

Abstract
Dopamine‐releasing neurons within the Substantia nigra (SN DA) are particularly vulnerable to degeneration compared to other dopaminergic neurons. The age‐dependent, progressive loss of these neurons is a pathological hallmark of Parkinson's disease (PD), as the resulting loss of striatal dopamine causes its major movement‐related symptoms. SN DA neurons release dopamine from their axonal terminals within the dorsal striatum, and also from their cell bodies and dendrites within the midbrain in a calcium‐ and activity‐dependent manner. Their intrinsically generated and metabolically challenging activity is created and modulated by the orchestrated function of different ion channels and dopamine D2‐autoreceptors. Here, we review increasing evidence that the mechanisms that control activity patterns and calcium homeostasis of SN DA neurons are not only crucial for their dopamine release within a physiological range but also modulate their mitochondrial and lysosomal activity, their metabolic stress levels, and their vulnerability to degeneration in PD. Indeed, impaired calcium homeostasis, lysosomal and mitochondrial dysfunction, and metabolic stress in SN DA neurons represent central converging trigger factors for idiopathic and familial PD. We summarize double‐edged roles of ion channels, activity patterns, calcium homeostasis, and related feedback/feed‐forward signaling mechanisms in SN DA neurons for maintaining and modulating their physiological function, but also for contributing to their vulnerability in PD‐paradigms. We focus on the emerging roles of maintained neuronal activity and calcium homeostasis within a physiological bandwidth, and its modulation by PD‐triggers, as well as on bidirectional functions of voltage‐gated L‐type calcium channels and metabolically gated ATP‐sensitive potassium (K‐ATP) channels, and their probable interplay in health and PD.
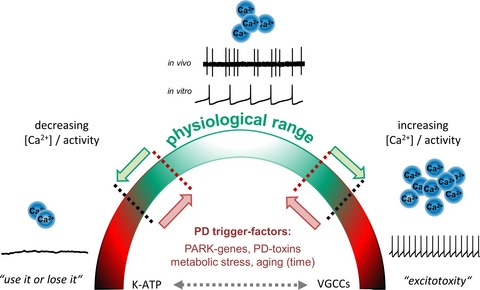
We propose that SN DA neurons possess several feedback and feed‐forward mechanisms to protect and adapt their activity‐pattern and calcium‐homeostasis within a physiological bandwidth, and that PD‐trigger factors can narrow this bandwidth. We summarize roles of ion channels in this view, and findings documenting that both, reduced as well as elevated activity and associated calcium‐levels can trigger SN DA degeneration.
This article is part of a special issue on Parkinson disease .
Keywords: ambroxol, A‐type Kv4.3/KChip3 potassium channels, Cav1.3 L‐type/Cav3.1 T‐type calcium channels, D2‐autoreceptor‐coupled GIRK2 channels, Kir6.2/SUR1 potassium channels, neuronal calcium sensor NCS‐1
Abbreviations used
- ABC‐transporter
ATP‐binding cassette transporter
- ALP
autophagy–lysosome pathway
- AMPA‐R
α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor
- BBB
blood–brain barrier
- CB
calbindin‐D28K
- CREB
cAMP response element‐binding protein
- D2‐AR
D2‐autoreceptor
- DA
dopaminergic
- DCC
deleted in colorectal cancer
- DHP
dihydropyridine
- DREAM
downstream regulatory element antagonist modulator
- ER
endoplasmic reticulum
- ETC
electron transfer chain
- GBA
glucocerebrosidase
- GDNF
glial‐derived neurothrophic factor
- GIRK2
G‐protein coupled inwardly rectifying potassium channel
- GRK2
G‐protein‐coupled receptor kinase 2
- HCN
hyperpolarization‐activated cyclic nucleotide‐gated cation channel
- HtrA2/Omi
high temperature requirement A2/Omi serine protease
- HVA
high voltage‐activated calcium channel
- IP3
inositol‐tris‐phosphate
- K‐ATP
ATP‐sensitive potassium channel
- KChip
Kv channel‐interacting protein
- Kir6.2
inwardly rectifying potassium channel
- KO
knockout
- L‐3
4‐dihydroxyphenylalanine
- LC
locus coeruleus
- L‐DOPA
Levodopa
- LETM1
leucine zipper EF hand containing transmembrane protein 1
- LMX1B
LIM homeobox transcription factor 1‐beta
- LRRK2
leucine‐rich repeat kinase 2
- LTCC
L‐type calcium channel
- LVA
low voltage‐activated calcium channel
- mCU
mitochondrial Ca2+ uniporter
- MEF‐2
myocyte enhancer factor‐2
- MNCX
mitochondrial sodium‐calcium exchanger
- mNOS
mitochondrial nitric oxide synthase
- MPTP
1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine
- NCS‐1
neuronal calcium sensor 1
- NFAT
nuclear factor of activated T‐cells
- NMDA‐R
N‐Methyl‐D‐aspartate receptor
- NO
nitric oxide
- NR1
NMDA‐R subunit 1
- PARK‐gene
Parkinson's disease‐related gene
- PD
Parkinson's disease
- PINK1
PTEN‐induced novel kinase 1
- PIP2
phosphoinositol‐bisphosphate
- PMCX
plasma membrane Ca2+ ATPases
- PPN
pedunculopontine nucleus
- RDP
rapid‐onset dystonia pakinsonism
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- RRF
retrorubal field
- RyR
ryanodine‐receptor
- SERCA
sarco‐endoplasmic reticulum Ca2+ ATPase
- SK
small conductance
- SN DA
dopaminergic neurons of the Substantia nigra pars compacta
- SOCCs
store‐operated calcium channels
- STIM
stromal interaction molecule
- STN
subthalamic nucleus
- SUR
sulfonylurea receptor
- T2D
type 2 diabetes
- TCA
tricarboxylic acid
- TRP
transient receptor potential
- TTCC
T‐type calcium channel
- UCP
uncoupling protein
- VGCC
voltage‐gated calcium channel
- VTA DA
dopaminergic neurons of the ventral tegmental area
- WT
wildtype
SN DA neurons – specific functions and fate in Parkinson's disease
Functional anatomy of the dopaminergic midbrain system
Dopamine‐releasing catecholaminergic neurons within the midbrain are part of the basal ganglia network, and are crucial for a variety of fundamental brain functions, such as voluntary movement, goal‐directed behavior and habit formation, and also motivation, emotion, cognition, reward, memory, associative learning, and decision making (Bjorklund and Dunnett 2007; Gerfen and Surmeier 2011; D'Ardenne et al. 2012; Pignatelli and Bonci 2015). Consequently, their loss or their dysfunction is associated with respective pathophysiologies, like schizophrenia, substance abuse, or Parkinson's disease (PD) (Wise 2009; Brisch et al. 2014; Kalia and Lang 2015).
