

Abstract
Staphylococcus aureus trigger inflammation through inflammasome activation and recruitment of neutrophils, responses critical for pathogen clearance but associated with substantial tissue damage. We postulated that necroptosis, cell death mediated by the RIPK1/RIPK3/MLKL pathway, would function to limit pathological inflammation. In models of skin infection or sepsis, Mlkl−/− mice had high bacterial loads, inability to limit IL.1b production and excessive inflammation. Similarly, mice treated with RIPK1 or RIPK3 inhibitors had increased bacterial loads in a model of sepsis. Ripk3−/− mice exhibited increased staphylococcal clearance and decreased inflammation in skin and systemic infection, due to direct effects of RIPK3 on IL-1b activation and apoptosis. In contrast to Casp1/4 −/− mice with defective S. aureus killing, the poor outcomes of Mlkl−/− mice could not be attributed to impaired phagocytic function. We conclude that necroptotic cell death limits the pathological inflammation induced by S. aureus.
Graphical Abstract
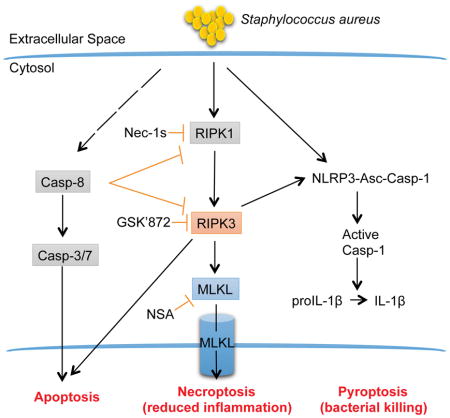
Introduction
Staphylococcus aureus cause pyogenic infections characterized by a robust inflammatory response both locally and systemically. This inflammation must be regulated to prevent host tissue damage. Multiple, redundant proinflammatory signaling cascades are activated by S. aureus through pathogen-associated molecular patterns and toxins. Several S. aureus toxins activate the NLRP3 inflammasome and caspase-1 to generate IL-1β, IL-18 and pyroptosis, a highly proinflammatory mechanism of cell death (Craven et al., 2009; Munoz-Planillo et al., 2009). Pyroptosis, along with the generation of reactive oxygen species (ROS) and the proteolytic activity of phagocytes, contributes to eradication of endocytosed pathogens (Miao et al., 2010; Sokolovska et al., 2013).
The balance between sufficient proinflammatory signaling to clear infection and the potentially lethal consequences of inflammasome activation has been extensively studied in the context of TNF- and LPS-induced mortality (Aziz et al., 2014; Duprez et al., 2011; Lawlor et al., 2015). Systemic effects of IL-1β and IL-18 are major factors in sepsis and endotoxemia associated mortality (Vanden Berghe et al., 2014a). Cell death through pyroptosis can also contribute to the control of infection, through release of endocytosed pathogens that are more efficiently killed by neutrophils (Miao et al., 2010).
Like immune cells, keratinocytes undergo caspase-1-mediated pyroptosis and IL-1β production in response to S. aureus, contributing to the eradication of skin infection (Soong et al., 2012; Soong et al., 2015). Keratinocytes follow a highly regulated pattern of proliferation, maturation, cornification and cell death through several mechanisms including apoptosis and necroptosis (Nestle et al., 2009; Vanden Berghe et al., 2014b). S. aureus through its pore-forming toxins induces necroptosis in immune cells (Gonzalez-Juarbe et al., 2015; Kitur et al., 2015). Necroptosis results from oligomerization of MLKL (Murphy et al., 2013), which is initiated by RIPK3-dependent phosphorylation (Cai et al., 2014; Sun et al., 2012) and RIPK1(Linkermann and Green, 2014). MLKL then forms a pore that leaks intracellular contents, with a resulting inflammatory response (Kaczmarek et al., 2013). Like caspase-1-mediated pyroptosis, necroptosis also results in the release of intracellular contents promoting inflammation. Necroptosis and pyroptosis share multiple effectors, with some of these components including caspase 8, RIPK1 and RIPK3 contributing to the processing and activation of IL-1β (Kaczmarek et al., 2013; Moriwaki et al., 2015; Pasparakis and Vandenabeele, 2015). Thus, both pyroptosis and necroptosis generate substantial inflammation. RIPK1/RIPK3/MLKL signaling can be activated by TLRs, TNFR and IFNAR mediated responses, indicating the close association between necroptosis and pathogen recognition (Chan et al., 2015). However, the consequences of necroptosis are not necessarily proinflammatory. Necroptosis leads to the elimination of cells that produce cytokines and inflammatory products and the release of bacteria to be cleared by neutrophils leading to an overall decrease in inflammation (Kearney et al., 2015; Stephenson et al., 2015).
Exactly how necroptosis and pyroptosis function in host defense from S. aureus infection is unclear, given that both signaling pathways appear to have redundant effects, stimulating inflammatory signaling as well as cell death (Kang et al., 2015; Kang et al., 2013). The successful control of S. aureus infection has been suggested to require two major host responses: first, the rapid suppression of S. aureus replication, and second, the rapid regulation of the hyper-inflammatory response that ensues (Powers et al., 2015; Stephenson et al., 2015). We postulated that pyroptosis initiates clearance of staphylococci and that necroptosis serves to eliminate cells that cause excessive inflammation. Using models of skin infection and sepsis, we delineate how these signaling pathways contribute to both of these components of successful host defense.
Results
MLKL contributes to the clearance of S. aureus from infected skin
Murine models of skin infection provide the opportunity to monitor both inflammation and bacterial killing in an in vivo setting (Miller et al., 2006). To determine the role of necroptosis during skin infection, we compared the responses of Mlkl−/− mice, which are unable to undergo necroptosis (Murphy et al., 2013), with wild type (WT) mice after a subcutaneous inoculation of USA300 S. aureus (Figure 1). In comparison to WT mice, there were significantly higher bacterial counts and more tissue damage in the Mlkl−/− mice (Figures 1A–C). We confirmed that S. aureus infection activates necroptosis by documenting phosphorylated MLKL in infected human keratinocytes (Figure S1) and demonstrating that S. aureus-induced cytotoxicity was decreased significantly by inhibitors of MLKL (NSA) in human keratinocytes and primary cultures (Figure 1D, 1E) and by siRNA knockdown of MLKL (Figure 1F, 1G) in keratinocytes. Thus, the ability of host cells to undergo necroptosis contributed to pathogen clearance.
