

Abstract
Hypoxia can act as an initial trigger to induce erythrocyte sickling and eventual end organ damage in sickle cell disease (SCD). Many factors and metabolites are altered in response to hypoxia and may contribute to the pathogenesis of the disease. Using metabolomic profiling, we found that the steady-state concentration of adenosine in the blood was elevated in a transgenic mouse model of SCD. Adenosine concentrations were similarly elevated in the blood of humans with SCD. Increased adenosine levels promoted sickling, hemolysis and damage to multiple tissues in SCD transgenic mice and promoted sickling of human erythrocytes. Using biochemical, genetic and pharmacological approaches, we showed that adenosine A2B receptor (A2BR)-mediated induction of 2,3-diphosphoglycerate, an erythrocyte-specific metabolite that decreases the oxygen binding affinity of hemoglobin, underlies the induction of erythrocyte sickling by excess adenosine both in cultured human red blood cells and in SCD transgenic mice. Thus, excessive adenosine signaling through the A2BR has a pathological role in SCD. These findings may provide new therapeutic possibilities for this disease.
SCD is a devastating inherited hemolytic disorder that affects red blood cells (RBCs) and is caused by a point mutation that results in the substitution of valine for glutamic acid at the sixth position of the β-globin chain of hemoglobin1–3. Despite our precise knowledge of the molecular defect that is associated with sickle hemoglobin (HbS) in RBCs1,4 and recent progress in understanding the molecular events that control polymerization of HbS and sickling of erythrocytes5–9, the specific factors and signaling pathways that are involved in this process are unclear. As a result, we lack effective preventative approaches or mechanism-specific treatment options for the disease. Thus, defining the molecular mechanisms underlying erythrocyte sickling is essential for advancing our understanding of the pathogenesis of SCD and for developing new treatment strategies.
Hypoxic conditions are known to promote deoxygenation and subsequent polymerization of HbS, resulting in RBC sickling, hemolysis and eventual end organ damage10,11. Many factors and metabolites are altered in response to hypoxia and may contribute to the pathogenesis of the disease. In an effort to identify metabolites that contribute to sickling and thereby exacerbate disease pathogenesis, we used liquid and gas chromatography coupled with mass spectral analysis to measure and compare metabolite profiles in the whole blood of control mice and SCD transgenic mice, an accepted animal model of SCD12,13.
Results
Excess adenosine contributes to mouse erythrocyte sickling
Metabolomic profiling revealed that adenosine was among the metabolites most highly elevated in the whole blood of SCD transgenic mice compared to controls (Supplementary Fig. 1). Adenosine concentration was also significantly elevated in the plasma of SCD transgenic mice (P < 0.05; Fig. 1a,b). The concentration of adenosine, a signaling nucleoside, increases under hypoxic conditions owing to the degradation of extracellular ATP derived from affected cells or tissues14. Hypoxia is the initial trigger that induces erythrocyte sickling, but it is unknown whether increased adenosine is involved in this process.
Figure 1.
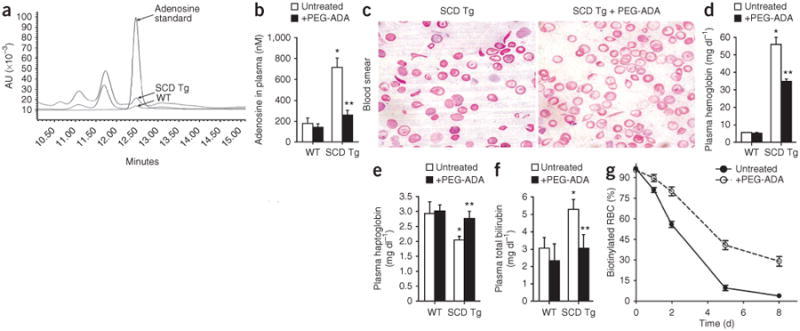
Increased adenosine levels contribute to sickling and hemolysis in SCD transgenic mice. (a) Representative HPLC profile showing adenosine concentrations in the plasma of wild-type (WT) and SCD transgenic (Tg) mice at steady state. (b) The effect of chronic PEG-ADA treatment on adenosine concentrations in the plasma of wild-type and SCD transgenic mice. (c) Blood smears of SCD transgenic mice without or with PEG-ADA enzyme therapy. (d–f) Effects of PEG-ADA treatment on plasma hemoglobin (d), plasma haptoglobin (e) and plasma total bilirubin (f) concentrations in wild-type and SCD transgenic mice. (g) Lifespan of RBCs in SCD transgenic mice treated with or without PEG-ADA. Mean ± s.e.m; n = 4–8 mice per group; *P < 0.05 versus wild type; **P < 0.05 versus untreated SCD transgenic mice.
To determine whether increased adenosine levels contribute to SCD in vivo, we treated SCD transgenic mice with polyethylene glycol–modified adenosine deaminase (PEG-ADA), a drug that has been successfully used for more than 20 years to lower adenosine concentrations in ADA-deficient humans15,16 (Fig. 1b). PEG-ADA treatment is well tolerated by ADA-deficient humans and mice15,17 and produced no obvious adverse side effects in SCD transgenic mice (data not shown). After 8 weeks of PEG-ADA treatment, blood smear analysis and reticulocyte flow cytometry showed that the percentages of sickled RBCs and reticulocytes were significantly reduced (P < 0.05; Fig. 1c and Table 1). This finding provides evidence that increased adenosine levels contribute to chronic sickling in SCD transgenic mice.
Table 1. Hematological parameters of wild-type and SCD transgenic mice treated with or without either PEG-ADA or PSB1115 for 8 weeks.
| RBC (×106 cells μl−1) | Hb (g dl−1) | HCT (%) | MCV (f) | MCH (pg) | MCHC (g dl−1) | RDW (%) | Reticulocyte (%) | Sickled cells (%) | WBC (×103 cells μl−1) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Wild type (untreated; n = 4) | 8.87 ± 0.307 | 13.75 ± 0.87 | 45.15 ± 2.44 | 49.47 ± 0.57 | 15.05 ± 0.23 | 30.47 ± 0.65 | 15.05 ± 0.63 | ND | ND | 3.25 ± 0.95 |
| Wild type (+PEG-ADA; n = 4) | 8.81 ± 0.18 | 13.28 ± 0.67 | 43.83 ± 1.16 | 49.75 ± 0.54 | 15.07 ± 0.46 | 30.27 ± 0.79 | 15.05 ± 0.75 | ND | ND | 3.27 ± 0.81 |
| Wild type (+PSB1115; n = 4) | 8.81 ± 0.39 | 13.28 ± 0.54 | 45.26 ± 1.64 | 49.12 ± 0.80 | 14.4 ± 0.29 | 29.34 ± 0.41 | 14.1 ± 0.67 | ND | ND | 3.89 ± 1.52 |
| SCD (untreated; n = 8) | 4.27 ± 1.08* | 5.05 ± 1.10* | 20.70 ± 4.16* | 49.88 ± 8.65 | 12.08 ± 1.96* | 24.31 ± 1.47* | 31.61 ± 0.63* | 62 ± 4.58 | 18.29 ± 0.59 | 26.03 ± 8.49* |
| SCD (+PEG-ADA; n = 8) | 6.15 ± 1.01** | 7.71 ± 2.64** | 30.15 ± 8.90** | 48.58 ± 8.79 | 12.38 ± 2.85 | 25.33 ± 1.21 | 28.03 ± 1.14** | 43 ± 4.52** | 10.35 ± 0.39** | 15.00 ± 2.79** |
| SCD (+PSB1115; n = 6) | 6.45 ± 0.45** | 8.25 ± 0.29** | 31.82 ± 3.43** | 49.17 ± 1.82 | 12.78 ± 0.46 | 26.02 ± 1.82 | 28.27 ± 1.36** | 37.15 ± 3.27** | 9.07 ± 0.75** | 11.95 ± 3.27** |
WT, wild type; SCD, SCD transgenic mice; Hb, hemoglobin; HCT, hematocrit; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; MCHC, mean corpuscular hemoglobin concentration.