The cell bodies of tyrosine hydroxylase‐positive dopaminergic (DA) neurons within the midbrain are mainly arranged within three nuclei in close proximity: the ventral tegmental area (VTA, or A10 group), the retrorubral field (RRF, or A8 group), and the Substantia nigra (SN, or A9 group) (Dahlstrom and Fuxe 1964; Bjorklund and Dunnett 2007). Simplified, according to their widespread axonal projections, DA midbrain neurons are subdivided into the mesocorticolimbic system (VTA and RRF DA neurons), and the mesostriatal system (SN DA neurons). However, in‐depth cell‐specific analyses of DA projections and inputs, biophysical properties, neuronal wiring, gene expression, and physiological functions have revealed a much more complex picture, defining a variety of different subpopulations of midbrain DA neurons (Bromberg‐Martin et al. 2010; Liss and Roeper 2010; Bourdy and Barrot 2012; Lammel et al. 2012; Ungless and Grace 2012; Watabe‐Uchida et al. 2012; Jhou et al. 2013; Roeper 2013; Volman et al. 2013; Poulin et al. 2014; Beier et al. 2015; Hoglinger et al. 2015; Lerner et al. 2015).
Electrical activity patterns of SN DA neurons in vivo and in vitro
The number of SN DA neurons ranges from about 10.000 (bilaterally) in adult mice to about 400.000 in adult humans (Damier et al. 1999a; Brichta and Greengard 2014), and they are predominantly found within the SN pars compacta. They receive direct excitatory inputs from the subthalamic nucleus, the pedunculopontine nucleus, as well as from somatosensory and motor cortices, and they receive inhibitory inputs particularly from the (dorsolateral) striatum (Grillner and Mercuri 2002; Henny et al. 2012; Watabe‐Uchida et al. 2012; Lerner et al. 2015).
In vivo, within the intact basal ganglia network, SN DA neurons predominantly display two types of firing patterns: tonic irregular single‐spike activity in the frequency range of 1–10 Hz (compare Fig. 2) (Hage and Khaliq 2015), or phasic so‐called burst activity at higher frequencies (~ 13–20 Hz in anesthetized animals, or up to 80 Hz in awake animals or humans), as originally described by the pioneering work of Grace and Bunney (Grace and Bunney 1984a,b) and Johnson (Johnson et al. 1992), reviewed, e.g. in (Johnson and Wu 2004; Bean 2007a; Deister et al. 2009; Lee and Tepper 2009; Paladini and Roeper 2014; Dragicevic et al. 2015). Burst activity of SN DA neurons causes a phasic supralinear increase in released dopamine, predominantly in response to novel, unexpected or salient events (Schultz et al. 2015), and it encodes the so‐called ‘Go’ signals that facilitate movement initiation and motor learning (Jin and Costa 2010; Costa 2011; Sommer et al. 2014). As illustrated in Figs 1 upper and 2, in vitro, even in complete synaptic isolation, SN DA neurons still display an intrinsically generated, very regular activity with relatively low frequencies (~ 0.5–4 Hz), the so‐called pacemaker activity (Grace and Onn 1989; Bean 2007a; Chan et al. 2007; Lammel et al. 2008; Margolis et al. 2010).
Figure 2.
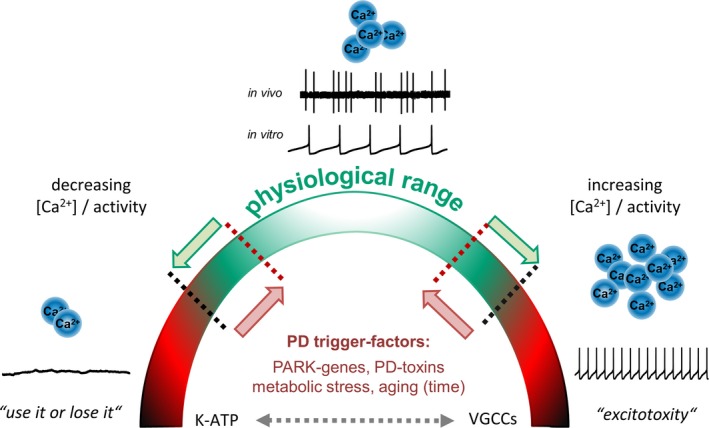
Double‐edged roles of activity and free intracellular Ca2+ levels for physiological functions of SN DA neurons and their vulnerability to PD‐triggers. Shown are typical in vivo and in vitro single‐spike activity patterns of SN DA neurons from adult mice (adapted from Dragicevic et al. 2015; methods detailed in Schiemann et al. 2012; Dragicevic et al. 2014). Note that the important – and energy demanding – burst activity mode of SN DA neurons is not shown. The cartoon summarizes that SN DA neurons possess several intrinsic feedback and feed‐forward mechanisms to protect and to adapt their activity pattern as well as their oscillatory calcium homeostasis (crucial for dopamine release, and metabolic homeostasis) in both directions within a physiological bandwidth (indicated by green arrows/color and black dotted lines). Both, reduced as well as elevated activity and associated calcium homeostasis can trigger SN DA degeneration, as indicated by ‘use it or lose it’ and ‘excitotoxicity’. PD‐trigger factors could narrow the physiological bandwidth of these two parameters, and thus facilitate pathophysiological and degenerative pathways (indicated by red arrows/color and red dotted lines). VGCCs as well as K‐ATP channels are from particular importance, as they have bidirectional physiological functions, can stimulate each other's activity (indicated by dotted gray double‐arrow), and they both can trigger selective SN DA degeneration and PD (compare figure 1, for details see text). Abbreviations: K‐ATP: ATP‐sensitive potassium channel; PARK‐gene: Parkinson's disease associated gene; PD: Parkinson's disease; VGCCs: voltage‐gated calcium channels.
Figure 1.
