT, also known as Tumor Necrosis Factor Superfamily member 14 (TNFSF14), is one of the costimulatory molecules that can regulate T-cell activation (Wang et al., 2009). LIGHT is predominantly expressed on immune cells, especially on the surface of immature Dendritic Cells (DCs) and activated T cells. Forced expression of LIGHT in tumor cells promotes the formation of lymphoid-like structures for direct T-cell sequestration and activation, leading to tumor regression (Yu et al., 2004; Yu et al., 2007). Furthermore, adoptive transfer of LIGHT-expressing mesenchymal stem cells can enhance T cell infiltration and efficiently control tumors (Zou et al., 2012).
LIGHT is a ligand protein that can bind to two different receptors, HerpesVirus Entry Mediator (HVEM), which is also known as tumor necrosis factor receptor superfamily member 14 and is encoded by TNFRSF14, and Lymphotoxin beta Receptor (LTβR), which is encoded by LTBR. The binding of LIGHT to HVEM delivers a co-stimulatory signal to T cells (Wang et al., 2009). In addition, LIGHT can bind to LTβR, which is commonly expressed on non-lymphoid cells, and is critical for the formation of secondary and tertiary lymphoid structures (Fu and Chaplin, 1999; Ware, 2005). LTβR plays a pivotal role in the formation of lymph nodes (LNs) and in the organization of distinct T-cell and B-cell zones in secondary lymphoid organs. Signaling via LTβR regulates the expression of various chemokines and adhesion molecules which control the migration and positioning of DCs and lymphocytes in the spleen (Cyster, 1999). Over-expression of lymphotoxin in non-lymphoid tissues is sufficient to promote functional lymphoid neogenesis (Ruddle, 1999). These activities indicate that activating LIGHT signaling might be an attractive approach to increase lymphocyte infiltration in tumor tissues. Based on these studies, we sought to test whether targeting LIGHT into tumor tissues could increase TIL numbers, and whether it could synergize with current checkpoint blockade therapies.
RESULTS
Higher T cell infiltration, but not PD-L1 level, is associated with responsiveness to checkpoint blockade
The mechanistic studies about whether and how TILs or PD-L1 are required for a positive response to checkpoint blockades has not been completely elucidated due to the lack of proper experimental models. To understand why some PD-L1+ tumors do not respond to PD-L1 blockade while other tumors do respond, we compared a series of well-established mouse tumor lines for their PD-L1 expression and responsiveness to anti-PD-L1 treatment (Table S1). Interestingly, the implanted tumor lines, such as MC38 and Ag104Ld, represent distinct models mirroring what have been observed in the clinic; specifically, MC38 and Ag104Ld both have similarly high levels of PD-L1 expression while having different responsiveness to anti-PD-L1 therapy (Figure 1A–1C, Table S1). When stimulated by IFN-γ, they both upregulated PD-L1 to similar levels, indicating there was no intrinsic defects in PD-L1 expression upon stimulation in both cell lines (Figure 1A). Mice bearing MC38 tumors were able to control their tumor burdens effectively with anti-PD-L1 treatment (Figure 1B). In contrast, mice bearing Ag104Ld tumors treated with the same anti-PD-L1 did not respond to treatment and the tumor burdens were not controlled (Figure 1C). Both ex vivo MC38 and Ag104Ld tumors expressed similar levels of PD-L1, as analyzed by flow cytometry (Figure 1D). In addition, there was no significant difference of PD-L1 expression in TILs (Figure S1A–S1C). These data suggest that factors other than PD-L1 expression inside the tumor environment might be essential for responsiveness to PD-L1 blockade therapy.
Figure 1. Significant lymphocyte infiltration is associated with responsiveness to PD-L1 blockade.

(A) MC38 and Ag104Ld cells were treated with or without 500 U/ml IFN-γ for 24 hr then stained with anti-PD-L1. The expression levels of PD-L1 were measured by flow cytometry. Mean fluorescent intensities (MFIs) of PD-L1 staining were compared. (B) WT B6 mice were inoculated subcutaneously with 1×106 MC38 cells on day 0. On day 7 and 10, mice were treated with 200 μg anti-PD-L1 or control Immunoglobulin G (IgG). Tumor growth was measured and compared twice weekly. (C) B6C3F1 mice were injected subcutaneously with 1×106 Ag104Ld cells and treated with 200 μg anti-PD-L1 or control IgG on day 7 and 10. (D) Tumor tissues were collected 7 days after inoculation. PD-L1 expression levels in CD45− cells were measured by flow cytometry. FMO, Fluorescence Minus One. (E) Tumor tissues were collected as in (D). Percentages of CD3+ among CD45+ cells (upper panel) and CD8+ among CD3+ cells (lower panel) were analyzed by flow cytometry. (F) WT B6 mice were injected subcutaneously with 1×106 MC38 cells on day 0 and treated with FTY720 from day 1. On day 7 and 10, mice were treated with 200 μg anti-PD-L1 or control IgG. Tumor growth was measured and compared twice weekly. Data indicate mean ± SEM and are representative of two (A, B, F) or three (C, D, E) independent experiments. **p < 0.01. See also Figure S1 and Table S1.
To determine whether MC38 and Ag104Ld might have different tumor microenvironments that could contribute to the differences in response to checkpoint blockade therapy, we decided to examine the tumors and look for the presence of TILs. To compare the levels of lymphocyte infiltration, tumor tissues were collected and analyzed by flow cytometry. Interestingly, MC38 tumors have much more (up to 5 fold) T cells (CD45+CD3+) than Ag104Ld tumors. Among the tumor-infiltrating T cells, the percentage of CD8+ T cells is also higher in MC38 (Figure 1E). Specifically, there is approximately 7~10 fold more CD8+ T cells in MC38 over Ag104Ld. These data raise the possibility that more CD8+ T cells inside MC38 tumors, not seen in Ag104Ld tumors, leads to its responsiveness to anti-PD-L1. To address the role of TILs for checkpoint blockade responsiveness, we sought two strategies to test: 1) whether a reduction of TILs in MC38 will diminish its response to PD-L1 blockade, and 2) whether an increase in TILs in Ag104Ld will induce its response to PD-L1 blockade.
To address whether higher number of TILs in MC38 is responsible for its responsiveness to PD-L1 blockade, we utilize FTY720 to block new lymphocyte infiltration. FTY720 is a small molecule analogue of Sphingosine 1-Phosphate (S1P). FTY720 treatment induces the internalization and degradation of S1P Receptor, thereby preventing lymphocyte egress from the lymph nodes (Thompson et al., 2010). After a single injection of FTY720, there was ~90% reduction in peripheral T cells (Figure S1D). Circulating T cell numbers gradually recovered after four days post injection. The concentration of FTY720 used does not induce cell death in vitro (Figure S1E–S1H). To test whether a higher number of TILs is required for the response to PD-L1 blockade, mice were treated with FTY720 after MC38 tumor inoculation. After tumors were established, mice were treated with anti-PD-L1. Strikingly, the antitumor effects of anti-PD-L1 were completely abrogated in the presence of FTY720 (Figure 1F). Those data suggest that a significant number of TILs is a prerequisite for the response to PD-L1 blockade.