P < 0.05 versus wild type;
P < 0.05 versus untreated SCD transgenic mice.
Erythrocyte sickling is the primary cause of intravascular hemolysis. Because PEG-ADA enzyme therapy reduced sickling in SCD transgenic mice, we tested whether chronic treatment with PEG-ADA would ameliorate hemolysis. We treated SCD transgenic mice with PEG-ADA for 8 weeks to lower adenosine concentrations and reduce erythrocyte sickling as described above. Intravascular hemolysis in these mice was significantly reduced by PEG-ADA treatment as demonstrated by decreased plasma hemoglobin concentration, increased plasma haptoglobin concentration and decreased total bilirubin concentration (P < 0.05; Fig. 1d–f). We also found that the half-life of RBCs in SCD transgenic mice increased from 2 d to 4 d after chronic PEG-ADA treatment (Fig. 1g). Complete blood count analysis showed that chronic PEG-ADA treatment significantly increased the total number of RBCs, hemoglobin concentration and hematocrit and lowered the total number of white blood cells (WBCs; P < 0.05; Table 1). The increase in hematocrit (Table 1) presumably reflects the corresponding increase in RBC numbers. Red cell distribution width (RDW) was also significantly reduced by PEG-ADA treatment (P < 0.05; Table 1), suggesting that the sizes of RBCs were more uniform and regular in treated than untreated mice. These results show that decreased sickling, reduced hemolysis and prolonged lifespan of RBCs in SCD transgenic mice treated with PEG-ADA resulted in significantly increased erythrocyte number and total hemoglobin content and decreased inflammatory response.
Elevated adenosine leads to multi-tissue damage in mice
In addition to hemolysis, sickling also led to tissue damage in SCD transgenic mice. Histological analysis showed that vascular congestion, vascular damage and necrosis in lung, liver and spleen of SCD transgenic mice were improved by chronic treatment with PEG-ADA (Fig. 2a). The ratio of spleen weight to body weight was significantly reduced, from 4.52 ± 0.24 to 2.48 ± 0.36, after chronic PEG-ADA treatment (P < 0.05, n = 8 mice per group). Semiquantification of histological changes showed that the pathological changes in SCD transgenic mice were significantly improved by PEG-ADA treatment (P < 0.05) as compared with untreated mice (Fig. 2b–d). Consistent with these histological improvements, PEG-ADA treatment of SCD transgenic mice significantly decreased the elevated heme content in all tissues examined including lung, liver and spleen (P < 0.05; Fig. 2e).
Figure 2. In vivo.
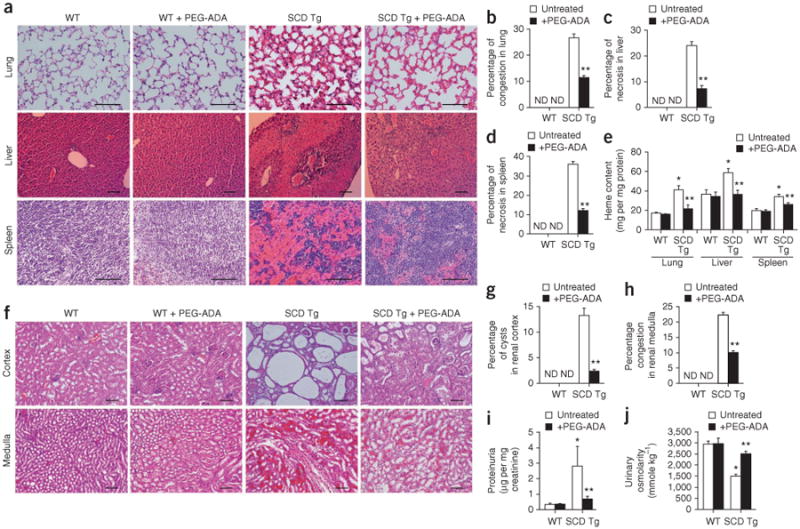
effects of PEG-ADA treatment on multiple organ damage and renal dysfunction in SCD transgenic mice. (a) H&E staining of lung, liver and spleen of wild-type and SCD transgenic mice after 8 weeks with or without PEG-ADA treatment. Substantial vascular congestion, vascular damage and necrosis in the lungs, livers and spleens observed in SCD transgenic mice was reduced by PEG-ADA treatment. For lung and spleen, scale bar, 20 μm; for liver, scale bar, 10 μm. (b–d) Semiquantitative analysis of histological changes in lung, liver and spleen of wild-type and SCD transgenic mice using Image-Pro Plus software (Media Cybernetics). (e) The effect of PEG-ADA enzyme therapy on heme content in lungs, livers and spleens of wild-type and SCD transgenic mice. (f) The effect of PEG-ADA enzyme therapy on microinfarction and cysts in renal cortex and on congestion in renal medulla of wild-type and SCD transgenic mice, as analyzed by H&E staining. Scale bar, 10 μm. (g,h) Semiquantitative analysis of histological changes in renal cortex and renal medulla of wild-type and SCD transgenic mice using Image-Pro Plus. (i,j) The effect of PEG-ADA enzyme therapy on proteinuria (i) and urine osmolarity (j) in wild-type and SCD transgenic mice. Mean ± s.e.m.; n = 4–8 mice per group; *P < 0.05 versus wild type; **P < 0.05 versus untreated SCD transgenic mice. ND, not detected.