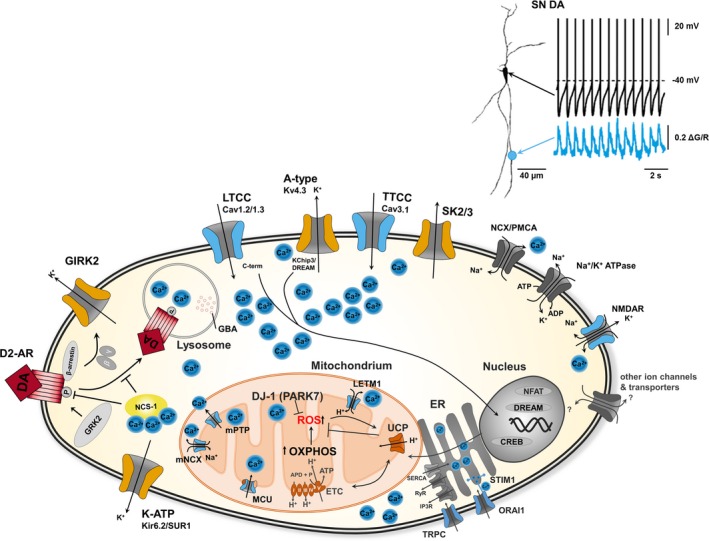
Converging pathways of ion channel activities, Ca2+ homeostasis and metabolic stress in Substantia nigra dopaminergic neurons in health and Parkinson's disease. Upper: Whole‐cell current clamp recording of a SN DA neuron (shown left as a projection image), illustrating the typical low‐frequency pacemaker activity (black trace, mV). Lower blue trace depicts parallel 2‐photon laser scanning fluo‐4 Ca2+ imaging of the same neuron, illustrating the dendritic Ca2+ oscillations (∆G/R), that are fully blocked by the L‐type Ca2+ channel blocker isradipine particularly in the proximal dendrites, while activity of SN DA neurons remains largely unaffected (figure adapted from and methods detailed in Guzman et al. 2010). Lower: Cartoon illustrating distinct ion channels, receptors and transporters that generate or modulate the activity patterns of SN DA neurons in vivo and in vitro, and that are associated with oscillating Ca2+ levels and signaling pathways, affecting mitochondrial and lysosomal function as well as gene expression in health and in Parkinson's disease, (see text for details). Note that only a selection of ion channels that are expressed in SN DA neurons is depicted. Voltage‐gated LTCCs (particular of the Cav1.3 type) as well as metabolically gated K‐ATP channels (of the Kir6.2/SUR1 type) seem to be crucial for physiological SN DA function, but have both also been linked to SN DA degeneration and PD. Abbreviations: A‐type Kv/KChip: A‐type voltage‐gated K+ channel; CREB: cAMP response element‐binding protein; D2‐AR: dopamine D2 autoreceptor; DJ‐1: PARK7 gene product; DREAM: DRE antagonist modulator; ER: endoplasmatic reticulum; ETC: electron transport chain; GBA: glucocerebrosidase; GIRK: G‐protein‐coupled inwardly rectifying K+ channel; GRK2: G‐protein‐dependent kinase 2; IP3R: inositol‐3‐phosphate receptor; K‐ATP: ATP‐sensitive K+ channel; LETM1: high Ca2+ affine leucine zipper EF‐hand containing transmembrane protein 1; LTCC (Cav1.3): Cav1.3 L‐type voltage‐gated Ca2+ channel; mCU: mitochondrial Ca2+ uniporter; MNCX: mitochondrial Na+/Ca2+ exchanger; mPTP: mitochondrial permeability transition pore; NCS‐1: neuronal Ca2+ sensor 1; NCX: Na+/Ca2+ exchanger; NMDA‐R: N‐methyl‐D‐aspartate glutamate receptor; OXPHOS: oxidative phosphorylation; P: phosphate; PD: Parkinson's disease; PMCA: plasma membrane Ca2+ ATPase; ROS: reactive oxygen species; RyR: ryanodine receptor; SERCA: sarcoplasmic/endoplasmic reticulum (ER) Ca2+ ATPase; SK: small conductance Ca2+ sensitive K+ channel; STIM: stromal interaction molecule; TCA: tricarboxylic acid cycle, TRP: transient receptor potential; TTCC: T‐type voltage‐gated Ca2+ channel; UCP: uncoupling protein.
SN DA neurons are particularly vulnerable to degenerative triggers
Although seemingly more homogeneous than the VTA/RRF DA neurons, recent work identified distinct subpopulations of DA neurons also within the SN: the medial and lateral SN DA neurons, with axonal projections to the dorsomedial and dorsolateral striatum, respectively (Bjorklund and Dunnett 2007; Schiemann et al. 2012; Lerner et al. 2015), as well as the ventral tier and dorsal tier SN DA neurons (Damier et al. 1999a; Watabe‐Uchida et al. 2012; Lehericy et al. 2014). However, dorsal tier SN DA neurons are more ‘VTA/RRF DA‐type’ like, in view of their electrophysiological properties, their expression of the cytosolic calcium‐binding protein calbindin‐D28K, the absence of DCC (deleted in colorectal cancer, the receptor for the axon guidance molecule netrin), and their lower susceptibility to degenerative triggers and to cell death in PD (Yamada et al. 1990; Damier et al. 1999a; Neuhoff et al. 2002; Lammel et al. 2008; Margolis et al. 2010; Brichta and Greengard 2014). In contrast, ventral tier SN DA neurons build the ‘classical’ calbindin‐D28K‐negative and DCC‐positive mesostriatal SN DA population (located particularly in the ventrolateral SN) that is crucial for control and modulation of voluntary movements and for habit learning/formation. These SN DA neurons are particularly vulnerable to degeneration in PD, and their loss causes the major motor symptoms of PD patients: bradykinesia, rigor, resting tremor, and postural instability (Fearnley and Lees 1991; Agid et al. 1993; Damier et al. 1999b; Braak et al. 2004; Osborne et al. 2005; Kordower et al. 2013; Brichta and Greengard 2014; Dopeso‐Reyes et al. 2014; Kalia and Lang 2015). It is noteworthy that other, non‐DA neurons are also progressively degenerating in PD, particularly noradrenergic neurons within the locus coeruleus (LC), but also neurons, e.g. within the pedunculopontine nucleus, or the dorsal motor nucleus (Jellinger 1991; Zarow et al. 2003; Del Tredici and Braak 2013; Goedert et al. 2013; Greene 2014; Sanchez‐Padilla et al. 2014).