Generation and selection of human LIGHT mutants that bind to mouse receptors with higher affinities
To test our hypothesis that increased TILs in non-T cell-inflamed tumors, such as Ag104Ld, are able to induce response to PD-L1 blockade, we sought to target LIGHT signaling to increase infiltration. Initial attempts to produce recombinant LIGHT protein to target tumor tissues were not successful since recombinant mouse LIGHT (mLIGHT) is not stable and tends to aggregate (data not shown, and (Del Rio et al., 2010)). On the other hand, human LIGHT (hLIGHT) is more stable but fails to bind mouse receptors, so it cannot be used in experimental mouse models. To verify the absence of cross-species binding, we performed binding experiments using a yeast-displayed version of wild-type (WT) hLIGHT that showed undetectable binding to mouse LTβR and HVEM (mLTβR and mHVEM), with positive binding to human LTβR and HVEM (hLTβR and hHVEM) (Figure 2A). To better evaluate the therapeutic efficacy of hLIGHT construct in experimental model systems, we engineered a LIGHT protein that has the capabilities of binding and activating both human and mouse receptors. HmLIGHT (human LIGHT mutant that can effectively bind both human and mouse receptors) was selected from a random error mutagenesis library of hLIGHT using yeast surface display. Engineered mutants of hLIGHT that had increased surface expression (indicative of protein stability) and binding to both mouse and human receptors were isolated (Figure 2A). Several of these mutants have been identified. One of them has been chosen for further studies due to its remarkable thermal stability (LTβR binding above 80 ºC, Figure 2B) and, similarly higher binding affinities to both mouse and human receptors in the yeast display system. Sequencing results showed the presence of 4 point mutations between hmLIGHT and WT hLIGHT (Figure 2C). This stable and higher affinity hmLIGHT may have an increased therapeutic efficacy in immunocompetent host, with the additional benefit of being suitable for both mouse and human experimental model systems.
Figure 2. Engineered LIGHT has increased stability and binding affinities to both human and mouse receptors.

(A) Flow cytometry histograms of yeast-displayed mLIGHT, hLIGHT, and hmLIGHT clones. All clones were stained with the indicated ligands and analyzed by flow cytometry. (B) Flow cytometry histograms from thermal denaturation experiments using yeast-displayed hLIGHT and hmLIGHT. Samples were heated at indicated temperatures for 30 minutes and stained with mLTβR. (C) Amino acid sequence alignment of hLIGHT, mLIGHT, and hmLIGHT. Residues highlighted by black indicate the differences between hmLIGHT and hLIGHT. Differences between mLIGHT and hLIGHT are highlighted by grey.
Production and characterization of antibody-LIGHT fusion protein in vitro
To confirm the functionality of the sequence isolated from the yeast display system, hmLIGHT was cloned to produce recombinant protein. First, we confirmed that hmLIGHT was capable of binding to both human and mouse receptors in ELISA assays with great sensitivity (Figure 3A). Second, we were able to demonstrate activation of signaling since stimulation of human T cells by hmLIGHT induced the production of IFN-γ in a dose-dependent manner (data not shown). HmLIGHT also induced the production of IFN-γ in mouse splenocytes (Figure 3B) and IL-6 in Mouse Embryonic Fibroblast (MEF) cells (Figure 3C), while hLIGHT only showed limited activity.
Figure 3. In vitro characterization of hmLIGHT fusion protein.

(A) Binding between recombinant hmLIGHT and human/mouse LTβR/HVEM were measured by ELISA. (B) Splenocytes from Rag1−/− mice were stimulated with 25, 5, or 1 nM of hLIGHT or hmLIGHT for 48 hr. IFN-γ levels in culture supernatants were measured by Cytometric Bead Array (CBA). (C) MEF cells were stimulated with hLIGHT or hmLIGHT for 24 hr. IL-6 levels were measured by CBA. (D) Schematic representation shows anti-EGFR-hmLIGHT fusion protein construction. (E) B16-EGFR cells were incubated with anti-EGFR-hmLIGHT, followed by mLTβR or mHVEM staining. A control antibody-hmLIGHT fusion protein was used as negative control. (F) Splenocytes from Rag1−/− mice were pretreated with or without mHVEM-Ig fusion protein before treated with 25, 5, or 1 nM anti-EGFR-hmLIGHT for 48 hr. IFN-γ levels were measured by CBA. Data indicate mean ± SEM and are representative of at least two independent experiments. Conc., Concentration. See also Figure S2.
Given the limitations of a therapeutic that requires local delivery to patients, and that systemic injections of immune cytokine can often lead to dose-dependent side effects, we wanted to develop a system that can provide targeted delivery of LIGHT (Yang et al., 2014). To study the mechanism of targeted LIGHT delivery, we took advantage of the inherent specificities of antibody fusion proteins. We generated an anti-EGFR-hmLIGHT fusion protein (Ab-LIGHT) to specifically target hmLIGHT to EGFR-expressing tumor tissues. In order to avoid aggregations, three units of hmLIGHT (3×hmLIGHT) were linked together by polypeptide linkers, and then fused to the N-terminal of antibody IgG Fc (Figure 3D). The resulting anti-EGFR-hmLIGHT fusion protein could specifically bind to both EGFR and mLTβR/mHVEM (Figure 3E and S2A–S2B). In vitro activities of the fusion protein were further confirmed by its ability to induce IFN-γ production in mouse splenocytes (Figure 3F). This stimulation could be abrogated by recombinant mHVEM-Ig. Furthermore, the fusion protein can specifically target to EGFR-positive tumor tissues when delivered systemically (Figure S2C). Together, these data suggest hmLIGHT’s functional capabilities to bind and specifically activate LIGHT receptors in vitro.