Because SCD transgenic mice show marked pathological changes in their kidneys13 and because approximately 25% of people with SCD develop renal dysfunction with proteinuria18, we investigated the potential therapeutic effect of chronic PEG-ADA treatment on renal damage and dysfunction. The increased microinfarction and cysts seen in the renal cortex and the vascular congestion in the renal medulla of SCD transgenic mice were reduced by chronic PEG-ADA treatment (Fig. 2f). However, chronic PEG-ADA enzyme therapy had no significant effect in control mice (Fig. 2f). Semiquantification of histological changes indicated that PEG-ADA treatment significantly (P < 0.05) reduced renal damage in SCD transgenic mice (Fig. 2g,h). Consistent with the improved histological appearance of kidneys after PEG-ADA therapy, chronic PEG-ADA treatment of SCD transgenic mice also decreased proteinuria and increased urine osmolality, both of which indicate improvements in kidney function (Fig. 2i,j). These studies provide additional evidence for the detrimental role of excess adenosine in the pathophysiology of SCD and for the beneficial effects of chronic PEG-ADA enzyme therapy.
The concentration of ATP inside RBCs is extremely high (∼5 mM). One factor contributing to elevation of plasma adenosine is probably hemolysis-mediated ATP release and subsequent conversion to adenosine. In support of this hypothesis, we found that ATP concentrations in the plasma of SCD transgenic mice were significantly elevated relative to those seen in plasma of wild-type mice (P < 0.05; Supplementary Fig. 2a). Plasma ADA activity was very low in SCD transgenic mice and not different from that in wild-type mice (Supplementary Fig. 2b), consistent with the extremely low level of ADA activity in erythrocytes17,19,20.
Role of elevated adenosine in mice with acute sickle crisis
It is accepted that hypoxia followed by reoxygenation triggers an acute sickle crisis in SCD21. As expected, we found that elevated steady-state plasma adenosine concentrations in SCD transgenic mice under normoxic conditions were further increased by exposing the mice to hypoxia for 2 h followed by reoxygenation for 4 h (Fig. 3a). Treatment with PEG-ADA before hypoxia-reoxygenation resulted in significantly lowered adenosine concentrations (Fig. 3a), reduced sickling (P < 0.05; Fig. 3b) and attenuated hemolysis, as evidenced by decreased plasma hemoglobin concentration (Fig. 3c) and plasma total bilirubin concentration (Fig. 3d). Pretreatment with PEG-ADA also significantly (P < 0.05) prevented the hypoxia-reoxygenation–induced additional decrease in RBC numbers, hemoglobin concentration and hematocrit and also attenuated the hypoxia-reoxygenation–induced additional increase in RDW and in WBC numbers (Supplementary Table 1). These findings provide strong evidence that elevated adenosine levels are responsible for increased erythrocyte sickling and hemolysis after an acute sickle crisis event triggered by hypoxia-reoxygenation.
Figure 3.
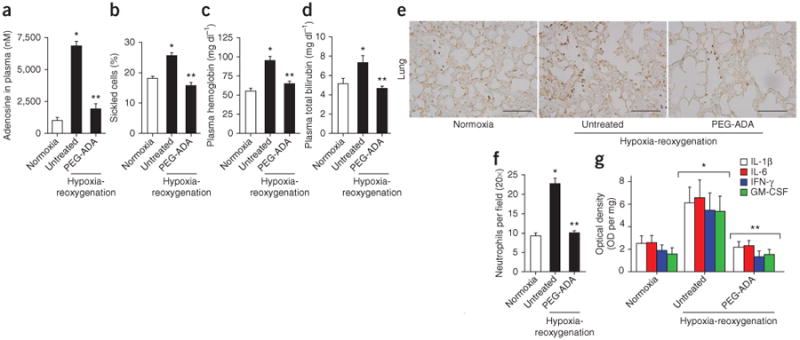
PEG-ADA treatment attenuates hypoxia-reoxygenation–induced acute sickle crisis in SCD transgenic mice. (a–d) The effects of hypoxia-reoxygenation, with or without PEG-ADA, on plasma adenosine concentration (a), percentage of sickled cells (b), plasma hemoglobin concentration (c) and plasma bilirubin concentration (d) in SCD transgenic mice. (e) Lung sections from SCD transgenic mice were stained with an antibody that recognizes Ly-6B.2 (a polymorphic 40-kDa antigen expressed by neutrophils) to visualize infiltrated tissue neutrophils (brown). Scale bar, 10 μm. (f) Quantification of neutrophil infiltration in the lungs of SCD transgenic mice by Image-Pro Plus software. (g) The effect of hypoxia-reoxygenation with and without PEG-ADA treatment on proinflammatory cytokine abundance (IFN-γ, IL-6, IL-1β and GM-CSF) in lung homogenates from SCD transgenic mice. Mean ± s.e.m. from 5–7 mice per group; *P < 0.05 relative to SCD transgenic mice under normoxic conditions and **P < 0.05 relative to untreated SCD transgenic mice under hypoxia-reoxygenation conditions.
In addition to sickling and hemolysis, vaso-occlusion is a key end-point of an acute crisis event11. We next assessed the effect of PEG-ADA therapy on hypoxia-reoxygenation–induced lung inflammation, a well-accepted measure of vaso-occlusion11,21. Immunostaining with neutrophil-specific antibodies showed that hypoxia-reoxygenation increased neutrophil infiltration in the lungs of SCD transgenic mice compared to normoxic conditions (Fig. 3e). PEG-ADA treatment reduced neutrophil infiltration in the lungs after hypoxia-reoxygenation (Fig. 3e). Image quantification analysis confirmed that PEG-ADA treatment significantly (P < 0.05) lowered hypoxia-reoxygenation–induced neutrophil infiltration (Fig. 3f). PEG-ADA treatment also significantly (P < 0.05) decreased the abundance of proinflammatory cytokines in the lungs of SCD transgenic mice, including interferon (IFN)-γ, interleukin (IL)-6, IL-1β and granulocyte-macrophage colony-stimulating factor (GM-CSF; Fig. 3g). These findings indicate that elevated adenosine levels not only underlie sickling induced by hypoxia-reoxygenation but also contribute to lung inflammation induced by hypoxia-reoxygenation, a major outcome of vaso-occlusion in acute sickle crisis.
2,3-DPG underlies adenosine-mediated mouse RBC sickling
To identify factors responsible for adenosine-induced erythrocyte sickling, we re-examined the metabolic profiles of control and SCD transgenic mice, with a particular focus on erythrocyte-specific metabolites. We found that 2,3-diphosphoglycerate (2,3-DPG), an erythrocyte-specific by-product of glycolysis, was increased in whole blood from SCD transgenic mice (Supplementary Fig. 1). We confirmed that 2,3-DPG concentrations were significantly elevated in RBCs from SCD transgenic mice at steady state under normoxic conditions compared to wild-type control mice (Fig. 4a). 2,3-DPG is an erythrocyte-specific metabolite that decreases the oxygen-binding affinity of hemoglobin22–25 and 2,3-DPG concentrations are increased in the RBCs of individuals with SCD and contribute to erythrocyte sickling under hypoxic conditions26–28. Because we found that adenosine levels were elevated at steady state (Fig. 1b) and responsible for sickling, it seemed possible that elevated 2,3-DPG concentration in SCD transgenic mice is a mediator of adenosine-induced sickling.