Figure 1. MLKL contributes to the clearance of S. aureus from infected skin.

(A–I) Mlkl−/− and wild type (WT) mice were infected subcutaneously with 2 × 106 CFU/mouse of S. aureus (SA) for 5 days and the area of infection biopsied for analysis.
(A) S. aureus colony forming units (CFU) recovered
(B) Representative images of mice showing skin lesions caused by S. aureus infection.
(C) Quantification of lesion sizes from (B).
(D and E) Cytotoxicity in HaCaT (C) or primary keratinocytes (HEKn) (D) pretreated with 10 μM necrosulfonamide (NSA) and infected with WT S. aureus.
(F) Cytotoxicity in MLKL-deficient HaCaT infected with S. aureus for 4 h.
(G) Immunoblot of HaCaT with MLKL knocked down by siRNA or scrambled control.
(H, I and J) Number of neutrophils (PMNs) (H), macrophages (Macs) (I), and gamma-delta (γδ) T cells (J) on Mlkl−/− and WT mice.
(K) Cytokines from S. aureus infected WT and Mlkl−/− mice (“nd” means “not detected”).
(L) Representative images of trichrome stain on skin biopsies of PBS or S. aureus-infected mice. Scale bars, 100 μm.
(M) Western blot showing cleaved caspase-1 (p20), full-length caspase-1, actin and ponceau on skin homogenate of infected WT and Mlkl−/− mice.
Each point represents a mouse and lines show median values. Data are represented as bar graphs with mean ± SEM. Results shown are pooled from at least two independent experiments. *p<0.05, **p<0.01. See also Figures S1 and S2; and Tables S1 and S2.
We next determined whether excessive inflammation was contributing to the defective bacterial clearance exhibited by the Mlkl−/− mice. We compared the ability of these Mlkl−/− mice to activate proinflammatory cytokine production and to recruit the immune cells necessary to clear S. aureus infection. No differences between the WT, Mlkl−/− mice were observed after 1 day of infection either in the clearance of S. aureus or recruitment of immune cells (Figures S2A, S2B). However, by day 5, Mlkl−/− mice had significantly more neutrophils, macrophages and γδ T cells recruited than WT mice at the site of infection (Figures 1H–J, Table S1). As early as one day post infection, Mlkl−/− mice had increased T cell cytokine expression (IP-10/CXCL10, Rantes and MIG) (Figure S2C, Table S2), cytokines that mediate inflammatory damage in the skin (McLoughlin et al., 2006). Consistent with the heightened immune cell recruitment, by day 5, Mlkl−/− mice had significantly increased proinflammatory cytokines (IL-6, TNF, IL-1β, KC, and MIP1α) (Figures 1K, Table S1).
The histopathological responses to infection also were different. Mlkl−/− mice exhibited increased necrosis and adiposity, a marker of inflammation (Rajala and Scherer, 2003; Schaffler and Scholmerich, 2010), in comparison to WT infected controls (Figure 1L). Mlkl−/− mice had hyperkeratosis and more immune cell infiltration as compared with the WT mice (Figure 1L). As increased IL-1β was observed in Mlkl−/− infections, tissue lysates were screened for the presence of active caspase-1 (Figure 1M). Substantially more caspase-1 p20 was found at both baseline and following infection in Mlkl−/− than in WT mice. The Mlkl−/− mice displayed increased caspase-1 activity, increased generation of proinflammatory cytokines including IL-1β, and more neutrophils, but were nonetheless unable to clear S. aureus as efficiently as the WT mice. Together, these results suggest that necroptosis participates in the regulation of excessive inflammation that causes tissue damage during S. aureus skin infection.
Lack of caspases-1/4 is detrimental in S. aureus skin infection
We wanted to understand whether caspase-1 activation is beneficial during S. aureus skin infection. Production of IL-1β, which is processed by caspase-1, is critical for clearance of S. aureus from skin (Miller et al., 2006; Sokolovska et al., 2013; Soong et al., 2012). The contribution of the inflammasome to bacterial clearance was tested in Casp1/4−/− mice infected with S. aureus (Figure 2). The Casp1/4−/− knockout mice exhibited significantly decreased bacterial clearance and increased lesion sizes at 5 days after infection (Figure 2A–C). Similar to Mlkl−/− mice, Casp1/4−/− mice had increased numbers of neutrophils and γδ T cells but not macrophages at the site of infection (Figure 2D, E, F). Unlike Mlkl−/− mice, which exhibited an increased in many pro-inflammatory cytokines, Casp1/4−/− mice had increased KC only and slightly but not significantly less IL-1β post-infection (Figure 2G, Table S3). Thus, caspase 1/4 activity is necessary for S. aureus clearance, and contributes substantially to the generation of IL-1β.
Figure 2. Lack of caspases-1/4 is detrimental in S. aureus skin infection.

(A–G) Casp1/4−/− and wild type (WT) mice were infected subcutaneously with 2 × 106 CFU/mouse of S. aureus (SA) for 5 days and the area of infection biopsied for analysis.
(A) S. aureus colony forming units (CFU) recovered
(B) Representative images of mice showing skin lesions caused by S. aureus infection.
(C) Quantification of lesion sizes from (B).
(D, E and F) Number of neutrophils (PMNs) (D), macrophages (Macs) (E), and gamma-delta (γδ) T cells (F) on Mlkl−/− and WT mice.
(G) Cytokines from S. aureus infected WT and Mlkl−/− mice (“nd” means “not detected”).
Each point represents a mouse and lines show median values. Data are represented as bar graphs with mean ± SEM. Results shown are pooled from at least two independent experiments. *p<0.05, **p<0.01. See also Table S3.
Pharmacological inhibition of RIPK1 impairs clearance and increases inflammation
To verify participation of necroptosis in staphylococcal clearance, we used a pharmacological inhibitor of the RIPK1 kinase activity necrostatin-1s (Nec-1s) (Degterev et al., 2008; Takahashi et al., 2012), which also obviates unanticipated effects of the germline mutation in the Mlkl−/− mice. After documenting that Nec-1s blocked S. aureus-induced keratinocyte cell death in vitro (Figure 3A), we treated WT mice with Nec-1s and monitored their response to S. aureus skin infection (Figure 3B–D). Similar to the Mlkl−/− mice, the Nec-1s-treated mice had significantly larger skin lesions, greater numbers of S. aureus recovered and greater recruitment of inflammatory cells (Figures 3E–G). The overall pattern of cytokine induction suggested that Nec-1s-treated mice had increased levels of proinflammatory cytokines over the DMSO control (Figures 3H, Table S4). RIPK1 inhibition was associated with increased inflammation seen in histological sections and increased adiposity (Figure 3I). Thus, inhibition of necroptosis by Nec-1s-treatment or Mlkl−/− deletion leads to increased inflammation but impaired staphylococcal killing.