Targeted delivery of LIGHT eradicates established tumors
To test the activity of hmLIGHT in targeting tumors in vivo, mice bearing established EGFR-expressing tumors were treated with anti-EGFR-hmLIGHT or control antibody. Ag104Ld is a highly progressive tumor model that is resistant to most immunotherapies through unknown mechanisms (Chen et al., 1994; Melero et al., 1997; Ward et al., 1989; Wick et al., 1997; Yu et al., 2005). We hypothesized that the lack of sufficient TILs makes Ag104Ld less responsive to most immunotherapies. Strikingly, anti-EGFR-hmLIGHT induced complete regression in established EGFR-expressing Ag104Ld tumors, while control antibody or anti-PD-L1 alone has no effect on tumor growth (Figure 4A and Figure 1C). No significant side effect was observed, as we didn’t see significant changes in either body weights or serum inflammatory cytokines (data not shown). The anti-tumor effects of anti-EGFR-hmLIGHT depend on EGFR-expression on tumor cells, as EGFR-negative tumor fails to response to the treatment (data not shown). To test whether LIGHT-mediated antitumor responses result in prolonged protective T cell immunity, mice that underwent complete tumor regression after anti-EGFR-hmLIGHT treatment were rechallenged with a lethal dose of Ag104Ld-EGFR cells. All the mice rejected rechallenged tumor (Figure 4B). Therefore, LIGHT is able to mediate rejection of a highly progressive tumor that is traditionally thought to be resistant to immunotherapies. In addition, LIGHT allows for the generation of memory cells that can mediate protection.
Figure 4. LIGHT delivered to tumor eradicates established tumors.

(A) B6C3F1 mice were inoculated subcutaneously with 2×106 Ag104Ld-EGFR cells and treated with 25 μg control IgG, anti-EGFR or anti-EGFR-hmLIGHT on day 7, 9, 11, and 13. Tumor growth was measured and compared twice weekly. (B) Two weeks after tumor eradication, mice treated with anti-EGFR-hmLIGHT from (A) were rechallenged with 1×107 Ag104Ld-EGFR cells. (C) Rag1−/− mice were inoculated subcutaneously with 1×106 MC38-EGFR cells, and 5×106 WT splenocytes were adoptively transferred on day 11. Mice were treated with 25 μg of control IgG, anti-EGFR, or anti-EGFR-hmLIGHT on day 12, 14, 16, and 18. (D) Rag1−/− mice were injected subcutaneously with 1×106 A431 cells. OT-1 lymph node (LN) cells (2×106) were adoptively transferred on day 13. Twenty-five microgram of control IgG, anti-EGFR, or anti-EGFR-hmLIGHT (Ab-homotrimer LIGHT from Abbvie) was administered intravenously daily from day 14 to 18. *p < 0.05, **p < 0.01. Data indicate mean ± SEM. One representative result of a total of three (A and D) or two (B and C) independent experiments is shown.
When tumor cells are initially transplanted into immunocompetent hosts, massive tumor cell necrosis leads to inflammation within the first few days (Dirkx et al., 2006). Therefore, it is possible that this initial inflammation might artificially increase the priming and recruiting of TILs. To avoid these extra priming and infiltrations, we set up a mouse tumor model without such priming of TILs. In this model, Rag1−/− mice were challenged with MC38-EGFR cells to allow the growth of tumor without T cell infiltration and priming. After tumors were established, WT splenocytes were transferred before immunotherapy treatment. Significantly, anti-EGFR-hmLIGHT showed superior antitumor effects over antibody control (Figure 4C).
One advantage of hmLIGHT is that it can be suitable for both human and mouse experimental models. To test the efficacy of anti-EGFR-hmLIGHT for controlling human tumor, and for possible future clinical implications, we developed a xenograft model using immune-reconstituted mice (Lee et al., 2009; Yang et al., 2013). Rag1−/− mice were inoculated with human A431 tumor cells that were previously established from an epidermoid carcinoma patient. After tumors were established, two million lymph node (LN) cells (~50% T cells) from Ovalbumin-specific class I-restricted T Cell Receptor (OT-1 TCR) transgenic mice were adoptively transferred. T cells from OT-1 mice have ~2% non-OT-1 T cells, some of which have the potential to recognize antigens from human tumors. A few hundreds of potential specific T cells is comparable to the number of tumor-reactive T cells observed in human patients. Furthermore, among the transferred T cells, ~98% of them are OT-1 specific T cells, which can prevent the homeostatic proliferation of tumor-reactive T cells. Without T cell transfer, A431 tumors grew aggressively. In the presence of T cells, anti-EGFR-hmLIGHT treatment induced much better antitumor effects compared to anti-EGFR control (Figure 4D). The same LIGHT treatment cannot control tumor growth in the absence of LN cells, indicating LIGHT-mediated antitumor effects are T cell-dependent. Taken together, these data showed that LIGHT is able to control tumor growth in different mouse and xenograft human tumor models, and to provide long-term immunological memory.
LIGHT-mediated antitumor immunity depends on LTβR signaling
LIGHT has two receptors, LTβR and HVEM. To further elucidate the essential contributions of LTβR and HVEM signaling in LIGHT-mediated antitumor immunity, tumor-bearing Rag1−/−;Ltbr−/− and Rag1−/−;Tnfrsf14−/− mice reconstituted with T cells were treated with anti-EGFR-hmLIGHT. Anti-EGFR-hmLIGHT fusion protein failed to control tumor growth in Rag1−/−;Ltbr−/− mice (Figure 5A). In contrast, anti-EGFR-hmLIGHT could control tumors in Rag1−/−;Tnfrsf14−/− mice as effectively as in Rag1−/− mice (Figure 5B and 4C). Similar to Ltbr−/− mice, Rag1−/−;Ltbr−/− mice have multiple immune abnormalities, including lack of secondary lymphoid structures (Zhu et al., 2010). To exclude the possibility that developmental defects dampen antitumor immune responses, WT mice bearing Ag104Ld-EGFR tumors were pretreated with LTβR-Ig before LIGHT treatment. The antitumor activities of anti-EGFR-hmLIGHT were completely abrogated in the presence of LTβR-Ig (Figure 5C). Consistently, depleting CD8+ T cells also eliminated the effects. We also determined that both MC38 and Ag104Ld tumors express similar level of LTβR (Figure S3). Together, these results suggest that LIGHT-mediated antitumor immunity mainly depends on LTβR signaling and T cells.
Figure 5. LIGHT-mediated antitumor immunity depends on LTβR-signaling.