Figure 4.

Excess adenosine acts through A2BRs to induce 2,3-DPG and subsequent sickling in SCD transgenic mice. (a) Concentration of 2,3-DPG in RBCs of wild-type and SCD transgenic mice with or without chronic PEG-ADA enzyme therapy. (b) In vivo measurement of oxygen saturation of hemoglobin in wild-type and SCD transgenic mice with or without PEG-ADA treatment for 8 weeks. (c) 2,3-DPG concentrations in isolated RBCs treated with NECA in the presence or absence of theophylline. (d) 2,3-DPG concentrations in RBCs isolated from wild-type or mice deficient in each of the four types of adenosine receptors cultured in the presence or absence of NECA. (e) Immunostaining of A2BRs in RBCs from wild-type and A2BR-deficient mice. (f) 2,3-DPG concentrations in RBCs isolated from wild-type and adenosine receptor–deficient mice under hypoxic conditions. (g) cAMP concentrations in RBCs isolated from wild-type and A2BR-deficient mice treated with or without NECA. (h) 2,3-DPG concentrations in RBCs isolated from wild-type mice treated with NECA in the presence or absence of the PKA-specific inhibitor H-89. (i) 2,3-DPG concentrations in RBCs of wild-type and SCD transgenic mice treated with or without PSB1115. (j) Lifespan of RBCs in SCD transgenic mice treated with or without PSB1115. Mean ± s.e.m.; *P < 0.05 relative to untreated controls, **P < 0.05 relative to treated or positive samples; Each experiment was repeated five to seven times. n = 5–8 mice per group.
In support of this idea, we found that lowering steady-state adenosine concentrations in SCD transgenic mice by chronic PEG-ADA treatment resulted in a decrease in 2,3-DPG concentrations in RBCs (Fig. 4a), increased hemoglobin oxygen-binding affinity measured by an increased percentage of saturated hemoglobin (Fig. 4b) and attenuated chronic sickling (Fig. 1c and Table 1). Similarly, 2,3-DPG levels were further elevated by hypoxia-reoxygenation and pretreatment with PEG-ADA inhibited this elevation (Supplementary Fig. 3), indicating that elevated adenosine levels were responsible for the induction of 2,3-DPG by hypoxia-reoxygenation. PEG-ADA treatment also prevented the induction by hypoxia-reoxygenation of an acute vascular crisis with increased sickling, hemolysis and pulmonary inflammation (Fig. 3b–g). These results indicate that adenosine is responsible for increased 2,3-DPG concentrations in erythrocytes from SCD transgenic mice and suggest that elevated 2,3-DPG concentrations contribute to both chronic sickling and acute sickle crisis in these mice.
Adenosine induces 2,3-DPG in mouse RBCs via PKA signaling
We next treated normal, mature primary mouse erythrocytes with 5′-N-ethylcarboxamidoadenosine (NECA), a potent, nonmetabo-lizable adenosine analog. NECA stimulated an increase in 2,3-DPG concentrations (Fig. 4c), indicating that adenosine can directly induce 2,3-DPG in mature mouse RBCs.
Adenosine is a potent signaling molecule that elicits many physiological and pathological effects by activating G protein–coupled receptors on target cells29. Four such receptors have been identified—A1R, A2AR, A2BR and A3R—and each has a distinct affinity for adenosine and a distinct cellular and tissue distribution29. NECA-mediated induction of 2,3-DPG in normal mouse RBCs was prevented by theophylline (Fig. 4c), a broad-spectrum adenosine receptor antagonist, indicating that induction of 2,3-DPG in erythrocytes by adenosine is mediated through adenosine receptors. To determine which adenosine receptor was responsible, we used RBCs from mice deficient in each of the four different adenosine receptors. RBCs isolated from mice genetically deficient in A1R, A2AR or A3R (encoded by Adora1, Adora2a and Adora3, respectively) or from WT mice showed similar increases in 2,3-DPG concentration after treatment with NECA. By contrast, NECA did not induce 2,3-DPG in RBCs from mice genetically deficient in A2BR (encoded by Adora2b) (Fig. 4d). Immunostaining with A2BR-specific antibodies confirmed that A2BR was expressed in isolated wild-type mouse erythrocytes but not in those from A2BR-deficient mice (Fig. 4e). Finally, hypoxia-mediated induction of 2,3-DPG was significantly decreased in RBCs from A2BR-deficient mice (Fig. 4f). These results provide strong genetic evidence that A2BR signaling is required for hypoxia-mediated induction of 2,3-DPG in normal mouse RBCs.
A2BR is commonly coupled to adenylyl cyclase by the stimulatory G-protein subunit (Gαs) and increases intracellular cAMP, which activates PKA30,31. We examined the role of cAMP-dependent activation of PKA in A2BR-mediated induction of 2,3-DPG and found that NECA induced cAMP production in RBCs from wild-type mice but not in those from A2BR-deficient mice (Fig. 4g). Moreover, treatment of normal mouse RBCs with H-89, a specific, potent PKA inhibitor, significantly inhibited NECA-mediated induction of 2,3-DPG (P < 0.05; Fig. 4h). These findings suggest that A2BR-mediated cAMP-dependent activation of PKA is responsible for adenosine-mediated induction of 2,3-DPG in normal mouse RBCs.
In vivo effects of A2BR antagonism in SCD transgenic mice
To determine the in vivo effect of A2BR-mediated induction of 2,3-DPG on sickling in SCD transgenic mice, we treated these mice for 8 weeks with the A2BR-specific antagonist PSB1115. Like PEG-ADA treatment (Fig. 4a), PSB1115 treatment reduced both 2,3-DPG concentrations in RBCs (Fig. 4i) and the percentage of cells that were sickled (Table 1). Moreover, chronic treatment with PSB1115 increased the half-life of RBCs in SCD transgenic mice from 2 d to 5.5 d (Fig. 4j), a slightly greater improvement than that seen in mice treated with PEG-ADA (Fig. 1g). In addition, complete blood count analysis showed significant improvement with PSB1115 treatment (P < 0.05), including increased total RBC numbers and hemoglobin concentration as well as decreased total WBC numbers, percentage of reticulocytes and RDW (Table 1). Much like chronic PEG-ADA enzyme therapy, chronic treatment with PSB1115 reduced vascular congestion, vascular damage and necrosis in multiple tissues (Supplementary Fig. 4). Overall, these studies provide in vivo evidence that A2BR-mediated induction of 2,3-DPG is involved in erythrocyte sickling and subsequent multiple organ damage in SCD transgenic mice.