Figure 3. Pharmacological blockade of necroptosis leads to worse outcome during S. aureus skin infection.

(A) Cytotoxicity in keratinocyte cell line (HaCaT) pretreated with necrostatin-1 stable (Nec-1s) and infected with S. aureus MOI10 for 4 h.
(B–I) Mice were infected subcutaneously with S. aureus (SA) and treated with 10mg/kg necrostatin-1 stable (Nec) or DMSO control for 5 days and their skin tissues biopsied for analysis.
(B) S. aureus CFU recovered on day 5 from Nec-1s (N) or DMSO-treated mice.
(C) Representative images of mice showing lesions on day 5 of skin infection.
(D) Quantification of lesion sizes on day 5.
(E–G) Number of neutrophils (PMNs) (E), gamma-delta (γδ) T cells (F), and macrophages (Macs) (G) on infected area.
(H) Cytokines from S. aureus-infected mice (“nd” means “not detected”).
(I) Representative images of trichrome stain on skin biopsies of PBS or S. aureus-infected mice. Scale bars, 100 μm.
Each point represents a mouse and lines show median values. Data are represented as bar graphs with mean ± SEM. Results shown are pooled from at least two independent experiments. *p<0.05, **p<0.01. See also Figure S3 and Table S4.
Ripk3−/− mice have enhanced S. aureus clearance from the skin
RIPK3 is a target of RIPK1 and a major effector of necroptosis (Christofferson et al., 2014; Vanden Berghe et al., 2014b). However, in contrast to MLKL which functions as an effector of necroptosis alone, RIPK3 can directly activate apoptosis and stimulate the NLRP3 inflammasome to generate IL-1β (Newton and Manning, 2016). After documenting that an inhibitor of RIPK3, GSK’872, blocked S. aureus induced cytotoxicity in keratinocytes (Figure 4A), we wanted to understand the role of RIPK3 during S. aureus skin infection. Using the model of cutaneous S. aureus infection, we noted significantly improved outcomes in the Ripk3−/− mice with decreased staphylococcal burden (Figure 4B–D). Significantly fewer inflammatory cells and decreased cytokine production, including IL-1β and G-CSF, were observed following infection, with similar baseline cytokine activity in Ripk3−/− mice compared to WT mice (Figures 4E–H, Table S5). In contrast to Mlkl−/− mice that had increased IL-1β production (Figure 1K), the Ripk3−/− mice had significantly less IL-1β (Figure 4H). Histopathology was notable for better preservation of skin architecture in the infected Ripk3−/− mice (Figure 4I). The interactions of RIPK3 with additional binding partners were evident, as we observed less cell death due to apoptosis, as shown by decreased Annexin V staining in both keratinocytes and immune cells in Ripk3−/− skin after infection (Figure 4J). Thus, deletion of Ripk3 inhibited excessive inflammatory signaling and enhanced eradication of S. aureus from infected skin.
Figure 4. Ripk3−/− mice have improved outcome in S. aureus skin infection.

(A) Cytotoxicity in HaCaT pretreated with 10 μM GSK‘872 and infected with S. aureus.
(B–J) Ripk3−/− and wild type (WT) mice were infected subcutaneously with S. aureus (SA) for 5 days and skin tissues biopsied for analysis.
(B) S. aureus CFU recovered on day 5 on day of infection.
(C) Representative images of mice showing lesions on day 5 of skin infection
(D) Quantification of lesion sizes on day 5.
(E–G) Number of neutrophils (PMNs) (E), gamma-delta (γδ) T cells (F), and macrophages (Macs) (G) on infected area.
(H) Cytokines after 5 days of infection.
(I) Representative images of trichrome stain on skin biopsies of S. aureus-infected mice. Scale bars, 100 μm.
(J) Number of total Annexin V+ cells and number of Annexin V+ immune cells (CD54+) after 5 days of infection.
Each point represents a mouse and lines show median values. Data are represented as bar graphs with mean ± SEM. Results shown are pooled from at least two independent experiments. *p<0.05, **p<0.01. See also Table S5.
Caspases -8, -3 and -7 are activated by S. aureus but do not contribute to cytotoxicity
Apoptosis and necroptosis share upstream effectors, with RIPK1-FADD-Caspase-8 cascade mediating apoptosis and RIPK1-RIPK3-MLKL pathway regulating necroptosis in the absence of active caspase-8 (Chan et al., 2015). Caspase-8 targets the deubiquitinase CYLD preventing RIPK1 initiation of necroptosis (Legarda et al., 2016). S. aureus has been shown previously to activate caspases-8, -3 and -7 in keratinocytes (Soong et al., 2012). Since apoptosis and necroptosis effectors are intertwined and since S. aureus has the ability to activate both mechanisms of cell death, we wanted to dissect which of the pathways predominate during infection. We observed that S. aureus activated caspases -8, -3 and -7 in macrophage and keratinocyte cell lines (Figures 5A–C). Wild type as well as toxin-deficient (agr mutant) strains of S. aureus activated caspases-3/7 to the same extent (Figure 5B). However, deletion of caspase-8, as tested in Casp8−/−Ripk3−/− BMDMs did not affect the overall cytotoxicity of S. aureus (Figure 5D), since they still express inflammasome components. Similarly, pharmacological inhibitors of the apoptosis executioners (caspases-3/7), which decreased the activation of these caspases (Figure 5C), did not prevent S. aureus induced cytotoxicity in THP-1 cells (Figure 5E). To document the induction of apoptosis in vivo, we demonstrated active caspase-3 in the sloughing corneocyte layer of keratinocytes on S. aureus-infected murine skin (Figure 5F). S. aureus toxins activate necroptosis (Kitur et al., 2015), as well as pyroptosis (Munoz-Planillo et al., 2009; Munoz-Planillo et al., 2013; Soong et al., 2012). Thus, while S. aureus can induce apoptosis executioners, nonetheless, cytotoxicity is mediated predominantly by the components of the inflammasome and the RIPK1/RIPK3/MLKL necroptosis pathways.