(A) Rag1−/−;Ltbr−/− or (B) Rag1−/−;Tnfrsf14−/− mice were inoculated subcutaneously with 1×106 MC38-EGFR cells. Five million splenocytes were adoptively transferred on day 11. Mice were treated with 25 μg of anti-EGFR or anti-EGFR-hmLIGHT on day 12, 14, 16, and 18. Tumor growth was measured and compared twice weekly. (C) B6C3F1 mice were inoculated subcutaneously with 2×106 Ag104Ld-EGFR cells and treated with 25 μg anti-EGFR or anti-EGFR-hmLIGHT on day 7, 9, 11, and 13. LTβR-Ig (100 μg/mouse) was administrated on day 4, 7, and 11. For CD8+ T cell depletion, mice were treated with 200 μg anti-CD8 on day 7 and 11. Tumor growth was measured and compared twice weekly. (D–I) B6C3F1 mice were inoculated subcutaneously with 2×106 Ag104Ld-EGFR cells and treated with 25 μg anti-EGFR or anti-EGFR-hmLIGHT on day 7, 9, 11, and 13. Tumor tissues were analyzed on day 25. (D) Percentages of CD3+ among CD45+ cells (left) and CD8+ among CD3+ cells (right) were analyzed by flow cytometry. (E) Frozen sections of the indicated tumor tissues were stained with hematoxylin and anti-CD3 or anti-CD8. Scale bar, 100 μm. (F) Splenocytes were collected and an IFN-γ ELISPOT assay was performed with or without SIY peptide restimulation. (G) Inflammatory cytokine levels in homogenates from tumor tissues were measured by CBA. (H) RNA was isolated from tumor tissues treated with anti-EGFR or anti-EGFR-hmLIGHT. Relative expression levels of chemokines in pooled samples were measured by RT2 PCR Profiler and calculated as (anti-EGFR-hmLIGHT/anti-EGFR). (I) Expressions of CCL21a, CCL21b, CXCL19, and GlyCAM-1 in individual sample were measured by quantitative RT-PCR. Shown is the representative of two independent experiments (A, B, C, E, F) or the pool of two independent experiments (D, G, I). Data indicate mean ± SEM. *p < 0.05, **p < 0.01. N.S., not significant. See also Figure S3 and Table S2.
Activation of LTβR signaling in non-lymphoid tissues promotes functional lymphoid neogenesis (Ruddle, 1999). To find out whether anti-EGFR-hmLIGHT activated LTβR to increase TILs, tumor tissues were collected and analyzed after fusion protein treatment. There was 300~500% increase of CD8+ T cells in tumor tissues treated with anti-EGFR-hmLIGHT as both CD3+ and CD8+ cells were significantly increased (Figure 5D). Tumor histology showed higher number of CD3+ and CD8+ cells after anti-EGFR-hmLIGHT treatment (Figure 5E). The increase in antigen-specific CD8+ T cells indicated that a sufficient number of CTLs might play important roles in the rejection of tumors. To track tumor antigen-specific T cell responses, we generated an Ag104Ld-EGFR-SIY tumor cell line using the SIY peptide to mimic mutated antigens. Twelve days after the last anti-EGFR-hmLIGHT treatment, splenocytes from tumor-bearing mice were collected, and an IFN-γ ELISPOT assay was performed in the presence or absence of SIY peptides. The number of SIY-specific T cells dramatically increased after anti-EGFR-hmLIGHT treatment (Figure 5F). Inflammatory cytokine profile analysis showed that there were significant increases in the levels of IFN-γ, TNF-α, and IL-12 (Figure 5G). Taken together, these data suggest that LIGHT can, not only increase TILs, but also induce tumor-specific T cell responses for tumor control.
LTβR signaling induces IKKα-dependent expression of lymphoid tissues chemokines and adhesion molecules, which are able to recruit lymphocytes (Dejardin et al., 2002). To compare the chemokine expression profiles after LIGHT treatment, RNA was isolated from tumor tissues and the expression levels of chemokines were analyzed by RT2 Profiler PCR array. Interestingly, most of the chemokines associated with T cell trafficking were significantly upregulated after anti-EGFR-hmLIGHT treatment (Figure 5H, and Table S2) (Bromley et al., 2008). To further confirm anti-EGFR-hmLIGHT-mediated tumor rejection is through activating LTβR signaling, real-time PCR was performed. The level of CCL21 was increased ~6 fold after anti-EGFR-hmLIGHT treatment (Figure 5I). CCL21 is a ligand for CCR7. It is important for the homing of T cells to both lymphoid and nonlymphoid tissues (Lo et al., 2003). Other LTβR-regulated chemokines, including CXCL13 and GlyCAM-1, were also upregulated. Collectively, these data suggest that LIGHT enhances the recruitment of lymphocytes into tumor tissues through LTβR activation, and can induce tumor-specific T cell responses for tumor control and rejection.
LIGHT overcomes tumor resistance to anti-PD-L1 by increasing T cell infiltration
When tumors became larger, the antitumor effects of LIGHT gradually reduce. One possibility that could explain why LIGHT treatment does not work on large tumors is due to the fact that LIGHT increases the level of IFN-γ (Figure 5G), which can also promote PD-L1 upregulation as part of the adaptive resistance mechanism (Blank et al., 2004). We hypothesized that LIGHT might trigger inhibitory signals as a negative feedback mechanism, which in turn can dampen the initial antitumor effects on large tumors. To test this notion, tumor-bearing mice were treated with anti-EGFR-hmLIGHT and CD45− cells from tumors were analyzed for PD-L1 expression. Indeed, LIGHT treatment significantly increased the expression level of PD-L1 (Figure 6A). When Ag104Ld-EGFR tumors reached the size > 120 mm3, anti-EGFR-hmLIGHT alone has limited effects on tumor growth (Figure 6B). Impressively, additional PD-L1 blockade following LIGHT completely eradicated tumors, while PD-L1 blockade or LIGHT treatment alone failed to control tumors. The same synergistic effect was also observed in MC38-EGFR tumors (Figure 6C). Together, these data suggest that a proper combination treatment that dampens PD-L1 inhibition while increasing new T cell infiltration can overcome checkpoint blockade resistance, thus resulting in better tumor control than either treatment alone.
Figure 6. LIGHT overcomes tumor resistance to checkpoint blockade.

(A) B6C3F1 mice were treated as in Figure 5D. PD-L1 expression in CD45− cells were compared by flow cytometry. (B) B6C3F1 mice were inoculated subcutaneously with 2×106 Ag104Ld-EGFR cells and treated with 25 μg anti-EGFR or anti-EGFR-hmLIGHT on day 14, 16, 18, and 20. A PD-L1 blocking antibody (200 μg/mouse) was administered on day 14, and 18. Tumor growth was measured and compared twice weekly. (C) WT B6 mice were injected subcutaneously with 2×106 MC38-EGFR cells and treated with 25 μg of anti-EGFR or anti-EGFR-hmLIGHT on days 7 and 9. Anti-PD-L1 (100 μg/mouse) was administered on day 7. Tumor growth was measured and compared twice weekly. (D) WT B6 mice were inoculated subcutaneously with 2×106 MC38-EGFR cells and treated with FTY720 from day 1 to 3. Twenty-five microgram of anti-EGFR or anti-EGFR-hmLIGHT was administered on day 7, 9, 11, and 13. For PD-L1 blockade, mice were treated with 200 μg anti-PD-L1 on day 7 and 11. (E) Mice were treated as in (D). Two days after last treatment, tumor tissues were collected and tumor infiltrating leukocytes (CD3+ among CD45+ cells) were measured by flow cytometry. (F) MFIs of PD-L1 staining in CD45− cells were compared. Data are representative of three (A, D) or two (B) independent experiments, or the pool of two independent experiments (C, E, F). Data indicate mean ± SEM. *p < 0.05. **p < 0.01. N.S., not significant.