In this review, we focus on the ‘classical’ mesostriatal (or nigrostriatal) SN DA neurons, and on their particularly high vulnerability to degeneration in PD. The cause for the differential vulnerability of the different types of SN DA and VTA DA neurons to degenerative triggers is still not clear; however, it is present not only in PD and its animal models, but it also occurs in the non‐PD brain during aging (Damier et al. 1999b; Hindle 2010; Collier et al. 2011; Reeve et al. 2014; Rodriguez et al. 2015). Indeed, aging is the most prominent risk factor for PD, and PD is discussed to reflect a form of premature or accelerated aging (Surmeier 2007; Hindle 2010; Collier et al. 2011).
Impaired calcium homeostasis, mitochondrial and lysosomal dysfunction, and metabolic stress are central trigger factors for PD
Although the cause of most PD cases is unclear (idiopathic), in addition to age, a variety of environmental and genetic (PARK‐genes) PD trigger factors have been identified, as detailed in a range of excellent reviews (Hirsch and Hunot 2009; Hardy 2010; Winklhofer and Haass 2010; Gasser et al. 2011; Deleidi and Gasser 2013; Kieburtz and Wunderle 2013; Ramanan and Saykin 2013; Singleton et al. 2013; Sulzer and Surmeier 2013). Mutations in PARK‐genes lead to familial Mendelian forms of PD. Particularly, ion channels and activity‐related oscillatory intracellular Ca2+ load (compare Fig. 1, upper insert), mitochondrial and lysosomal dysfunction, oxidative and metabolic stress, as well as toxic protein propagation, protein mishandling, impairment of the ubiquitin–proteasome pathway and inflammation contribute to SN DA degeneration and sporadic as well as familial PD (Gonzalez‐Hernandez et al. 2010; Guzman et al. 2010; Obeso and Lanciego 2011; Coskun et al. 2012; Sulzer and Surmeier 2013; Beilina and Cookson 2015; Gan‐Or et al. 2015; Pacelli et al. 2015; Roselli and Caroni 2015). In view of PD as a multifactorial disease, there is increasing evidence that all these intrinsic as well as extrinsic factors seem to intermingle and aggravate each other, and affect particularly SN DA neurons and their vulnerability in PD, as discussed in the next paragraphs.
Mitochondrial (dys‐)function and metabolic stress in SN DA neurons
SN DA neurons appear to be intrinsically particularly vulnerable to metabolic stress and mitochondrial dysfunction. Thus, it is not too surprising that inhibitors of the complex I of the mitochondrial electron transfer chain (ETC), like MPTP, induce selective SN DA neuron degeneration and parkinsonism in humans and animals, and are widely used to generate PD‐animal models (Langston and Ballard 1983; Schapira 2010; Blesa and Przedborski 2014). The dopamine metabolism itself already seems to cause intrinsically high levels of metabolic stress and free reactive oxygen and nitrogen species (ROS/RNS) in SN DA neurons (Berman and Hastings 1999; Sulzer 2007; Surmeier et al. 2011). ROS/RNS can damage proteins, lipids, and DNA of mitochondria, leading to their dysfunction, while mitochondrial dysfunction in turn provides a source of metabolic stress and ROS/RNS – propagating a self‐accelerating cycle of increasing metabolic stress in DA neurons (Henchcliffe and Beal 2008; Schapira 2008; Vila et al. 2008). Mitochondria build a dynamic network (Youle and van der Bliek 2012), where important metabolic pathways take place, like the tricarboxylic acid (TCA) cycle and the oxidative phosphorylation within the ETC, and thus ATP production, as well as control of metabolic stress and cell death, e.g. via apoptosis (Perier et al. 2012). Consequently, cells possess defense responses against mitochondrial dysfunction. This is particularly important for the highly vulnerable SN DA neurons, which are subjected to continued metabolic stress. In this context, a double‐edged role of neuromelanin is discussed. It is build by catecholamine metabolites, and accumulates particularly in highly vulnerable SN DA (and LC) neurons with age. On one hand, neuromelanin seems to protect SN DA neurons from (iron‐catalyzed) dopamine oxidation, ROS production, metabolic stress, and from degeneration, while its neuronal release seems to have opposite effects on SN DA neurons, as it can propagate microglia activation, neuroinflammation, and neurodegeneration (Hirsch et al. 1988; Zucca et al. 2015). Additional defense mechanisms in SN DA neurons against mitochondrial dysfunction and metabolic stress are mild mitochondrial uncoupling by uncoupling proteins (UCPs), and a variety of other antioxidative mechanisms, mediated e.g. by PARK‐gene functions (Liss et al. 2005; Guzman et al. 2010; Sanchez‐Padilla et al. 2014). The PARK7 gene product DJ‐1 (Bonifati et al. 2003) for instance suppresses mitochondrial metabolic stress and ROS levels particularly in SN DA neurons (Guzman et al. 2010). However, despite their defense mechanisms and their DNA‐repair machinery (Alexeyev et al. 2013), mitochondrial DNA damage accumulates in SN DA neurons with age, likely contributing to their already high metabolic stress levels, and to their progressive degeneration (Bender et al. 2006; Kraytsberg et al. 2006; Reeve et al. 2008).
Lysosomal (dys‐)function and metabolic stress in SN DA neurons
As a further protection mechanism against mitochondrial dysfunction and metabolic stress, defective mitochondria are disposed of by mitophagy, an autophagic‐lysosomal elimination (Pilsl and Winklhofer 2012; Ryan et al. 2015). This dynamic process seems to be particularly important for SN DA neurons, as a variety of PARK‐gene products are involved in this pathway, like the mitochondrial kinase PINK1/PARK6 (Valente et al. 2004; Hao et al. 2010), the mitochondrial protease HtrA2/Omi/PARK13 (Plun‐Favreau et al. 2007), the E3‐ubiquitin ligase parkin/PARK2, (Kitada et al. 1998), and the F‐box only protein 7/PARK15 (Di Fonzo et al. 2009). Loss of function mutations in these PARK‐genes trigger SN DA degeneration and cause autosomal recessive familial forms of PD (Gasser et al. 2011; van Brug et al. 2015). A variety of further PARK‐genes are associated with the autophagy–lysosome pathway (Bourdenx et al. 2014; Gan‐Or et al. 2015). For instance, the lysosomal p‐type ATPase13A2/PARK13 (Ramirez et al. 2006; Kruger et al. 2011), or alpha‐synuclein (PARK1, PARK4), which is partly degraded by lysosomes (Dehay et al. 2013; Hunn et al. 2015), the leucine‐rich repeat kinase LRRK2/PARK8 (Funayama et al. 2002; Khan et al. 2005), the LIM homeodomain transcription factor LMX1b, crucial not only for SN DA development and maintenance but also for autophagy–lysosome pathway function (Laguna et al. 2015), and the lysosomal glucocerebrosidase (GBA) (Siebert et al. 2014; Schapira 2015).