A2BR-mediated 2,3-DPG induction in human RBC sickling
To determine the pathophysiological significance of adenosine signaling in humans with SCD, we used HPLC to measure adenosine and 2,3-DPG in the blood of both healthy individuals and those with SCD (Supplementary Table 2). Adenosine concentrations were significantly (P < 0.05) higher in the blood of individuals with SCD than in the blood of control subjects (Fig. 5a). Consistent with earlier studies26,28 and with our in vivo findings in SCD transgenic mice (Fig. 4a), we found that the concentration of 2,3-DPG was significantly higher in RBCs from individuals with SCD than in RBCs from control individuals (Fig. 5b). Much as we had found in mice, we found that the A2BR is expressed on human erythrocytes (Supplementary Fig. 5a) and that NECA stimulated 2,3-DPG in a dose- and time-dependent manner in primary cultured human RBCs from healthy individuals (Supplementary Fig. 5b-c). NECA-stimulated induction of 2,3-DPG was inhibited by theophylline, indicating that adenosine receptor signaling is required (Supplementary Fig. 5d). Supporting our genetic studies in mice, we found that NECA-mediated induction of 2,3-DPG in RBCs from healthy individuals was inhibited by an A2BR antagonist (MRS1754) but not by other adenosine receptor antagonists tested (PSB36, SCH442416 and MRS3777, antagonists of A1R, A2AR and A3R, respectively; Supplementary Fig. 5d). In agreement with these findings, we found that the A2BR agonist BAY 60-6583, but not the A2AR agonist CGS21680, induced 2,3-DPG production in a dose-dependent manner in RBCs (Supplementary Fig. 5e,f). Thus, as in mice, the A2BR is required for adenosine-mediated induction of 2,3-DPG in normal human RBCs.
Figure 5.
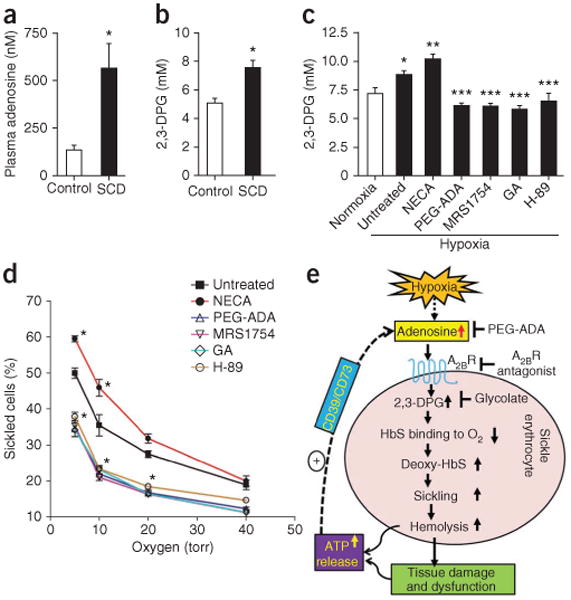
Adenosine levels are elevated in individuals with SCD and A2BR-mediated elevation of 2,3-DPG concentrations is required for hypoxia-induced human erythrocyte sickling. (a) Average adenosine concentrations in plasma from individuals with SCD (SCD, n = 12) and healthy volunteers (control, n = 11). (b) 2,3-DPG concentration in RBCs isolated from controls (n = 11) and individuals with SCD (n = 12). In a and b, *P < 0.05. (c) Changes in 2,3-DPG concentrations in isolated SCD RBCs after incubation under hypoxic conditions in the absence or presence of PEG-ADA, MRS1754 (A2BR antagonist), H-89 (PKA-specific inhibitor) or glycolate (GA; promotes degradation of 2,3-DPG). *P < 0.05 versus normoxic condition; **P < 0.05 versus untreated samples under hypoxic condition; ***P < 0.05 versus NECA-treated samples under hypoxic condition. (d) Changes in the percentage of sickled cells in isolated SCD RBCs after incubation under varying oxygen concentrations in the absence or presence of PEG-ADA, MRS1754, H-89 or glycolic acid. *P < 0.05 versus untreated cells. In c and d, each experiment was repeated four to six times. Data in a–d are mean ± s.e.m. (e) Working model of excessive adenosine signaling in erythrocyte sickling and potential mechanism-based therapies in SCD. Under hypoxic conditions, increased adenosine-mediated 2,3-DPG production induced by activation of A2BRs decreases the O2 binding affinity of HbS, resulting in increased amounts of deoxy-HbS and increased sickling, hemolysis and multiple tissue damage and dysfunction. Without interference, hemolysis and multiple tissue injury lead to increased release of ATP, which is converted to adenosine by the combined action of the ectonucleotidases CD39 and CD73. The use of PEG-ADA to lower adenosine concentrations or an A2BR antagonist to block receptor activation would reduce the production of erythrocyte 2,3-DPG and reduce sickling.
To test the pathophysiological significance of the induction of 2,3-DPG by adenosine for erythrocyte sickling in humans, we incubated primary erythrocytes purified from individuals with SCD under hypoxic conditions (2% oxygen) to induce sickling in the presence or absence of PEG-ADA, NECA, MRS1754, H-89 or glycolate (an agent which depletes 2,3-DPG26–28). Hypoxic conditions resulted in an induction of 2,3-DPG which was attenuated by PEG-ADA, MRS1754, H-89 or glycolate treatment to a similar degree (Fig. 5c), indicating that adenosine induces 2,3-DPG through A2BR signaling. By contrast, NECA stimulated a further increase in 2,3-DPG under hypoxic conditions (Fig. 5c). These findings show that A2BR-mediated activation of PKA is required for hypoxia to induce 2,3-DPG in human sickle erythrocytes.
Finally, to determine the functional role of A2BR-mediated induction of 2,3-DPG in sickling, we tested the effect of oxygen pressure on the sickling of RBCs from individuals with SCD in the presence or absence of NECA, PEG-ADA, MRS1754, H-89 or glycolate. We found that the percentage of sickled cells was inversely dependent on oxygen concentration (Fig. 5d). Treatment with PEG-ADA, MRS1754, H-89 or glycolate significantly reduced the percentage of sickled cells, whereas NECA treatment significantly enhanced the percentage of sickled cells under hypoxic conditions (Fig. 5d). Overall, these findings show that the induction of 2,3-DPG by adenosine-mediated A2BR activation followed by downstream signaling through PKA is a major underlying mechanism in hypoxia-mediated erythrocyte sickling in RBCs from individuals with SCD.
Discussion
Using a high-throughput metabolomic screen, we found that adenosine and 2,3-DPG levels are elevated in the blood of SCD transgenic mice at steady state. These findings led us to discover that elevated adenosine signaling through A2BR promotes sickling by inducing 2,3-DPG production. In addition, we showed that cAMP-dependent PKA activation functions downstream of A2BR signaling and underlies the adenosine-induced increase in 2,3-DPG concentrations in erythrocytes. Lowering adenosine concentrations or interfering with activation of A2BR reduced sickling, hemolysis and tissue injury in SCD transgenic mice both at steady state and during an acute crisis event. Finally, both adenosine and 2,3-DPG concentrations were elevated in the blood of individuals with SCD, and we showed that adenosine signaling through the A2BR increases 2,3-DPG concentrations and induces sickling of RBCs derived from humans with SCD. Hence, our findings provide evidence for the pathogenic consequences of excessive adenosine signaling in SCD and suggest that interfering with adenosine signaling (particularly with A2BR activation on erythrocytes) may be an effective mechanism-based therapy for preventing sickling and hemolysis in individuals with SCD and ultimately for reducing the life-threatening complications associated with SCD (Fig. 5e).