Since our data showed that LIGHT is efficient in recruiting TILs, we wonder whether LIGHT is able to rescue the responsiveness to checkpoint blockade in non-T cell-inflamed tumor. To test this hypothesis, MC38-EGFR tumor bearing mice were treated with FTY720 to block lymphocyte trafficking until four days before treatment. Mice were then treated with either anti-EGFR-hmLIGHT or anti-PD-L1 alone, or together (Figure 6D). Interestingly, although mice were only treated with FTY720 for the first few days, the antitumor effect of anti-PD-L1 was still completely lost. Strikingly, targeting tumor with LIGHT restored its ability to respond to anti-PD-L1 for tumor burden control (Figure 6D). Flow cytometry analysis showed that there was a significant reduction of TILs after FTY720 blockade (Figure 6E). Anti-EGFR-hmLIGHT treatment increased the number of TILs to a level comparable to control mice. Interestingly, LIGHT also increased the level of PD-L1. This observation could explain why LIGHT alone was not sufficient for tumor control while PD-L1 blockade with LIGHT allowed for synergistic responses (Figure 6F). Taken together, these data indicate that significant lymphocyte infiltration is critical for tumor responsiveness to checkpoint blockade immunotherapy. They also show that activation of LTβR signaling by LIGHT is able to overcome resistance to checkpoint blockade by increasing enough lymphocyte infiltration to the tumor tissues.
DISCUSSION
Immune checkpoint blockade is one of the most remarkable progresses in recent cancer therapy; however, objective responses are only achieved in a small portion of patients. Both PD-L1 expression and the presence of TILs have been implicated to correlate with responses to PD-L1 blockade (Herbst et al., 2014; Topalian et al., 2015; Topalian et al., 2012; Tumeh et al., 2014). Furthermore, the relative contribution of PD-L1 on tumor cells and non-tumor cells, such as DCs, remains to be determined (Curiel et al., 2003; Herbst et al., 2014). In the current study, we showed that sufficient T cell infiltration, and not PD-L1 expression, is essential for tumor responses to checkpoint blockade. Specifically, a PD-L1+ tumor with an insufficient number of TILs is unresponsive to anti-PD-L1 immunotherapy. In contrast, a PD-L1+ tumor with sufficient number of TILs can be well controlled by the same immunotherapy. Furthermore, prevention of T cells from entering the tumor microenvironment can transform a checkpoint blockade responsive tumor into an unresponsive tumor. Unfortunately, increasing TILs within established tumor has been very difficult. In order to develop approaches to effectively increase TILs, we produced an Ab-LIGHT fusion protein to specifically target LIGHT to tumor tissues. In three different tumor models, we were able to show that Ab-LIGHT therapy can control established tumors. We found that Ab-LIGHT activates LTβR signaling to induce the production of chemokines and adhesion molecules in tumor tissues. These chemokines attract lymphocyte to the local tumor tissues, thus resulting in control and rejection of tumors. Therefore, we have developed a strategy to overcome tumor resistance to checkpoint blockade by increasing lymphocyte infiltration.
In order to increase the response rate to checkpoint blockade, several combination therapies have been developed (Ai and Curran, 2015). Among them, the combination with anti-PD-1 and anti-Cytotoxic T Lymphocyte-associated Antigen-4 (CTLA-4) has shown the best improvement in clinical trials (Hammers et al., 2014; Postow et al., 2015; Wolchok et al., 2013). CTLA-4 blockade induces the expansion of tumor infiltrating T cells inside the tumor tissue, which is critical for the efficacy of combination therapy (Cha et al., 2014). However, anti-CTLA-4 might only expand T cells already present inside tumors. The antitumor effects are completely abrogated when initial lymphocyte infiltration is blocked. Specifically, blocking lymphocyte trafficking at a later time point has no effects on the synergy (Spranger et al., 2014). In significant contrast, LIGHT increases TILs by recruiting naïve T cells from the periphery (Yu et al., 2004). Spontaneous tumor infiltrating T cells in established tumors are usually exhausted or anergic due to the inhibitory microenvironment, and they are difficult to be re-activated (Crespo et al., 2013). By contrast, newly recruited T cells have less chance to be suppressed, and might be easier to be activated. Furthermore, recruiting naïve T cells from periphery by LIGHT gives us the potential to maximize the effects of checkpoint blockade therapies in treating tumors without pre-existing lymphocyte infiltrations.
The presence of spontaneous TILs correlates with better prognosis, especially for tumor immunotherapies (Woo et al., 2015). Recently, consistent with our observations, two exceptional studies have shown that both mouse and human tumor cells can program to suppress chemokine productions that can limit immune infiltrates leading to resistance to PD-1 blockade therapies (Peng et al., 2015; Spranger et al., 2015). Unfortunately, limited approaches are available to increase lymphocyte infiltration without severe side effects. Interferons (IFN) have been considered as such candidates, and our lab has shown that an antibody-IFN-β fusion protein can synergize with PD-L1 blockade in B16 tumor model (Yang et al., 2014). However, increased TILs were only observed in some tumor models but not the others, probably due to the multiple effects downstream of IFNs (unpublished observations). B-raf inhibitors represent another candidate that could increase TILs (Wilmott et al., 2012). But these drugs only work on B-raf mutated tumors, and side effects usually occur during treatment, both of which limit their applications (Boussemart et al., 2013). Some viral vectors, such as adenovirus, have been shown to enhance lymphocyte infiltration and improve efficacy of adoptive T cell therapy (Tähtinen et al., 2015). Previous studies in our lab have used adenovirus expressing LIGHT for tumor immunotherapy (Lee et al., 2009; Yu et al., 2007). However, those therapies usually require intratumoral injection of the virus, which is not feasible for the majority of patients. Furthermore, safety is another concern for using viral vectors (Pesonen et al., 2010). Recently, Rosa et al. have demonstrated that inhibition of dipeptidylpeptidase 4 (DPP4) preserves active chemokine CXCL10, which can lead to increased lymphocyte infiltration to tumor tissues (da Silva et al., 2015). However, combination therapy with DPP4 inhibitor and checkpoint blockade only had a marginal improvement when compare to checkpoint blockade alone (da Silva et al., 2015). Their study implies that targeting a single chemokine may not be sufficient to recruit enough lymphocytes for complete tumor control. In fact, when comparing chemokine profiles in MC38 and Ag104Ld tumors, we found that several chemokines related to T cell trafficking, besides CXCL10, were significantly higher in MC38 (data not shown). In contrast to targeting one specific chemokine, LIGHT activates LTβR signaling in tumor tissues, which induces the expression of multiple chemokines and adhesion molecules for effective T cell recruitment to the tumor tissue (Figure 5H and 5I). The induction of multiple chemokines makes LIGHT more efficient for lymphocyte recruitment and activation.