Heterozygous mutations in the GBA gene occur in about 10% of ‘sporadic’ PD cases and have just recently emerged as a major risk factor for PD (Mazzulli et al. 2011; Schapira and Gegg 2013). Reduced GBA function triggers accumulation of misfolded alpha‐synuclein and PD (Asselta et al. 2014; Schapira 2015). Mutant GBA‐protein is trapped in the endoplasmic reticulum (ER), causing not only elevated metabolic stress but also elevated Ca2+ release from the ER (Westbroek et al. 2011; Tan et al. 2014; Kilpatrick et al. 2015). Furthermore, dopaminergic neurons derived from pluripotent stem cells of GBA‐associated PD patients not only display lysosomal and autophagic dysfunction but also elevated intracellular Ca2+ levels and impaired calcium homeostasis (Schondorf et al. 2014). These findings point to another factor that contributes to metabolic stress, as well as to mitochondrial and lysosomal (dys)function in SN DA neurons: their calcium homeostasis.
Calcium homeostasis and metabolic stress in SN DA neurons
Intracellular Ca2+ levels, Ca2+ microdomains, Ca2+ buffering, and the mode of Ca2+ entry are tightly controlled in SN DA neurons, as Ca2+ modulates a variety of cellular functions, like excitability, neurotransmitter release, ATP‐production, apoptosis, and general regulation of enzymes and gene expression (Dolmetsch 2003; Brini et al. 2014; Burgoyne and Haynes 2014). In view of SN DA neuron vulnerability to metabolic stress, Ca2+ regulates mitochondrial motility (Wang and Schwarz 2009), stimulates the mitochondrial nitric oxide synthase, leading to elevated levels of ROS/RNS (Traaseth et al. 2004; Murphy 2009; Sanchez‐Padilla et al. 2014), and it stimulates enzymes of the TCA cycle and the ETC, and thus ATP production, thereby further boosting ROS generation (Gleichmann and Mattson 2011). Ca2+ also stimulates calpain, a protease that is involved in alpha‐synuclein processing, SN DA degeneration, and PD (Crocker et al. 2003; Dufty et al. 2007; Diepenbroek et al. 2014; Games et al. 2014). Furthermore, when mitochondrial Ca2+ levels rise above a certain threshold, opening of the mitochondrial permeability transition pore, and thus neurodegeneration is triggered (Martin et al. 2014). In view of differential vulnerability of DA neurons to degenerative triggers, physiological mitochondrial matrix Ca2+ levels appear to be higher in highly vulnerable SN DA neurons compared to more resistant VTA DA neurons (J. Surmeier, personal communication), possibly contributing to their differential vulnerability to metabolic stress and PD‐trigger factors.
As illustrated in Fig. 1, Ca2+ can enter SN DA neurons from the extracellular space via channels and transporters located in their plasma membrane. More precisely, via voltage‐gated calcium channels (VGCCs) and/or glutamate receptors like NMDA‐R or AMPA‐R (Bezprozvanny and Mattson 2008), or by store operated calcium channels, like STIM/ORAI and transient receptor potential channel complexes (Dietrich et al. 2014; Majewski and Kuznicki 2015). Ca2+ is also released from mitochondria (e.g. via the mitochondrial sodium–calcium exchanger) and the ER (via RyR and IP3 receptors) (Brini 2003; Malli et al. 2003). Spatial and temporal dynamics of cytosolic Ca2+ levels in SN DA neurons are further controlled by its extrusion, e.g. via plasma membrane Ca2+ ATPases, or sodium–calcium exchangers (NCX), and by intracellular Ca2+‐buffering (Schwaller 2010; Hajnoczky et al. 2014). Because of low levels of Ca2+‐binding proteins in SN DA neurons, like calbindin, Ca2+ is mainly buffered by uptake into the ER, via the sarco‐ER Ca2+ ATPases (SERCAs), and by mitochondria (Verkhratsky 2005; Cali et al. 2012; Hoppins and Nunnari 2012). Mitochondria take up Ca2+ directly via a mitochondrial Ca2+ uniporter mCU, or a mitochondrial Ca2+/H+ exchanger Letm1 (Baughman et al. 2011; De Stefani et al. 2011; Waldeck‐Weiermair et al. 2011), or indirectly via the ER‐mitochondria pathway (Rizzuto et al. 2009; Drago et al. 2011; Bakowski et al. 2012). This pathway is involved in defining SN DA vulnerability and in PD (Cali et al. 2014; Guardia‐Laguarta et al. 2015), and survival of cultured SN DA neurons is stimulated by modulating Ca2+ release from the ER (Guerreiro et al. 2015), while MPTP can induce mitochondrial as well as ER stress in DA neurons (Selvaraj et al. 2012).
Besides the ER‐mitochondria compartment, lysosomes also buffer cytosolic Ca2+ – via uptake by a yet unidentified Ca2+/H+ exchanger (Gomez‐Suaga et al. 2012; Osellame and Duchen 2014). The Ca2+ concentration is assumed to be about 5000‐fold higher in lysosomes than in the cytoplasm (Christensen et al. 2002; Xu and Ren 2015). In view of novel neuroprotective PD‐therapies, ambroxol, a well‐established drug for treating conditions with productive cough or viscous mucus (Weiser 2008), has recently emerged as a promising new tool. Ambroxol, which can pass the blood–brain barrier, reduces neuronal metabolic stress and was shown to improve GBA‐activity and lysosomal function in human fibroblasts derived from PD‐patients (Bendikov‐Bar et al. 2013; Albin and Dauer 2014; McNeill et al. 2014; Siebert et al. 2014). Ambroxol normalizes PD‐associated GBA dysfunction (and associated calcium homeostasis) seemingly by increasing its activity and its expression (Maegawa et al. 2009; McNeill et al. 2014; Ambrosi et al. 2015; Schapira 2015). Furthermore, recent findings indicate that Ambroxol can accumulate in lysosomes, and directly affect their H+ and Ca2+ homeostasis, and it stimulates exocytosis from secretory lysosomes via pH‐dependent Ca2+ release (Fois et al. 2015).