It is well known that the concentration of adenosine, a metabolic signaling molecule, increases under energy depletion and ischemic or hypoxic conditions14,32,33. It has been speculated that increased adenosine levels under hypoxic conditions may be beneficial by increasing blood flow to ischemic or hypoxic tissue owing to its potent vasodilatory effect. However, our findings indicate that increased adenosine levels are detrimental in SCD because adenosine promotes sickling through the induction of 2,3-DPG in erythrocytes. Production of 2,3-DPG occurs only in RBCs and decreases the oxygen binding affinity of hemoglobin22–25. Previous studies showed that erythrocyte 2,3-DPG is elevated in individuals with SCD and is involved in erythrocyte sickling under hypoxic conditions26,28. To our knowledge, our findings provide the first evidence that elevated adenosine levels contribute to the induction of 2,3-DPG in RBCs, which leads to chronic sickling by decreasing the oxygen binding affinity of HbS. Acute hypoxia-reoxygenation treatment of SCD transgenic mice resulted in a further increase in the concentration of circulating adenosine and in erythrocyte 2,3-DPG concentrations, as well as in increased sickling and lung inflammation, presumed to result from acute vaso-occlusive events. Thus, although increased adenosine production is generally viewed as a beneficial response to hypoxia, it has detrimental effects in the setting of SCD. Our findings from both mouse and human studies support a model in which, under normal physiological conditions, adenosine regulates 2,3-DPG production to control the oxygen binding affinity of hemoglobin. However, in individuals with SCD, this normally beneficial effect becomes detrimental by decreasing the oxygen binding affinity of HbS, increasing polymerization of deoxy-HbS and subsequently inducing RBC sickling (Fig. 5e).
Although SCD is the first genetic disorder for which a causative mutation was identified at the molecular level and is the most prevalent autosomal recessive disorder worldwide34–36, we remain unable to effectively control the polymerization of HbS and erythrocyte sick-ling, events that are central to the pathogenesis and pathophysiology of the disease37. Previously, we found that that increased adenosine levels in the penile tissues of SCD transgenic mice lead to increased corporal cavernosal relaxation in vitro and probably contribute to priapism, a dangerous and painful erectile disorder that is prevalent among men with SCD38. Our current findings highlight the possibility of preventing and treating this dangerous, painful and life-threatening hemolytic disease by interfering with an adenosine-induced sickling pathway. PEG-ADA has been used effectively for more than two decades to lower elevated adenosine concentrations in ADA-deficient individuals and mice15,17 and in this way prevent unwanted pathological effects caused by elevated adenosine. We show here that PEG-ADA can be used to reduce the deleterious effects of elevated adenosine in SCD transgenic mice. In contrast to these harmful effects of elevated adenosine concentrations, recent studies have shown that inhibition of invariant natural killer T cells (iNKT cells) through activation of A2AR decreases pulmonary dysfunction in the NY1DD mouse model of SCD39. In view of the potentially protective effects of adenosine-mediated A2AR activation on iNKT cells, an A2BR antagonist may be a better therapeutic choice than PEG-ADA, because specific antagonism of A2BR will not prevent the potentially beneficial effects of adenosine in activating other types of adenosine receptors on cell types other than RBCs. By preventing erythrocyte sickling, interference with A2BR signaling has the potential to reduce the morbidity and mortality associated with SCD.
Online Methods
Human subjects
We took blood samples with informed consent. Relevant clinical features of study subjects are presented in Supplementary Table 2. The research protocol was approved by the University of Texas Health Science Center at Houston Committee for the Protection of Human Subjects.
Mice
Berkley SCD transgenic mice expressing exclusively human sickle hemoglobin (HbS) were purchased from The Jackson Laboratory12. Adenosine receptor–deficient mice were obtained from the following sources: A1R-deficient mice (J. Schnermann); A2AR-deficient mice (J.-F. Chen); A2BR-deficient mice (M.R. Blackburn) and A3R-deficient mice (M. Jacobson). Research protocols were reviewed and approved by the University of Texas Health Science Center at Houston Animal Welfare Committee.
Metabolomic profiling
Unbiased metabolomic profiling of whole blood from control and SCD transgenic mice was performed using liquid/gas chromatography coupled to mass spectrometry (LC/GC-MS) as described40. Specifically, a Thermo Fisher linear ion-trap mass spectrometer with Fourier transform and a Mat-95 XP mass spectrometer were used to analyze 251 named metabolites. The combinations of groups were analyzed using Welch's two-sample t test, following log transformation and imputation with minimum observed values for each compound. P < 0.05 was considered significant.
Adenosine analysis
Adenosine concentrations in whole blood and plasma were measured by HPLC as described41 with modification. More information is available in the Supplementary Methods.
Chronic PEG-ADA or PSB1115 therapy
SCD transgenic mice, at 8 weeks of age, were divided into two groups. One group was injected with PEG-ADA (2.5 U per week) or PSB1115 (200 μg per day; Tocris Bioscience) for 8 weeks, and the second was injected with normal saline for 8 weeks. Wild-type mice (C57BL/6J) used as control were injected with saline or PEG-ADA as for the SCD transgenic mouse groups.
Hypoxia-reoxygenation of SCD transgenic mice
SCD transgenic mice were subjected to 2 h of hypoxia with 8% oxygen. After hypoxia, mice were returned to normoxic conditions for 4 h21. At the end of the experiments, mice were killed and blood collected for adenosine measurement, sickling and hemolytic analysis (for details see Supplementary Methods).
Measurement of lifespan of RBCs in SCD transgenic mice
SCD transgenic mice were treated with or without PEG-ADA (2.5 U per week) for 8 weeks. After 6 weeks of treatment, RBCs were labeled in vivo with N-hydroxysuccinimide (NHS) biotin, and the lifespan of circulating red blood cells was measured as described41 (more information is available in the Supplementary Methods). The percentage of biotinylated RBCs was calculated by determining the fraction of peripheral blood cells labeled with Ter-119 (to identify RBCs) that were also labeled with a streptavidin-conjugated fluorochrome by flow cytometry.
Morphological study of erythrocytes and quantification of reticulocytes by flow cytometry
Blood smears were stained using the WG16-500ml kit (Sigma-Aldrich) for sickled cells and observed using Olympus BX60 microscope, and irreversibly sickled cells were counted26. The percentages of sickled cells among the counted cells were calculated. Reticulocytes were labeled by Retic-COUNT Reagent (Becton Dickinson) and quantified by flow cytometry42.