Figure 5. Activation of caspases-8, -3 and -7 by S. aureus.

(A) Western blot showing caspase-8 activation in THP-1 cells after S. aureus infection.
(B) Caspases-3/7 activation in HaCaT cells pretreated with 20 μM caspase 3/7 inhibitor or DMSO for 1 h and stimulated with WT or agr S. aureus strains for 4 h.
(C) Caspases-3/7 activation in THP-1 cells pretreated with 20 μM caspase 3/7 inhibitor or DMSO for 1 h and stimulated with S. aureus for 2 h.
(D) Cytotoxicity in WT, Casp8−/−Ripk3−/− and Mlkl−/− BMDMs after S. aureus infection.
(E) Cytotoxicity in THP-1 cells treated with DMSO or caspases-3/7 inhibitor and infected with S. aureus for 2 h.
(F) Confocal images of skin biopsies obtained from S. aureus-infected WT and Ripk3−/− mice and stained for pan cytokeratin (Red) and cleaved caspase-3 (Green, arrow). Magnification of 200x; Scale bars, 100 μm.
Each point represents a mouse and lines show median values. Data are represented as bar graphs with mean ± SEM. Results shown are pooled from at least two independent experiments. *p<0.05, **p<0.01.
Caspase-1 signaling, but not necroptosis, contributes to S. aureus killing
In vitro assays were performed to establish whether necroptosis participates in S. aureus killing by human keratinocytes or immune cells. A gentamicin protection assay performed using human keratinocytes in primary culture demonstrated that concentrations of NSA that prevent cytotoxicity fail to inhibit S. aureus killing in contrast to the pan-caspase inhibitor, ZVAD (Figures 1E, 6A). Peritoneal exudate cells (PECs) from the Ripk3−/− and Mlkl−/− mice had no defect in their ability to phagocytose and kill S. aureus, whereas the Casp1/4−/− PECs had significantly impaired bacterial killing ability (Figures 6B, 6C). These results were confirmed with bone marrow-derived macrophages (BMDMs), demonstrating a lack of effect of the Ripk3 or Mlkl mutation on either staphylococcal killing or the generation of IL-1β, in contrast to the significant contribution of caspase-1 in these processes (Figure 6D–F). Lack of caspase-1 did not completely abrogate the generation of IL-1β, suggesting that multiple pathways are involved in the production of this cytokine (Figure 6F). To determine the contribution of apoptosis to bacterial killing, we incubated BMDMs from Casp8−/−Ripk3−/− mice with S. aureus. Casp8−/−Ripk3−/− BMDMs had no defect in either bacterial uptake or killing abilities (Figure 6G, 6H). Casp8−/−Ripk3−/− BMDMs produce more IL-1β compared to WT BMDMs (Figure 6I), potentially because the lack of both necroptosis and apoptosis components is compensated by the upregulation of the inflammasome products. Thus, caspase-1 mediated responses, but not necroptosis or apoptosis, contribute to staphylococcal death within immune cells and keratinocytes.
Figure 6. Caspases-1/4 are necessary for S. aureus killing by host cells.

(A) S. aureus killing by primary keratinocytes (HEKn cells) pretreated with 10 μM NSA or 50 μM ZVAD, infected with S. aureus for 2 h and treated with gentamicin for 4 h.
(B) S. aureus uptake by mouse peritoneal exudate cells (PECs) infected with S. aureus for 1 h.
(C) S. aureus killing by PECs infected with S. aureus for 24 h.
(D) S. aureus uptake by BMDMs from mice after infection for 1 h.
(E) Gentamicin protection assay showing S. aureus killing by BMDMs from WT, Ripk3−/−, Mlkl−/− and Casp1/4−/− mice.
(F) IL-1β released by BMDMs from WT, Ripk3−/−, Mlkl−/− and Casp1/4−/− mice after stimulation for 4h with S. aureus.
(G) S. aureus uptake by Casp8−/−Ripk3−/− (C8−/−R3−/−) BMDMs from mice after infection for 1 h.
(H) Gentamicin protection assay showing S. aureus killing by BMDMs from WT and Casp8−/−Ripk3−/− mice.
(I) IL-1β released by BMDMs from WT and Casp8−/−Ripk3−/− mice after stimulation for 4h with S. aureus.
Data are represented as bar graphs with mean ± SEM. Results shown are pooled from at least two independent experiments. *p<0.05.
MLKL and caspase-1 enhance survival in a murine model of S. aureus sepsis
Our results using the skin model of infection reflect participation of RIPK1 and MLKL in staphylococcal clearance by limiting local inflammation. As systemic inflammation is a major factor in the morbidity associated with S. aureus infections, we used a murine model of bacteremia to compare bacterial clearance when necroptosis is inhibited with Nec-1s and to highlight the participation of immune cells. Significantly greater numbers of S. aureus were recovered from the blood 24 h after infection in the Nec-1s treated mice than from controls (Figure 7A). To confirm the results obtained with Nec-1s, we used GSK3002963, a potent and specific inhibitor of RIP1 kinase activity (Berger et al., 2015), in our mouse model of sepsis. Like Nec-1s-treated mice, GSK3002963-treated mice had a decreased ability to clear S. aureus in the blood compared to mice treated with a vehicle control (Figure 7B). Mortality studies were then performed using the Mlkl−/−, Ripk3−/− and Casp1/4−/− mice in a model of sepsis. We predicted that the Mlkl−/− mice, which are unable to activate necroptosis as a mechanism to control inflammatory responses, would have worse outcomes and observed their increased mortality (with median survival of 4 days) compared to the WT mice (Figure 7C). Although the Ripk3−/− mice are also unable to activate necroptosis, their limited ability to stimulate the inflammasome was predicted to similarly provide protection from excessive inflammation. Survival of the Ripk3−/− mice was equivalent to that of the WT mice and significantly improved as compared to Mlkl−/− mutants (Figure 7C). Casp1/4−/− mice, which have a staphylococcal killing defect as well as deficient inflammasome activation had significantly increased mortality (median survival of 2 days) (Figure 7C). The outcomes from S. aureus sepsis model are consistent with those observed in the skin, in that mutations in the necroptosis pathway associated with excessive inflammatory responses, are also associated with negative outcomes.
Figure 7. Necroptosis and caspase-1/4 are necessary for bacterial clearance and survival in a murine model of S. aureus sepsis.