In conclusion, all these findings illustrate a complex homeostatic interplay of calcium homeostasis, lysosomal and mitochondrial function and metabolic stress, as well as PARK‐gene products in SN DA neurons, whose physiological action is already metabolically challenging (see below). Accordingly, dysfunction of this complex interplay triggers degeneration, particularly of metabolically compromised SN DA neurons, and it constitutes a converging pathological PD‐pathway. Consequently, its pharmacological modulation offers novel strategies for neuroprotective PD therapies – as currently under investigation for Ambroxol.
The electrical activity of SN DA neurons causes particular metabolic stress and triggers dopamine release
There is one central factor that critically modulates the complex interplay of calcium, mitochondrial and lysosomal function particularly in SN DA neurons that we have not yet considered: their electrical activity. Neuronal activity per se implies high energy demand and metabolic stress, mainly because of stimulation of the Na+/K+ ATPase, that is necessary to maintain the asymmetric ion distribution after action potentials (intracellular high K+, low Na+) and that is consuming about 50% and more ATP in active neurons (Laughlin et al. 1998; Howarth et al. 2012; Johar et al. 2014). Again, SN DA neurons seem to be particularly dependent on proper Na+/K+ ATPase activity, as mutations in its ATP1A3 subunit causes a form of rapid‐onset dystonia parkinsonism, RDP (Heinzen et al. 2014). This might be attributed to the fact that the metabolic cost of SN DA neuron activity is particularly high, as specific voltage‐gated calcium channels (VGCC, see below) are active during their physiological action, causing an activity‐related, oscillatory increase in intracellular Ca2+ levels (compare Fig. 1, upper insert). These oscillatory Ca2+ changes cause related oscillatory changes of mitochondrial membrane potentials, ROS‐levels, and of plasma membrane Ca2+ ATPase activity (Fig. 1) (Guzman et al. 2010; Surmeier et al. 2011; Pissadaki and Bolam 2013). It is important to emphasize that more resistant VTA DA neurons display an electrical activity that is similar to that of highly vulnerable SN DA neurons; however, it is not associated with oscillatory elevated Ca2+ and metabolic stress levels (Guzman et al. 2010; Khaliq and Bean 2010). Highly vulnerable locus coeruleus neurons however display similar activity‐dependent Ca2+ oscillations and related metabolic stress levels – that are interestingly sensitive to orexinergic peptides (orexins) (Sanchez‐Padilla et al. 2014).
Taken together, the specific mode of electrical activity of SN DA neurons generates periodically elevated Ca2+ and metabolic stress levels that likely render them particularly vulnerable to additional metabolic stressors and PD‐triggers. Not only because of the general metabolic cost of neuronal activity, but predominantly as a result of their Ca2+ channel activity. In conclusion, the elevated activity‐related Ca2+ and metabolic stress levels in SN DA neurons provide a molecular rationale for their particularly high vulnerability to mitochondrial, lysosomal, and PARK‐gene dysfunction, and thus for the differential vulnerability of SN DA and VTA DA neurons to degeneration in PD. Moreover, given its high metabolic costs, the oscillatory Ca2+ signal in SN DA neurons must be crucial for their specific physiological functions. Indeed, as SN DA neurons release dopamine in a calcium‐ and activity‐dependent manner (Rice et al. 2011; Brimblecombe et al. 2015) – from presynaptic axonal sites into the striatum (De‐Miguel and Nicholls 2015), as well as from somatodendritic sites into the midbrain (Ford et al. 2010; Rice and Patel 2015) – action potential frequency and its pattern, as well as intracellular presynaptic and somatodendritic Ca2+ levels in fact mediate eventually all physiological functions of SN DA neurons.
Ion channels define activity pattern and Ca2+ homeostasis of SN DA neurons
Given the central role of SN DA neurons for voluntary movement control, and thus, e.g. context‐dependent fight‐or‐flight responses, it is not surprising that a variety of intrinsic and extrinsic factors and mechanisms are crucial for generating, maintaining, and modulating activity patterns, calcium homeostasis, and dopamine release of SN DA neurons within their physiological bandwidth (Guzman et al. 2009; Surmeier and Schumacker 2013; Dragicevic et al. 2015; Kimm et al. 2015). Particularly important in this context are ion channels and receptors, as they not only generate but also homeostatically shape SN DA activity patterns to physiological demands; because of their specific expression pattern, their regulations and their complex interplay, as illustrated in Fig. 1 and as reviewed, e.g. in (Bean 2007a; Michel et al. 2007; Guzman et al. 2009; Liss and Roeper 2010; Paladini and Roeper 2014; Dragicevic et al. 2015). Here, we focus on how distinct ion channels (within a complex feedback and feed‐forward signaling network) modulate activity patterns, intracellular calcium homeostasis, and metabolic sensitivity specifically of SN DA neurons and thus define not only their physiological functions, but also their vulnerability to degeneration in PD.