2,3-DPG analysis
2,3-DPG concentrations in RBCs were measured with a commercially available 2,3-DPG kit (catalog no. 1014833400; Roche)43.
Statistical analysis
All data are expressed as mean ± s.e.m. Data were analyzed for statistical significance using GraphPad Prism 4 software (GraphPad Software)44. Student's t tests (paired or unpaired as appropriate) were applied in two-group analysis. Differences between the means of multiple groups were compared by the one-way analysis of variance, followed by a Tukey's multiple comparisons test. A value of P < 0.05 was considered significant and was the threshold to reject the null hypothesis.
Additional methods
Detailed methodology is described in the Supplementary Methods.
Supplementary Material
Acknowledgments
Supported by US National Institute of Health grants DK077748 (to Y.X.), DK083559 (to Y.X.), HL070952 (to M.R.B.) and HL092188 (to H.K.E.) and by China National Science Foundation Scholarship Council 2008637068 (to J.W.). We thank T. Krahn (Bayer HealthCare AG) for the adenosine receptor A2BR agonist BAY 60-6583. Adenosine receptor–deficient mice were obtained from the following sources: A1R-deficient mice (J. Schnermann, National Institute of Diabetes and Digestive and Kidney Disease, National Institutes of Health); A2AR-deficient mice (J.-F. Chen, Boston University School of Medicine); A2BR-deficient mice (M.R. Blackburn, University of Texas–Houston Medical School); and A3R-deficient mice (M. Jacobson, Merck Research Laboratories).
Footnotes
Note: Supplementary information is available on the Nature Medicine website.
Author Contributions: Y.Z. carried out the measurement of adenosine and 2,3-DPG in humans and mice, analysis of sickling, hemolysis and lifespan of mouse RBCs, histological analysis of multiple tissues and image quantification, ELISA analysis of inflammatory cytokines in the lung homogenates and immunostaining of lung tissues with neutrophil markers, human erythrocyte culture and analysis of sickling under hypoxic conditions, and immunostaining of A2BRs on human and mouse RBCs, and contributed to generation of figures. Y.D. conducted PEG-ADA purification, treatment of mice with PEG-ADA or PSB1115 and proteinuria measurement, and isolation of multiple organs and blood from mice. J.W. was involved in the purification of PEG-ADA and treatment of mice with PEG-ADA or PSB1115, and performed heme content measurement and histological analysis of kidneys. W.Z. was involved in the purification of PEG-ADA; treated mice with PEG-ADA or PSB1115 and contributed to immunostaining of lung tissues with neutrophil markers. A.G. treated normal human erythrocyte cultures with A2BR or A2AR agonists. D.C.A. and M.V.M. conducted metabolomic screens in blood of wild-type and SCD transgenic mice. L.C.-D. provided expertise in confocal analysis of A2BR expression on RBCs. D.E.L. provided expertise in flow cytometry to measure the lifespan of RBCs. W.Z. assisted with urine osmolality analysis. H.S., L.T. and G.L. provided expertise in hemolytic disorders and kidney dysfunction. H.K.E. assisted A.G. with experiments on the effects of A2AR and A2BR agonists on 2,3-DPG induction in normal RBCs. R.E.K. provided expertise in adenosine signaling and helped edit the manuscript; M.R.B. provided mice deficient in each of the four types of adenosine receptors; H.S.J. provided expertise in hemolytic disorders, procured human subjects' approval and maintained the database of de-identified human subject information. Y.X. was the principal investigator, oversaw the design of experiments and interpretation of results, wrote and organized the manuscript, including the text and figures, and edited the manuscript.
Competing Financial Interests: The authors declare no competing financial interests.
References
- 1.Ingram VM. A specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobin. Nature. 1956;178:792–794. doi: 10.1038/178792a0. [DOI] [PubMed] [Google Scholar]
- 2.Madigan C, Malik P. Pathophysiology and therapy for haemoglobinopathies. Part I: sickle cell disease. Expert Rev Mol Med. 2006;8:1–23. doi: 10.1017/S1462399406010659. [DOI] [PubMed] [Google Scholar]
- 3.Urbinati F, Madigan C, Malik P. Pathophysiology and therapy for haemoglobinopathies. Part II: thalassaemias. Expert Rev Mol Med. 2006;8:1–26. doi: 10.1017/S1462399406010805. [DOI] [PubMed] [Google Scholar]
- 4.Christoph GW, Hofrichter J, Eaton WA. Understanding the shape of sickled red cells. Biophys J. 2005;88:1371–1376. doi: 10.1529/biophysj.104.051250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Montalembert M. Management of sickle cell disease. BMJ. 2008;337:a1397. doi: 10.1136/bmj.a1397. [DOI] [PubMed] [Google Scholar]
- 6.de Montalembert M. Advances in sickle cell disease. Bull Acad Natl Med. 2008;192:1375–1381. discussion 1381. [PubMed] [Google Scholar]
- 7.Chui DH, Dover GJ. Sickle cell disease: no longer a single gene disorder. Curr Opin Pediatr. 2001;13:22–27. doi: 10.1097/00008480-200102000-00004. [DOI] [PubMed] [Google Scholar]
- 8.Bunn HF, et al. Molecular and cellular pathogenesis of hemoglobin SC disease. Proc Natl Acad Sci USA. 1982;79:7527–7531. doi: 10.1073/pnas.79.23.7527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferrone FA. Polymerization and sickle cell disease: a molecular view. Microcirculation. 2004;11:115–128. doi: 10.1080/10739680490278312. [DOI] [PubMed] [Google Scholar]
- 10.Hebbel RP. Beyond hemoglobin polymerization: the red blood cell membrane and sickle disease pathophysiology. Blood. 1991;77:214–237. [PubMed] [Google Scholar]
- 11.Hebbel RP, Osarogiagbon R, Kaul D. The endothelial biology of sickle cell disease: inflammation and a chronic vasculopathy. Microcirculation. 2004;11:129–151. [PubMed] [Google Scholar]
- 12.Pászty C. Transgenic and gene knock-out mouse models of sickle cell anemia and the thalassemias. Curr Opin Hematol. 1997;4:88–93. doi: 10.1097/00062752-199704020-00003. [DOI] [PubMed] [Google Scholar]
- 13.Pászty C, et al. Transgenic knockout mice with exclusively human sickle hemoglobin and sickle cell disease. Science. 1997;278:876–878. doi: 10.1126/science.278.5339.876. [DOI] [PubMed] [Google Scholar]
- 14.Fredholm BB. Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ. 2007;14:1315–1323. doi: 10.1038/sj.cdd.4402132. [DOI] [PubMed] [Google Scholar]
- 15.Hershfield MS. PEG-ADA replacement therapy for adenosine deaminase deficiency: an update after 8.5 years. Clin Immunol Immunopathol. 1995;76:S228–S232. doi: 10.1016/s0090-1229(95)90306-2. [DOI] [PubMed] [Google Scholar]