(A) S. aureus CFU recovered from the blood of mice treated with Nec-1s and infected with 1 × 108 CFU/mouse of S. aureus via retro-orbital route. Mice were sacrificed 24 hours or 48 hours after infection.
(B) S. aureus CFU recovered from the blood of mice treated with GSK3002963 and infected with 1 × 108 CFU/mouse of S. aureus via retro-orbital route. Mice were sacrificed 24 hours after infection.
(C) Kaplan-Meier survival curves of WT (n=19), Ripk3−/− (n=8), Mlkl−/− (n=9) and Casp1/4−/− (n=5) mice after retro-orbital injection with 1 × 108 CFU/mouse of S. aureus.
Each point represents a mouse and lines show median values. Results shown are pooled from at least two independent experiments. *p<0.05, **p<0.01.
Discussion
Our findings suggest that necroptosis in the setting of either local or systemic S. aureus infection markedly contributes to improved outcome, not by participating in bacterial death, but by limiting the damage caused by excessive inflammation. The importance of IL-1 signaling (Miller and Cho, 2011) and the critical role of caspase-1 in S. aureus staphylococcal killing (Cohen and Prince, 2013; Sokolovska et al., 2013) are well established. The mechanisms in place to regulate the toxic effects of these cytokines are less well understood. In skin and systemic models of infection, lack of caspase-1 resulted in significantly increased staphylococcal survival, greater neutrophil recruitment and increased local pathology. However, the inability to activate necroptosis and the associated increased caspase-1 activity and IL-1β production were also associated with excessive local pathology and impaired staphylococcal clearance.
Several lines of evidence indicate a major role of necroptosis in regulating inflammation. Germline mutations in RIPK1/3, Caspase-8 and FADD that interrupt the normal regulation of cell death pathways lead to excessive skin inflammation (Dannappel et al., 2014), even in the absence of a potent stimulus such as staphylococci. Both Mlkl−/− and Nec-1s-treated mice demonstrate increased S. aureus persistence, despite increased phagocyte recruitment and cytokine production. Moreover, Mlkl−/− immune cells had no defect in staphylococcal killing. These findings in the setting of common and clinically important infection are consistent with recent reports indicating that cell death through necroptosis provides a major immune suppressive function by eliminating the source of cytokine production (Kearney et al., 2015; Stephenson et al., 2015).
Components of the necroptosis cascade are shared among the major cell death pathways and cell fates determined by the nature of the activating stimulus and the local cytokine milieu (Legarda et al., 2016). As we observed, Caspase-8 is activated by S. aureus, as are the executioner caspases 3/7, as well as the RIPK1/RIPK3/MLKL and caspase-1. However, the apoptosis pathway does not participate directly in staphylococcal clearance, nor does it appear to control IL-1β production, as the Casp8−/−Ripk3−/− BMDMs did not have impaired generation of this cytokine, that is critical for S. aureus clearance from skin (Abtin et al., 2014; Miller and Cho, 2011; Miller et al., 2006; Nestle et al., 2009). The toxin-mediated cell death pathways, necroptosis and pyroptosis, drive cytotoxicity. Our data further document that S. aureus activation of caspase-1 and IL-1β production contributes to S. aureus killing, as well as to the pathological consequences of inflammation. This is in contrast to the major role of apoptosis in host defense from intracellular pathogens such as Yersinia pestis (Weng et al., 2014), which lack the numerous toxins expressed by S. aureus that activate necroptosis (Gonzalez-Juarbe et al., 2015; Kitur et al., 2015) and inflammasome mediated pyroptosis (DuMont et al., 2013; Munoz-Planillo et al., 2009; Munoz-Planillo et al., 2013).
Our data also illustrate the involvement of RIPK3 in multiple cell death and inflammatory responses activated by S. aureus. As has been reported by other groups, we observed the effects of RIPK3 (independently of MLKL) in stimulating apoptosis, the NLRP3 inflammasome and IL-1β production (Lawlor et al., 2015; Newton and Manning, 2016; Pasparakis and Vandenabeele, 2015). The interruption of necroptosis through deletion of Ripk3 did not reproduce the phenotypes observed in either the Mlkl−/− or Nec-1s-treated mice. Instead, the improved outcome of the Ripk3−/− mice in both skin and sepsis models was similar to our previous observations in a model of S. aureus pneumonia (Kitur et al., 2015). Lack of Ripk3 correlated with decreased production of IL-1β and activation of apoptosis, which afforded protection from S. aureus-induced inflammatory damage.
The importance of necroptosis in regulating excessive inflammatory signaling was also evident in a model of sepsis. In the absence of Mlkl, lethality occurred at a rate significantly greater than in the WT mice in which the immunoregulatory functions of necroptosis remained intact. Necrostatin-1s-treatment similarly resulted in significantly increased rates of bacteremia early in infection, despite a lack of impairment of S. aureus killing by cells unable to undergo necroptosis. The sepsis model also provided the opportunity to observe the importance of caspase1/4 and its participation in staphylococcal clearance, particularly at the early stages of infection. This was in contrast to the consequences of Mlkl deletion that were more apparent over time.
Our findings add to a growing body of literature demonstrating that excessive inflammatory signaling contributes significantly to the morbidity and mortality caused by S. aureus both systemically and at a local site such as the skin (Kitur et al., 2015; Muller et al., 2015). Our results show that the induction of necroptosis during S. aureus infection is an essential component of the host response necessary to limit inflammation. Targeting this pathway or identifying mutations that are associated with its function may provide therapeutic strategies to improve outcome from this common cause of infection.
Experimental Procedures
Bacteria
S. aureus strain MRSA USA300 LAC was grown overnight in LB Broth at 37°C as previously described (Kitur et al., 2015) and suspended in PBS to achieve the required concentration. S. aureus were heat-killed by at 65°C for 90 mins.
Cell culture
Peritoneal exudate cells (PECs) were collected from mice infected intraperitoneally with 107 CFU of heat-killed S. aureus 48 h and 24 h (Reyes-Robles et al., 2013). S. aureus MOI 10 were opsonized for 30 min with 50% normal mouse serum (NMS, Innovative Research) in RPMI 1640 medium (Gibco), washed and suspended in medium. PECs were infected with opsonized S. aureus MOI 1 in coated plates for 1 h and 24 h. PECs were lysed with 0.1% saponin and recovered bacteria (CFU) enumerated by serial dilutions.