GIRK channel‐coupled D2‐autoreceptors mediate calcium‐ and dopamine‐dependent activity control of SN DA neurons
The activity of SN DA neurons in vitro and in vivo is inhibited by dopamine itself in a negative feedback loop via dopamine autoreceptors of the D2‐subtype (D2‐AR) (De Mei et al. 2009; Beaulieu et al. 2015). D2‐ARs belong to the seven‐transmembrane G‐protein‐coupled receptor family, and are located at the presynapses as well as on the somatodendritic compartment of SN DA neurons. Upon dopamine binding, the G‐protein beta‐gamma subunit gets released from the D2 autoreceptor, and directly opens G‐protein‐coupled inwardly rectifying potassium channels (GIRK2) (Luscher and Slesinger 2010). The GIRK2 activation in turn leads to hyperpolarization of the SN DA neuron membrane potential and thereby inhibits their electrical activity (Bozzi and Borrelli 2006; De Mei et al. 2009; Ford 2014; Dragicevic et al. 2015). In essence, in a negative feedback loop and via D2‐ARs, dopamine itself modulates and adapts its own activity‐dependent, presynaptic, and somatodendritic release from SN DA neurons, and consequently its physiological functions (De‐Miguel and Nicholls 2015; Rice and Patel 2015). It is noteworthy, that this D2‐AR/GIRK2 feedback regulation is particularly pronounced in highly vulnerable mature SN DA neurons, compared to more resistant VTA DA neurons (Lammel et al. 2008; Brichta and Greengard 2014; Dragicevic et al. 2015). Moreover, within the population of SN DA neurons, D2‐AR/GIRK2 activity control displays a homeostatic plasticity (Fig. 1). D2‐AR responses of SN DA neurons are sensitized in a dopamine‐, calcium‐, and age‐dependent manner: their dopamine autoinhibition – in response to phasically elevated in vivo dopamine levels (e.g. caused by cocaine or L‐DOPA), during maturation, and/or because of elevated free intracellular Ca2+ levels – is strengthened by functional coupling to the neuronal calcium sensor NCS‐1 (Borgkvist et al. 2014; Dragicevic et al. 2014; Ng et al. 2016; Poetschke et al. 2015). As illustrated in Fig. 1, NCS‐1 binds to D2‐receptors at their third intracellular loop in a calcium‐dependent fashion, and thereby prevents G‐protein‐coupled receptor kinase (GRK2) mediated phosphorylation, and thus beta‐arrestin mediated internalization and D2‐receptor desensitization (Kabbani et al. 2012; Beaulieu et al. 2015). We showed that this calcium‐dependent D2‐AR sensitization preferentially depends on VGCCs of the Cav1.3 channel subtype (that are discussed below) (Dragicevic et al. 2014).
In view of SN DA vulnerability in PD, sensitized D2‐AR responses appear less pronounced in PARK7 KO (DJ‐1) and in PARK8 transgenic (LRRK2‐R1441C) mice, and their mature SN DA neurons display juvenile‐like desensitizing D2‐AR responses (Goldberg et al. 2005; Tong et al. 2009; Tong and Shen 2012). Moreover, remaining human SN DA neurons from PD patients display a transcriptional dysregulation of the NCS‐1/D2‐AR/GIRK2 signaling network (Dragicevic et al. 2014). These findings indicate that dopamine‐ and calcium‐mediated sensitization of D2‐AR responses also adapts SN DA neuron activity in vivo and in PD. In context of SN DA degeneration, sensitized D2‐AR/GIRK2 responses will reduce elevated activity, and thus oscillatory calcium load, and related metabolic stress in SN DA neurons, and thus could protect them from overexcitability and excitotoxic cell death (compare Fig. 2). SN DA neurons are particularly prone to excitotoxicity, as it is triggered by elevated activity, perturbed calcium homeostasis, high levels of ROS/RNS, and metabolic stress (Prentice et al. 2015).
In conclusion, flexible dopamine‐dependent D2‐AR/GIRK2 responses in SN DA neurons allow modulation and homeostatic adaptation of their activity, their oscillatory calcium signaling and their dopamine release to physiological needs. Beyond that, D2‐AR/GIRK2 sensitization, e.g. as a result of pathophysiologically elevated SN DA activity and/or calcium overload, will counteract excitotoxic cell death, and thus represents another defense mechanism against metabolic stress. As summarized next, in this view, two different classes of ion channels have emerged to be particularly relevant for SN DA neuron activity, related calcium and metabolic homeostasis, as well as for PD‐pathophysiology: voltage‐gated calcium channels (VGCCs), and metabolically gated ATP‐sensitive potassium (K‐ATP) channels.
Bidirectional roles of voltage‐gated Ca2+ channels in SN DA neurons
A family of voltage‐gated Ca2+ channels is expressed in SN DA neurons
Voltage‐gated calcium channels (VGCCs) are transmembrane proteins that mediate membrane potential‐dependent extracellular Ca2+ entry into cells, and their dysfunction is associated with a variety of diseases (Catterall et al. 2005; Simms and Zamponi 2014; Zamponi et al. 2015). VGCC subtypes exhibit distinct spatial, temporal, and disease‐related expression patterns (Perez‐Reyes 2003; Catterall et al. 2005; Adams and Snutch 2007). They are tightly regulated by Ca2+, calmodulin, and related Ca2+ sensor proteins, which cause facilitation and inactivation of channel activity (Zuhlke et al. 1999; Catterall et al. 2013). In first instance, their open probability increases with depolarization of neuronal membrane potentials, and they undergo not only voltage‐dependent but also Ca2+‐dependent channel inactivation. This provides an important autoregulatory feedback mechanism (Cens et al. 2006; Christel and Lee 2012; Catterall and Zheng 2015). Depending on their membrane potential activation thresholds, VGCCs are divided into high voltage‐activated (HVA) and low voltage‐activated (LVA) channels (Flockerzi et al. 1991; Snutch et al. 2001; Perez‐Reyes 2003; Durante et al. 2004). Members of the HVA channel family require a stronger membrane depolarization for activation (mainly caused by action potentials), while LVA channels are already active at subthreshold membrane potentials, before the threshold for activation of voltage‐gated sodium channels is reached and action potentials are generated (Armstrong and Matteson 1985; Simms and Zamponi 2014; Zamponi et al. 2015). The family of HVA channels consists of the so‐called L‐type calcium channels (LTCCs), with channel pores build by Cav1.1‐1.4 alpha1‐subunits, the P/Q‐type (or Cav2.1), the N‐type (Cav2.2), and the R‐type channels (Cav2.3). LVA channels are constituted by T‐type channels (TTCCs), build by Cav3.1–3.3 pore‐forming alpha‐subunits (Catterall et al. 2005; Simms and Zamponi 2014; Zamponi et al. 2015).