- 16.Chan B, et al. Long-term efficacy of enzyme replacement therapy for adenosine deaminase (ADA)-deficient severe combined immunodeficiency (SCID) Clin Immunol. 2005;117:133–143. doi: 10.1016/j.clim.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 17.Blackburn MR, et al. The use of enzyme therapy to regulate the metabolic and phenotypic consequences of adenosine deaminase deficiency in mice. Differential impact on pulmonary and immunologic abnormalities. J Biol Chem. 2000;275:32114–32121. doi: 10.1074/jbc.M005153200. [DOI] [PubMed] [Google Scholar]
- 18.Scheinman JI. Sickle cell disease and the kidney. Nat Clin Pract Nephrol. 2009;5:78–88. doi: 10.1038/ncpneph1008. [DOI] [PubMed] [Google Scholar]
- 19.Carbonaro DA, et al. Neonatal bone marrow transplantation of ADA-deficient SCID mice results in immunologic reconstitution despite low levels of engraftment and an absence of selective donor T lymphoid expansion. Blood. 2008;111:5745–5754. doi: 10.1182/blood-2007-08-103663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carbonaro DA, et al. In vivo transduction by intravenous injection of a lentiviral vector expressing human ADA into neonatal ADA gene knockout mice: a novel form of enzyme replacement therapy for ADA deficiency. Mol Ther. 2006;13:1110–1120. doi: 10.1016/j.ymthe.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 21.Wallace KL, et al. NKT cells mediate pulmonary inflammation and dysfunction in murine sickle cell disease through production of IFN-gamma and CXCR3 chemokines. Blood. 2009;114:667–676. doi: 10.1182/blood-2009-02-205492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Narita H, Yanagawa S, Sasaki R, Chiba H. Synthesis of 2,3-bisphosphoglycerate synthase in erythroid cells. J Biol Chem. 1981;256:7059–7063. [PubMed] [Google Scholar]
- 23.Sasaki R, Chiba H. Functions and metabolism of 2,3-bisphosphoglycerate in erythroid cells. Tanpakushitsu Kakusan Koso. 1983;28:957–973. [PubMed] [Google Scholar]
- 24.Sasaki R, Chiba H. Role and induction of 2,3-bisphosphoglycerate synthase. Mol Cell Biochem. 1983;53–54:247–256. doi: 10.1007/BF00225257. [DOI] [PubMed] [Google Scholar]
- 25.Chiba H, Sasaki R. Functions of 2,3-bisphosphoglycerate and its metabolism. Curr Top Cell Regul. 1978;14:75–116. doi: 10.1016/b978-0-12-152814-0.50007-1. [DOI] [PubMed] [Google Scholar]
- 26.Poillon WN, Kim BC, Labotka RJ, Hicks CU, Kark JA. Antisickling effects of 2,3-diphosphoglycerate depletion. Blood. 1995;85:3289–3296. [PubMed] [Google Scholar]
- 27.Poillon WN, Kim BC. 2,3-Diphosphoglycerate and intracellular pH as interdependent determinants of the physiologic solubility of deoxyhemoglobin S. Blood. 1990;76:1028–1036. [PubMed] [Google Scholar]
- 28.Poillon WN, Kim BC, Welty EV, Walder JA. The effect of 2,3-diphosphoglycerate on the solubility of deoxyhemoglobin S. Arch Biochem Biophys. 1986;249:301–305. doi: 10.1016/0003-9861(86)90006-8. [DOI] [PubMed] [Google Scholar]
- 29.Fredholm BB, AP IJ, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- 30.Stiles GL. Adenosine receptor subtypes: new insights from cloning and functional studies. In: Jacobson KA, Jarvis MF, editors. Purinergic Approaches in Experimental Therapeutics. Wiley-Liss; New York: 1997. pp. 29–37. [Google Scholar]
- 31.Palmer TM, Stiles GL. Identification of an A2a adenosine receptor domain specifically responsible for mediating short-term desensitization. Biochemistry. 1997;36:832–838. doi: 10.1021/bi962290v. [DOI] [PubMed] [Google Scholar]
- 32.Eltzschig HK, et al. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood. 2004;104:3986–3992. doi: 10.1182/blood-2004-06-2066. [DOI] [PubMed] [Google Scholar]
- 33.Eltzschig HK, et al. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med. 2003;198:783–796. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sebastiani P, et al. A network model to predict the risk of death in sickle cell disease. Blood. 2007;110:2727–2735. doi: 10.1182/blood-2007-04-084921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fung EB, et al. Morbidity and mortality in chronically transfused subjects with thalassemia and sickle cell disease: A report from the multi-center study of iron overload. Am J Hematol. 2007;82:255–265. doi: 10.1002/ajh.20809. [DOI] [PubMed] [Google Scholar]
- 36.Lanzkron S, Haywood C, Jr, Segal JB, Dover GJ. Hospitalization rates and costs of care of patients with sickle-cell anemia in the state of Maryland in the era of hydroxyurea. Am J Hematol. 2006;81:927–932. doi: 10.1002/ajh.20703. [DOI] [PubMed] [Google Scholar]
- 37.Ballas SK. Current issues in sickle cell pain and its management. Hematology Am Soc Hematol Educ Program. 2007;2007:97–105. doi: 10.1182/asheducation-2007.1.97. [DOI] [PubMed] [Google Scholar]
- 38.Mi T, et al. Excess adenosine in murine penile erectile tissues contributes to priapism through A2B adenosine receptor signaling. J Clin Invest. 2008;118:1491–1501. doi: 10.1172/JCI33467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wallace KL, Linden J. Adenosine A2A receptors induced on iNKT and NK cells reduce pulmonary inflammation and injury in mice with sickle cell disease. Blood. 2010;116:5010–5020. doi: 10.1182/blood-2010-06-290643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sreekumar A, et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature. 2009;457:910–914. doi: 10.1038/nature07762. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 41.Knudsen TB, et al. Effects of (R)-deoxycoformycin (pentostatin) on intrauterine nucleoside catabolism and embryo viability in the pregnant mouse. Teratology. 1992;45:91–103. doi: 10.1002/tera.1420450109. [DOI] [PubMed] [Google Scholar]
- 42.Perumbeti A, et al. A novel human gamma-globin gene vector for genetic correction of sickle cell anemia in a humanized sickle mouse model: critical determinants for successful correction. Blood. 2009;114:1174–1185. doi: 10.1182/blood-2009-01-201863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ericson A, de Verdier CH. A modified method for the determination of 2,3-diphosphoglycerate in erythrocytes. Scand J Clin Lab Invest. 1972;29:84–90. [PubMed] [Google Scholar]
- 44.Zhou CC, et al. Angiotensin receptor agonistic autoantibodies induce pre-eclampsia in pregnant mice. Nat Med. 2008;14:855–862. doi: 10.1038/nm.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.