Gentamicin protection assays were performed in HaCaT or BMDMs isolated from mice and differentiated in the presence of 60 ng/ml of mouse M-CSF (Peprotech). After stimulation, HaCaT 2 hours, BMDMs for 20 minutes with S. aureus MOI 100, cells were washed and treated with gentamicin (500 μg/mL) for 1 h or 24 h. Cells were washed in PBS, lysed with 0.1% saponin and bacteria enumerated by serial dilution on agar plates.
Cytotoxicity assays
HaCaT and HEKn cells were grown to confluence in Dulbecco modified Eagle medium (DMEM, Gibco) and THP-1 cells in RPMI Medium 1640 (Gibco) with 10% fetal bovine serum and 1% penicillin and streptomycin at 37°C and 5% CO2 in a humidified environment. Cells were maintained in no antibiotic medium 24 hours prior to infection. Cells were pretreated 1 hour prior to infection with necrostatin-1 stable (Nec-1s, Enzo Life Technologies), 10 μM necrosulfonamide (NSA, Calbiochem), 10 μM GSK’872 (Calbiochem), 20 μM caspase 3/7 inhibitor (Promega) or DMSO control (v/v). For siRNA knockdown, HaCaT were grown to confluence and transfected with 100 nM siMLKL (human MLKL siRNA pools of three targets, Santa Cruz Biotechnology) or scrambled siRNA control pool using Lipofectamine RNAiMAX (Life Technologies) as previously described (Kitur et al., 2015). Cell were infected after 3 days of siRNA treatment. S. aureus multiplicity of infection (MOI) 10 were added to the cells for 4 hours for keratinocytes and 2 hours for macrophages. Lysates were used for immunoblotting and caspases-3/7 activation assays and supernatants for cytotoxicity assays. LDH cytotoxicity was performed as per manufacturer’s instructions (Roche) and as described previously (Kitur et al., 2015). Caspases-3/7 activation were performed as per manufacturer’s instructions (Caspase-Glo, Promega).
Model of skin infection
Six- to eight-week-old sex-matched wild type C57BL/6J (Jackson Laboratory), Casp1/4−/− (B6N.129S2-Casp1tm1Flv/J, Jackson Laboratory) (Kuida et al., 1995), Ripk3−/− (Vishva Dixit, Genentech) (Newton et al., 2004) and Mlkl−/− (John Silke, WEHI via Douglas R. Green, St. Jude Children’s Research Hospital) (Murphy et al., 2013) were used. Knockouts were backcrossed to C57BL/6J background and equal sex ratios were used for experiments. Mice were anesthetized, shaved and inoculated subcutaneously on the back with 2 × 106 CFU/mouse of S. aureus in 100 μl of PBS or with 100 μl PBS (control group) using 265/8G sterile needle. Lesions were measured daily and mice were sacrificed 1 day or 5 days after infection.
Two 3-mm punch biopsy (Premier Uni-Punch) skin specimens were obtained from the lesions of infected mice, half of the tissue was fixed in 4% paraffin for 1 day and stored in 70% ethanol for histology, performed by the Histology Core at Columbia University Medical Center. Trichrome staining was done and images were taken on Zeiss Axiocam MRc 5 (Zeiss).
The other half of the specimens was homogenized using a cell strainer to obtain single cell suspension in 400 μl PBS. Recovered bacteria were enumerated by serial dilutions. Cells were spun down and homogenate used for ELISA and cells stained for FACS as described below.
Model of sepsis
1 × 108 CFU of S. aureus in 100 μl PBS was delivered intravenously through the retro-orbital injection route to six- to eight-week-old anesthetized mice and survival monitored daily. For necrostatin-1 stable (Nec-1s) sepsis experiments, mice were treated with 10mg/kg Nec-1s or DMSO control 24 hours prior, during infection and 4 hours (for 24-hour infection) or 24 hours (for 48-hour infection) after S. aureus infection. For GSK3002963 sepsis experiments, mice were treated with 10mg/kg GSK3002963 reconstituted with 10% DMSO v/v and 20% ethanol v/v in PBS (or reconstitution solution for vehicle control) during infection and 4 hours after S. aureus infection. Mice were sacrificed 24 hours or 48 hours after infection and bacteria in blood enumerated and cytokines in blood analyzed as described below.
FACS analysis
Homogenized cells were prepared as previously detailed (Kitur et al., 2015). Combinations of phycoerythrin (PE) anti-CD54 (YN1/1.7.4, Biolegend), fluorescein isothiocyanate-labelled (FITC) anti-Ly-6G (Gr-1; RB6-8C5; Biolegend), peridinin chlorophyll (PerCP)-Cy5.5-labelled anti-CD11c (N418; Biolegend), allophycocyanin (APC)-labelled anti-MHC II (I-A/I-E; Biolegend) and APC-labelled anti-TCR γδ T cells (γδ T cells) were used. Neutrophils (PMNs; Ly6G+/MHCII−), macrophages (Macs; CD11C+/MHCIIlow-mid) and γδ T cells were enumerated using FlowJo V10.
Cytokine analysis
Eve Technologies Corporation (Calgary, Canada) performed Multiplex ELISA on skin homogenate to determine mouse cytokine levels. Only cytokines that induced by infection are shown. IL-1β ELISA immunoblotting were performed as previously described (Kitur et al., 2015).
Immunoblotting and Immunohistochemistry
Cells were lysed using RIPA buffer as previously described (Kitur et al., 2015). Immuno-detection was done using anti-Caspase-1 (p20) (Casper-1, Adipogen), anti-MLKL (Abcam), anti-caspase-8 (Cell Signaling) and β-actin (Sigma-Aldrich) followed by secondary antibodies conjugated to horseradish peroxidase (Santa Cruz Biotechnology Inc.). Equal loading were confirmed by staining with 0.1% Ponceau S (w/v) in 5% acetic acid (Sigma-Aldrich).
For immunohistochemistry, sections were stained with cleaved caspase-3 (Cell Signaling) and anti-pan cytokeratin (Santa Cruz Biotechnology) followed by peroxidase staining using the Immunocruz ABC Staining kit (Santa Cruz Biotechnology Inc.). Control sections were stained with secondary antibodies only.
Ethics Statement
Animal experiments were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, the Animal Welfare Act and U.S. federal law. The Institutional Animal Care and Use Committee (IACUC) of Columbia University approved the protocol (AC-AAAH2350 and AC-AAAG7408).