LTβR signaling plays an important role in the organization of lymphocytes during lymphoid neogenesis (Ruddle, 1999), so it is an attractive target for modulating lymphocyte infiltration. Previous attempts have been made to activate LTβR signaling through agonist antibodies for tumor immunotherapy (Lukashev et al., 2006). However, the effects were marginal, possibly due to wide expression of LTβR, which makes it hard to specifically activate signaling in tumor tissues. Another approach to activate LTβR signaling is through engagement with LIGHT. Our lab has tried to produce recombinant mLIGHT protein, but it is characteristically unstable and tends to aggregate (data not shown, and (Del Rio et al., 2010)). Human LIGHT is more stable but does not cross-react with mouse receptors. In the past, such human molecules can only be evaluated in xenograft models, which lack the adaptive immune system. However, given the roles of LIGHT on adaptive immunity, it is important to evaluate LIGHT in immunocompetent hosts. By combining yeast surface display system and random error mutagenesis, we were able to engineer human LIGHT (hmLIGHT) to bind to both human and mouse receptors while remaining stable. We further showed that hmLIGHT is able to induce tumor regression in both mouse and human tumor models (Figure 4). Our study provides the proof-of-concept for the development of LIGHT for tumor immunotherapy.
Overall, our study proposes several interesting mechanisms that can be used to further cancer immunotherapy. First, our data suggest that a significant T cell-inflamed tumor microenvironment is critical for positive responses to checkpoint blockade. Blocking lymphocyte infiltration will abrogate the responsiveness in an originally anti-PD-L1 responding tumor. Second, we show that targeting LIGHT can induce antitumor immunity in both mouse and human tumor models by increasing lymphocyte infiltration. These data suggest that LIGHT, either alone or together with other immunotherapies, might be an effective strategy for cancer therapy. Third, in tumors resistant to checkpoint blockade therapy due to a lack of lymphocyte infiltration, we prove that additional LIGHT treatment can promote the efficacy of checkpoint blockade therapies. In other words, our study indicates that unresponsiveness to checkpoint blockade can be due to a lack of sufficient lymphocyte infiltration; and LIGHT could be used to increase the response rates to checkpoint blockades, and other immunotherapies, in commonly found non-T cell-inflamed tumors.
EXPERIMENTAL PROCEDURES
Mice
C57BL/6J, B6C3F1, Rag1−/−, and OT-1 CD8+ TCR-Transgenic mice were purchased from Jackson Laboratory. Ltbr−/− and Tnfrsf14−/− mice were kindly provided by Dr. K. Pfeffer (Heinrich-Heine-Universitat Dusseldorf, Dusseldorf, Gemany). Ltbr−/− and Tnfrsf14−/− mice were crossed to Rag1−/− mice to obtain Rag1−/−;Ltbr−/− and Rag1−/−;Tnfrsf14−/− mice. All mice were maintained under specific pathogen-free conditions at the University of Chicago. Animal experiment protocols were consistent with National Institutes of Health guideline. All studies were approved by the Animal Care and Use Committee of the University of Chicago.
In vitro evolution of human LIGHT
Engineered hLIGHT with increased affinities to both human and mouse receptors was selected using yeast surface display as previously described (Boder and Wittrup, 1997). Briefly, WT hLIGHT gene was inserted into the T7/pCT302 yeast display vector. The construct was used as template for error-prone PCR. Mutagenized PCR product and digested vector were co-electroporated into EBY100 yeast to generate libraries. The resulting library was cultured and induced for surface LIGHT expression. It was then stained by mouse/human LTβR-Ig/HVEM-Ig, followed by PE-conjugated goat anti-human IgG (Jackson Immu). Yeast clones with higher receptor binding affinities and species cross-reactivity were selected by alternating rounds of selection by flow cytometric sorting. Thermal stability was assessed by incubating yeast for 30 min at 37 ºC or 80 ºC, followed by mLTβR-Ig staining.
Tumor growth and treatments
1×106 MC38 or Ag104Ld cells were subcutaneously injected into the right flank of mice. Mice were treated intraperitoneally with 200 μg anti-PD-L1 (10F.9G2) on day 7 and 10. Tumor volumes were measured twice weekly and calculated as (Length×Width×Height/2). To block lymphocyte trafficking, mice were injected intravenously with 25 μg FTY720 on day 1 after tumor inoculation. Five microgram of FTY720 was given every day to maintain blockade. In some experiments, FTY720 was given on day 1 to 3 after tumor inoculation. For LIGHT treatment, 2×106 Ag104Ld-EGFR cells were inoculated subcutaneously into mice. Twenty-five microgram of anti-EGFR-hmLIGHT or anti-EGFR was injected intratumorally (or intravenously when specified) at indicated time points. Mouse LTβR-Ig (100 μg/mouse) was administrated on day 4, 7, and 11. To deplete CD8 T cells, mice were injected intraperitoneally with 200 μg anti-CD8 (YTS 169.4.2) on day 7 and 11. For immune-reconstituted models, Rag1−/− mice were inoculated subcutaneously with 1×106 MC38-EGFR or A431 cells. After tumors established, mice were adoptively transferred with 5×106 WT splenocytes or 2×106 OT-1 LN cells before treated with anti-EGFR-hmLIGHT.
Statistical Analysis
Mean values were compared using an unpaired Student’s two-tailed t test.
Supplementary Material
SIGNIFICANCE.
PD-1/PD-L1 blockade can produce positive responses in cancer patients. Most studies aiming to increase response rates to immune checkpoint blockade focus on combining PD-1/PD-L1 blockade with other checkpoints therapies for T cell activation. However, we demonstrate that increasing tumor infiltrating T cells in unresponsive tumors can also promote responses to checkpoint blockade, and conversely, inhibiting T cell infiltration in responsive tumors can diminish the efficacy. We generated an antibody-guided LIGHT fusion protein that is able to create a T cell-inflamed tumor microenvironment. We further demonstrate that antibody-LIGHT is able to overcome tumor resistance to checkpoint blockade by increasing T cell infiltration. Our study has created a strategy that could potentially increase the response rates to checkpoint blockades in cancer patients.
HIGHLIGHTS.
Sufficient T cell infiltration is essential for response to PD-L1 blockade
Targeting tumors with LIGHT creates a T cell-inflamed tumor microenvironment
LIGHT overcomes tumor resistance to checkpoint blockade by increasing TILs
This study provides a strategy to increase response rate to checkpoint blockade
Acknowledgments
We thank Ting Xu for providing heterodimeric Fc sequence. We thank Xuanming Yang, Liufu Deng, Heng Ru, Wenguang Liang, and Weifeng Liu for helpful scientific discussions. This work was in part supported by the U.S. National Institutes of Health through National Cancer Institute grants CA141975, grant from AbbVie, grants from the Chinese Academy of Sciences (XDA09030303), and the Chinese Ministry of Science and Technology (2012ZX10002006) to Y.X.F. H.T. is supported by the Cancer Research Institute Irvington Fellowship.
Footnotes
AUTHOR CONTRIBUTIONS
H.T. and Y.-X.F. designed experiments, analyzed data, and wrote the manuscript. H.T., Y.W., and L.K.C. performed experiments. J.G., W.L., J.W., and X.W. provided reagents. Y.Z. contributed to manuscript preparation. Y.-X.F. supervised the project.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ai M, Curran MA. Immune checkpoint combinations from mouse to man. Cancer Immunology, Immunotherapy. 2015:1–8. doi: 10.1007/s00262-014-1650-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, Gajewski TF. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer research. 2004;64:1140–1145. doi: 10.1158/0008-5472.can-03-3259. [DOI] [PubMed] [Google Scholar]
- Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nature biotechnology. 1997;15:553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- Boussemart L, Routier E, Mateus C, Opletalova K, Sebille G, Kamsu-Kom N, Thomas M, Vagner S, Favre M, Tomasic G. Prospective study of cutaneous side-effects associated with the BRAF inhibitor vemurafenib: a study of 42 patients. Annals of oncology. 2013;24:1691–1697. doi: 10.1093/annonc/mdt015. [DOI] [PubMed] [Google Scholar]
- Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K. Safety and activity of anti–PD-L1 antibody in patients with advanced cancer. New England Journal of Medicine. 2012;366:2455–2465. doi: 10.1056/NEJMoa1200694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromley SK, Mempel TR, Luster AD. Orchestrating the orchestrators: chemokines in control of T cell traffic. Nature immunology. 2008;9:970–980. doi: 10.1038/ni.f.213. [DOI] [PubMed] [Google Scholar]
- Cha E, Klinger M, Hou Y, Cummings C, Ribas A, Faham M, Fong L. Improved survival with T cell clonotype stability after anti–CTLA-4 treatment in cancer patients. Science translational medicine. 2014;6:238ra270–238ra270. doi: 10.1126/scitranslmed.3008211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, McGowan P, Ashe S, Johnston J, Li Y, Hellström I, Hellström K. Tumor immunogenicity determines the effect of B7 costimulation on T cell-mediated tumor immunity. The Journal of experimental medicine. 1994;179:523–532. doi: 10.1084/jem.179.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Current opinion in immunology. 2013;25:214–221. doi: 10.1016/j.coi.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, Krzysiek R, Knutson KL, Daniel B, Zimmermann MC. Blockade of B7-H1 improves myeloid dendritic cell–mediated antitumor immunity. Nature medicine. 2003;9:562–567. doi: 10.1038/nm863. [DOI] [PubMed] [Google Scholar]
- Cyster JG. Chemokines and cell migration in secondary lymphoid organs. Science. 1999;286:2098–2102. doi: 10.1126/science.286.5447.2098. [DOI] [PubMed] [Google Scholar]
- da Silva RB, Laird ME, Yatim N, Fiette L, Ingersoll MA, Albert ML. Dipeptidylpeptidase 4 inhibition enhances lymphocyte trafficking, improving both naturally occurring tumor immunity and immunotherapy. Nature immunology. 2015 doi: 10.1038/ni.3201. [DOI] [PubMed] [Google Scholar]
- Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, Li ZW, Karin M, Ware CF, Green DR. The lymphotoxin-β receptor induces different patterns of gene expression via two NF-κB pathways. Immunity. 2002;17:525–535. doi: 10.1016/s1074-7613(02)00423-5. [DOI] [PubMed] [Google Scholar]
- Del Rio M, Lucas C, Buhler L, Rayat G, Rodriguez-Barbosa J. HVEM/LIGHT/BTLA/CD160 cosignaling pathways as targets for immune regulation. Journal of leukocyte biology. 2010;87:223–235. doi: 10.1189/jlb.0809590. [DOI] [PubMed] [Google Scholar]
- Dirkx AE, oude Egbrink MG, Wagstaff J, Griffioen AW. Monocyte/macrophage infiltration in tumors: modulators of angiogenesis. Journal of leukocyte biology. 2006;80:1183–1196. doi: 10.1189/jlb.0905495. [DOI] [PubMed] [Google Scholar]
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nature medicine. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- Fu YX, Chaplin DD. Development and maturation of secondary lymphoid tissues. Annual review of immunology. 1999;17:399–433. doi: 10.1146/annurev.immunol.17.1.399. [DOI] [PubMed] [Google Scholar]
- Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nature immunology. 2013;14:1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M, Berger A, Wind P. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- Hammers HJ, Plimack ER, Infante JR, Ernstoff MS, Rini BI, McDermott DF, Razak AR, Pal SK, Voss MH, Sharma P. Phase I study of nivolumab in combination with ipilimumab in metastatic renal cell carcinoma (mRCC). Paper presented at: ASCO Annual Meeting Proceedings.2014. [Google Scholar]
- Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WT, Adams SF, Tahirovic E, Hagemann IS, Coukos G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: a meta-analysis. Gynecologic oncology. 2012;124:192–198. doi: 10.1016/j.ygyno.2011.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proceedings of the National Academy of Sciences. 2002;99:12293–12297. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Auh SL, Wang Y, Burnette B, Wang Y, Meng Y, Beckett M, Sharma R, Chin R, Tu T. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: changing strategies for cancer treatment. Blood. 2009;114:589–595. doi: 10.1182/blood-2009-02-206870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Chin RK, Christiansen P, Sun Y, Tumanov AV, Wang J, Chervonsky AV, Fu YX. Recruitment and activation of naive T cells in the islets by lymphotoxin β receptor-dependent tertiary lymphoid structure. Immunity. 2006;25:499–509. doi: 10.1016/j.immuni.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Lo JC, Chin RK, Lee Y, Kang HS, Wang Y, Weinstock JV, Banks T, Ware CF, Franzoso G, Fu YX. Differential regulation of CCL21 in lymphoid/nonlymphoid tissues for effectively attracting T cells to peripheral tissues. Journal of Clinical Investigation. 2003;112:1495. doi: 10.1172/JCI19188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashev M, LePage D, Wilson C, Bailly V, Garber E, Lukashin A, Ngam-ek A, Zeng W, Allaire N, Perrin S. Targeting the lymphotoxin-β receptor with agonist antibodies as a potential cancer therapy. Cancer research. 2006;66:9617–9624. doi: 10.1158/0008-5472.CAN-06-0217. [DOI] [PubMed] [Google Scholar]