Statistical analysis
Samples without normal distribution were analyzed using the nonparametric Mann-Whitney test and mouse survival analyzed by log-rank (Mantel-Cox) test for comparison of survival curves. Samples with normal distribution were analyzed with two-tailed Student’s t test and one-way ANOVA followed by Bonferroni Corrections to correct for multiple comparisons. p < 0.05 between groups were considered significant. Outliers were determined by Grubb’s test and removed. Statistical analysis was performed using GraphPad Prism Version 4.00 (GraphPad, La Jolla, USA). Data are presented as single points with lines representing median values or as bar graphs with mean ± SEM.
For more experimental procedures, see Supplemental Experimental Procedures in Supplemental Information.
Supplementary Material
Acknowledgments
Special thanks to John Silke (WEHI) and Douglas R. Green (St. Jude Children’s Research Hospital) for providing us with Mlkl−/− mice and Vishva Dixit (Genentech Inc.) for Ripk3−/− mice. We are grateful to Scott B. Berger and Peter J. Gough (GlaxoSmithKline plc) for providing us with GSK3002963. Much thanks to John Silke (WEHI), Douglas R. Green (St. Jude Children’s Research Hospital) and Razqallah Hakem (UHN, University of Toronto) for providing us with Casp8−/−Ripk3−/− mice bone marrows. Research reported in this publication was performed in the CCTI Flow Cytometry Core, supported in part by the Office of the Director, National Institutes of Health (NIH) under awards S10RR027050. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. This work was supported by FONDECYT 1140010, Comisión Nacional de Investigación Científica y Tecnológica (CONICYT), P09-016-F from the Millennium Institute in Immunology and Immunotherapy of the Ministry of Economy of Chile and Biomedical Research Consortium 13CTI-21526-P5 from INNOVA-CORFO Chile to S.B. and NIH grants 5R01HL079395 and 5R01AI103854 to A.P.
Footnotes
Author Contributions. K.K. and A.P. conceived and designed the experiments. K.K., S.W., A.B., F.P., M.W., G.S., H.F.P. and D.P. performed the experiments. K.K., S.W., A.B., F.P., M.W., G.S., H.F.P. S.B. and D.P. analyzed the data. K.K. and A.P. wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abtin A, Jain R, Mitchell AJ, Roediger B, Brzoska AJ, Tikoo S, Cheng Q, Ng LG, Cavanagh LL, von Andrian UH, et al. Perivascular macrophages mediate neutrophil recruitment during bacterial skin infection. Nat Immunol. 2014;15:45–53. doi: 10.1038/ni.2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aziz M, Jacob A, Wang P. Revisiting caspases in sepsis. Cell Death Dis. 2014;5:e1526. doi: 10.1038/cddis.2014.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SB, Bertin J, Gough PJ. Drilling into RIP1 biology: what compounds are in your toolkit? Cell Death Dis. 2015;6:e1889. doi: 10.1038/cddis.2015.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, Liu J, Ward Y, Wu LG, Liu ZG. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55–65. doi: 10.1038/ncb2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan FK, Luz NF, Moriwaki K. Programmed necrosis in the cross talk of cell death and inflammation. Annu Rev Immunol. 2015;33:79–106. doi: 10.1146/annurev-immunol-032414-112248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofferson DE, Li Y, Yuan J. Control of life-or-death decisions by RIP1 kinase. Annu Rev Physiol. 2014;76:129–150. doi: 10.1146/annurev-physiol-021113-170259. [DOI] [PubMed] [Google Scholar]
- Cohen TS, Prince AS. Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia. J Clin Invest. 2013;123:1630–1637. doi: 10.1172/JCI66142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven RR, Gao X, Allen IC, Gris D, Bubeck Wardenburg J, McElvania-Tekippe E, Ting JP, Duncan JA. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS One. 2009;4:e7446. doi: 10.1371/journal.pone.0007446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannappel M, Vlantis K, Kumari S, Polykratis A, Kim C, Wachsmuth L, Eftychi C, Lin J, Corona T, Hermance N, et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature. 2014;513:90–94. doi: 10.1038/nature13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, Abbott D, Cuny GD, Yuan C, Wagner G, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuMont AL, Yoong P, Day CJ, Alonzo F, 3rd, McDonald WH, Jennings MP, Torres VJ. Staphylococcus aureus LukAB cytotoxin kills human neutrophils by targeting the CD11b subunit of the integrin Mac-1. Proc Natl Acad Sci U S A. 2013;110:10794–10799. doi: 10.1073/pnas.1305121110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duprez L, Takahashi N, Van Hauwermeiren F, Vandendriessche B, Goossens V, Vanden Berghe T, Declercq W, Libert C, Cauwels A, Vandenabeele P. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35:908–918. doi: 10.1016/j.immuni.2011.09.020. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Juarbe N, Gilley RP, Hinojosa CA, Bradley KM, Kamei A, Gao G, Dube PH, Bergman MA, Orihuela CJ. Pore-Forming Toxins Induce Macrophage Necroptosis during Acute Bacterial Pneumonia. PLoS Pathog. 2015;11:e1005337. doi: 10.1371/journal.ppat.1005337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38:209–223. doi: 10.1016/j.immuni.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Kang S, Fernandes-Alnemri T, Rogers C, Mayes L, Wang Y, Dillon C, Roback L, Kaiser W, Oberst A, Sagara J, et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat Commun. 2015;6:7515. doi: 10.1038/ncomms8515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity. 2013;38:27–40. doi: 10.1016/j.immuni.2012.09.015. [DOI] [PubMed] [Google Scholar]