- Mahmoud SM, Paish EC, Powe DG, Macmillan RD, Grainge MJ, Lee AH, Ellis IO, Green AR. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. Journal of Clinical Oncology. 2011;29:1949–1955. doi: 10.1200/JCO.2010.30.5037. [DOI] [PubMed] [Google Scholar]
- Melero I, Shuford WW, Newby SA, Aruffo A, Ledbetter JA, Hellström KE, Mittler RS, Chen L. Monoclonal antibodies against the 4–1BB T-cell activation molecule eradicate established tumors. Nature medicine. 1997;3:682–685. doi: 10.1038/nm0697-682. [DOI] [PubMed] [Google Scholar]
- Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–322. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, Sun Y, Zhao E, Vatan L, Szeliga W. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015 doi: 10.1038/nature15520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesonen S, Kangasniemi L, Hemminki A. Oncolytic adenoviruses for the treatment of human cancer: focus on translational and clinical data. Molecular pharmaceutics. 2010;8:12–28. doi: 10.1021/mp100219n. [DOI] [PubMed] [Google Scholar]
- Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. New England Journal of Medicine. 2015 doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruddle NH. Lymphoid neo-organogenesis: lymphotoxin’s role in inflammation and development. Immunologic research. 1999;19:119–125. doi: 10.1007/BF02786481. [DOI] [PubMed] [Google Scholar]
- Shin DS, Ribas A. The evolution of checkpoint blockade as a cancer therapy: what’s here, what’s next? Current opinion in immunology. 2015;33:23–35. doi: 10.1016/j.coi.2015.01.006. [DOI] [PubMed] [Google Scholar]
- Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic [bgr]-catenin signalling prevents anti-tumour immunity. Nature. 2015 doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8 (+) T cells directly within the tumor microenvironment. J Immunother Cancer. 2014;2:3. doi: 10.1186/2051-1426-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sznol M, Chen L. Antagonist antibodies to PD-1 and B7-H1 (PD-L1) in the treatment of advanced human cancer. Clinical Cancer Research. 2013;19:1021–1034. doi: 10.1158/1078-0432.CCR-12-2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tähtinen S, Grönberg-Vähä-Koskela S, Lumen D, Merisalo-Soikkeli M, Siurala M, Airaksinen AJ, Vähä-Koskela M, Hemminki A. Adenovirus improves the efficacy of adoptive T-cell therapy by recruiting immune cells to and promoting their activity at the tumor. Cancer immunology research, canimm. 2015 doi: 10.1158/2326-6066.CIR-14-0220-T. 0220.2014. [DOI] [PubMed] [Google Scholar]
- Thompson ED, Enriquez HL, Fu YX, Engelhard VH. Tumor masses support naive T cell infiltration, activation, and differentiation into effectors. The Journal of experimental medicine. 2010;207:1791–1804. doi: 10.1084/jem.20092454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Drake CG, Pardoll DM. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer cell. 2015;27:450–461. doi: 10.1016/j.ccell.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. New England Journal of Medicine. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhu M, Miller M, Fu YX. Immunoregulation by tumor necrosis factor superfamily member LIGHT. Immunological reviews. 2009;229:232–243. doi: 10.1111/j.1600-065X.2009.00762.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward PL, Koeppen H, Hurteau T, Schreiber H. Tumor antigens defined by cloned immunological probes are highly polymorphic and are not detected on autologous normal cells. The Journal of experimental medicine. 1989;170:217–232. doi: 10.1084/jem.170.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware CF. Network communications: lymphotoxins, LIGHT, and TNF. Annu Rev Immunol. 2005;23:787–819. doi: 10.1146/annurev.immunol.23.021704.115719. [DOI] [PubMed] [Google Scholar]
- Wick M, Dubey P, Koeppen H, Siegel CT, Fields PE, Chen L, Bluestone JA, Schreiber H. Antigenic cancer cells grow progressively in immune hosts without evidence for T cell exhaustion or systemic anergy. The Journal of experimental medicine. 1997;186:229–238. doi: 10.1084/jem.186.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, Kefford RF, Hersey P, Scolyer RA. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clinical cancer research. 2012;18:1386–1394. doi: 10.1158/1078-0432.CCR-11-2479. [DOI] [PubMed] [Google Scholar]
- Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K. Nivolumab plus ipilimumab in advanced melanoma. New England Journal of Medicine. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SR, Corrales L, Gajewski TF. Innate immune recognition of cancer. Annual review of immunology. 2015;33:445–474. doi: 10.1146/annurev-immunol-032414-112043. [DOI] [PubMed] [Google Scholar]
- Yang X, Zhang X, Fu ML, Weichselbaum RR, Gajewski TF, Guo Y, Fu YX. Targeting the tumor microenvironment with interferon-β bridges innate and adaptive immune responses. Cancer Cell. 2014;25:37–48. doi: 10.1016/j.ccr.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Zhang X, Mortenson ED, Radkevich-Brown O, Wang Y, Fu YX. Cetuximab-mediated tumor regression depends on innate and adaptive immune responses. Molecular Therapy. 2013;21:91–100. doi: 10.1038/mt.2012.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P, Lee Y, Liu W, Chin RK, Wang J, Wang Y, Schietinger A, Philip M, Schreiber H, Fu YX. Priming of naive T cells inside tumors leads to eradication of established tumors. Nature immunology. 2004;5:141–149. doi: 10.1038/ni1029. [DOI] [PubMed] [Google Scholar]
- Yu P, Lee Y, Liu W, Krausz T, Chong A, Schreiber H, Fu YX. Intratumor depletion of CD4+ cells unmasks tumor immunogenicity leading to the rejection of late-stage tumors. The Journal of experimental medicine. 2005;201:779–791. doi: 10.1084/jem.20041684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P, Lee Y, Wang Y, Liu X, Auh S, Gajewski TF, Schreiber H, You Z, Kaynor C, Wang X. Targeting the primary tumor to generate CTL for the effective eradication of spontaneous metastases. The Journal of Immunology. 2007;179:1960–1968. doi: 10.4049/jimmunol.179.3.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Brown NK, Fu YX. Direct and indirect roles of the LTβR pathway in central tolerance induction. Trends in immunology. 2010;31:325–331. doi: 10.1016/j.it.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W, Zheng H, He TC, Chang J, Fu YX, Fan W. LIGHT delivery to tumors by mesenchymal stem cells mobilizes an effective antitumor immune response. Cancer research. 2012;72:2980–2989. doi: 10.1158/0008-5472.CAN-11-4216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.