- Kearney CJ, Cullen SP, Tynan GA, Henry CM, Clancy D, Lavelle EC, Martin SJ. Necroptosis suppresses inflammation via termination of TNF- or LPS-induced cytokine and chemokine production. Cell Death Differ. 2015;22:1313–1327. doi: 10.1038/cdd.2014.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitur K, Parker D, Nieto P, Ahn DS, Cohen TS, Chung S, Wachtel S, Bueno S, Prince A. Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PLoS Pathog. 2015;11:e1004820. doi: 10.1371/journal.ppat.1004820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuida K, Lippke JA, Ku G, Harding MW, Livingston DJ, Su MS, Flavell RA. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, D’Cruz AA, Hall C, Kaur Spall S, Anderton H, Masters SL, et al. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat Commun. 2015;6:6282. doi: 10.1038/ncomms7282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legarda D, Justus SJ, Ang RL, Rikhi N, Li W, Moran TM, Zhang J, Mizoguchi E, Zelic M, Kelliher MA, et al. CYLD Proteolysis Protects Macrophages from TNF-Mediated Auto-necroptosis Induced by LPS and Licensed by Type I IFN. Cell Rep. 2016 doi: 10.1016/j.celrep.2016.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370:455–465. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLoughlin RM, Solinga RM, Rich J, Zaleski KJ, Cocchiaro JL, Risley A, Tzianabos AO, Lee JC. CD4+ T cells and CXC chemokines modulate the pathogenesis of Staphylococcus aureus wound infections. Proc Natl Acad Sci U S A. 2006;103:10408–10413. doi: 10.1073/pnas.0508961103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol. 2010;11:1136–1142. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LS, Cho JS. Immunity against Staphylococcus aureus cutaneous infections. Nat Rev Immunol. 2011;11:505–518. doi: 10.1038/nri3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LS, O’Connell RM, Gutierrez MA, Pietras EM, Shahangian A, Gross CE, Thirumala A, Cheung AL, Cheng G, Modlin RL. MyD88 mediates neutrophil recruitment initiated by IL-1R but not TLR2 activation in immunity against Staphylococcus aureus. Immunity. 2006;24:79–91. doi: 10.1016/j.immuni.2005.11.011. [DOI] [PubMed] [Google Scholar]
- Moriwaki K, Bertin J, Gough PJ, Chan FK. A RIPK3-caspase 8 complex mediates atypical pro-IL-1beta processing. J Immunol. 2015;194:1938–1944. doi: 10.4049/jimmunol.1402167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller S, Wolf AJ, Iliev ID, Berg BL, Underhill DM, Liu GY. Poorly Cross-Linked Peptidoglycan in MRSA Due to mecA Induction Activates the Inflammasome and Exacerbates Immunopathology. Cell Host Microbe. 2015;18:604–612. doi: 10.1016/j.chom.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Planillo R, Franchi L, Miller LS, Nunez G. A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. J Immunol. 2009;183:3942–3948. doi: 10.4049/jimmunol.0900729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142–1153. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, Lewis R, Lalaoui N, Metcalf D, Webb AI, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39:443–453. doi: 10.1016/j.immuni.2013.06.018. [DOI] [PubMed] [Google Scholar]
- Nestle FO, Di Meglio P, Qin JZ, Nickoloff BJ. Skin immune sentinels in health and disease. Nat Rev Immunol. 2009;9:679–691. doi: 10.1038/nri2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Manning G. Necroptosis and Inflammation. Annu Rev Biochem. 2016 doi: 10.1146/annurev-biochem-060815-014830. [DOI] [PubMed] [Google Scholar]
- Newton K, Sun X, Dixit VM. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol. 2004;24:1464–1469. doi: 10.1128/MCB.24.4.1464-1469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517:311–320. doi: 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- Powers ME, Becker RE, Sailer A, Turner JR, Bubeck Wardenburg J. Synergistic Action of Staphylococcus aureus alpha-Toxin on Platelets and Myeloid Lineage Cells Contributes to Lethal Sepsis. Cell Host Microbe. 2015;17:775–787. doi: 10.1016/j.chom.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajala MW, Scherer PE. Minireview: The adipocyte--at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology. 2003;144:3765–3773. doi: 10.1210/en.2003-0580. [DOI] [PubMed] [Google Scholar]
- Reyes-Robles T, Alonzo F, 3rd, Kozhaya L, Lacy DB, Unutmaz D, Torres VJ. Staphylococcus aureus leukotoxin ED targets the chemokine receptors CXCR1 and CXCR2 to kill leukocytes and promote infection. Cell Host Microbe. 2013;14:453–459. doi: 10.1016/j.chom.2013.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffler A, Scholmerich J. Innate immunity and adipose tissue biology. Trends Immunol. 2010;31:228–235. doi: 10.1016/j.it.2010.03.001. [DOI] [PubMed] [Google Scholar]
- Sokolovska A, Becker CE, Ip WK, Rathinam VA, Brudner M, Paquette N, Tanne A, Vanaja SK, Moore KJ, Fitzgerald KA, et al. Activation of caspase-1 by the NLRP3 inflammasome regulates the NADPH oxidase NOX2 to control phagosome function. Nat Immunol. 2013;14:543–553. doi: 10.1038/ni.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soong G, Chun J, Parker D, Prince A. Staphylococcus aureus activation of caspase 1/calpain signaling mediates invasion through human keratinocytes. J Infect Dis. 2012;205:1571–1579. doi: 10.1093/infdis/jis244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soong G, Paulino F, Wachtel S, Parker D, Wickersham M, Zhang D, Brown A, Lauren C, Dowd M, West E, et al. Methicillin-resistant Staphylococcus aureus adaptation to human keratinocytes. MBio. 2015;6 doi: 10.1128/mBio.00289-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson HN, Herzig A, Zychlinsky A. Beyond the grave: When is cell death critical for immunity to infection? Curr Opin Immunol. 2015;38:59–66. doi: 10.1016/j.coi.2015.11.004. [DOI] [PubMed] [Google Scholar]
- Sun L, Wang H, Wang Z, He S, Chen S, Liao D, Wang L, Yan J, Liu W, Lei X, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB, Goossens V, Roelandt R, Van Hauwermeiren F, Libert C, et al. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012;3:e437. doi: 10.1038/cddis.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Berghe T, Demon D, Bogaert P, Vandendriessche B, Goethals A, Depuydt B, Vuylsteke M, Roelandt R, Van Wonterghem E, Vandenbroecke J, et al. Simultaneous targeting of IL-1 and IL-18 is required for protection against inflammatory and septic shock. Am J Respir Crit Care Med. 2014a;189:282–291. doi: 10.1164/rccm.201308-1535OC. [DOI] [PubMed] [Google Scholar]
- Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol. 2014b;15:135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- Weng D, Marty-Roix R, Ganesan S, Proulx MK, Vladimer GI, Kaiser WJ, Mocarski ES, Pouliot K, Chan FK, Kelliher MA, et al. Caspase-8 and RIP kinases regulate bacteria-induced innate immune responses and cell death. Proc Natl Acad Sci U S A. 2014;111:7391–7396. doi: 10.1073/pnas.1403477111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.