All these VGCC‐classes are functionally expressed in SN DA neurons: Cav1.2 and Cav1.3 generate LTCC currents (both expressed in several splice‐variants, and in complex with yet to be fully defined accessory β and α2‐δ subunits), and Cav3.1 is the predominant form of T‐type channels (Olson et al. 2005; Striessnig et al. 2006; Lipscombe et al. 2013; Dragicevic et al. 2014; Lieb et al. 2014; Poetschke et al. 2015; Scharinger et al. 2015). In view of their emerging role in SN DA neurons and in PD, it is important to note that Cav1.3 LTCCs activate at more negative (subthreshold) membrane potentials compared to the widely expressed Cav1.2 and other HVA channels (Koschak et al. 2001; Lipscombe et al. 2004; Lieb et al. 2014). The distinct types of VGCCs in SN DA neurons are crucial for their presynaptic as well as for their (mechanistically still not fully understood) somatodendritic dopamine release (Rice et al. 2011; Brimblecombe et al. 2015; Rice and Patel 2015). VGCC‐activity causes Ca2+ mediated slow oscillatory membrane potentials in SN DA neurons (Chan et al. 2007; Puopolo et al. 2007; Guzman et al. 2009; Putzier et al. 2009), and an oscillatory, metabolically challenging Ca2+ influx, associated with their electrical activity, as described above (Bean 2007b).
PD‐protective effects of voltage‐gated L‐type Ca2+ channel blockers
LTCCs in SN DA neurons, particularly those of the Cav1.3 subtype, have received much attention in recent years, since blood–brain barrier (BBB) permeable LTCC blockers of the dihydropyridine‐type [DHPs, like isradipine (Striessnig et al. 1998)] apparently reduce the risk for developing PD by about 30% in humans, as epidemiologic data of retrospective studies indicate (Becker et al. 2008; Ritz et al. 2010; Marras et al. 2012; Pasternak et al. 2012; Gudala et al. 2015; Lang et al 2015). This PD‐protective effect is assumed (but not yet clearly demonstrated) to be caused by inhibition of LTCCs in SN DA neurons, most likely of Cav1.3 channels, which in turn abolishes dendritic Ca2+ oscillations and associated oscillatory metabolic stress in mice (Guzman et al. 2010; Kang et al. 2013; Surmeier and Schumacker 2013; Huang et al. 2014; Ortner et al. 2014). These Ca2+ oscillations get stronger and more sensitive to DHPs with increasing distance from the soma (Wilson and Callaway 2000; Guzman et al. 2010; Jang et al. 2011; Dryanovski et al. 2013), and in distal dendrites, they are completely suppressed by low‐doses of DHPs (Guzman et al. 2010; Dryanovski et al. 2013; Surmeier and Schumacker 2013). BBB permeable DHP‐based LTCC blockers are already well‐established drugs, commonly prescribed to treat hypertension (Epstein et al. 2007; Coca et al. 2013). Consequently, after a phase II safety study, they are already in an ongoing clinical phase III study (2013–2018; ClinicalTrials.gov Identifier: NCT00909545) to test their efficacy as neuroprotective PD therapy (Simuni et al. 2010, Parkinson Study 2013). Furthermore, given the widespread expression of Cav1.2 LTCCs but not of Cav1.3 throughout the body, in particular in the cardiovascular system (Sinnegger‐Brauns et al. 2004; Hofmann et al. 2014), novel Cav1.3 selective drugs are under development (Kang et al. 2013; Huang et al. 2014; Ortner et al. 2014). However, the specific physiological functions of Cav1.2 and Cav1.3 and of associated Ca2+ oscillations in SN DA neurons are still not clear. This is an important issue in view of the aimed chronic pharmacological Cav1.3 block as novel therapeutic avenue for PD, as LTCCs seem to have bidirectional and context‐dependent functions in SN DA neurons (Chan et al. 2007; Guzman et al. 2009; Dragicevic et al. 2014; Poetschke et al. 2015).
Cav1.3 L‐type Ca2+ channels stabilize and stimulate activity of SN DA neurons
While originally thought to be crucial for the generation of intrinsic pacemaker activity in SN DA neurons (Nedergaard et al. 1993; Mercuri et al. 1994; Chan et al. 2007; Puopolo et al. 2007), it is now widely accepted that LTCCs are not essential for SN DA pacemaker activity generation, but are stabilizing pacemaker activity and its precision (Guzman et al. 2009; Drion et al. 2011; Branch et al. 2014; Dragicevic et al. 2014; Poetschke et al. 2015). SN DA neurons are able to compensate pharmacological LTCC block as well as germline loss of Cav1.3 LTCCs [Cav1.3 KO mice (Platzer et al. 2000; Striessnig and Koschak 2008)] by other ion channels, e.g. by functional down‐regulation of inhibitory Kv1 potassium channels within minutes after DHP‐mediated distortion of SN DA activity, or functional up‐regulation of either hyperpolarization‐activated cyclic nucleotide‐gated (HCN) cation channels, or of TTCCs (Cav3.1) on longer time scales (Chan et al. 2007; Guzman et al. 2009; Poetschke et al. 2015). HCN channels (built by HCN2‐4) facilitate SN DA pacemaker generation and its frequency (Franz et al. 2000; Neuhoff et al. 2002) in interplay with voltage‐gated sodium channels (Bean 2007a). Cav3.1 TTCCs stabilize SN DA pacemaker frequency and its precision in an age‐dependent manner (Poetschke et al. 2015), most likely by functional coupling to Ca2+ sensitive small conductance potassium channels (SK3), and thereby shaping SN DA action potential after‐hyperpolarization (Wolfart et al. 2001; Wolfart and Roeper 2002; Stocker 2004; Bond et al. 2005; Gueguinou et al. 2014; Poetschke et al. 2015).
In addition to stabilizing SN DA activity, LTCCs allow maintenance of neuronal electrical activity, particularly in metabolic demand situations, because of their Ca2+‐mediated stimulation of the TCA cycle and the ETC, and thus ATP production that is crucial for Na+/K+ ATPase activity and neuronal function. On the other hand, LTCC‐mediated dendritic Ca2+ oscillations also stimulate Ca2+‐related detrimental pathways, as discussed above. Thus, to avoid a vicious LTCC activity‐dependent feed‐forward spiral toward cell death, SN DA neurons are in need of respective protective feedback control mechanisms.
VGCCs can also inhibit SN DA activity, in interplay with K+ channels
Such a protection mechanism is provided by the Ca2+‐dependent sensitization of inhibitory D2‐AR/GIRK2 responses via NCS‐1, that are sensitive to isradipine and depend on Cav1.3 function, as described above (Dragicevic et al. 2014). Hence, Cav1.3 channels have bidirectional functions in SN DA neurons: on one hand, they stimulate their electrical activity, their ATP production, and also their metabolic stress levels, while on the other hand, they can inhibit SN DA activity via stimulation of Ca2+‐ and NCS‐1‐dependent D2/GIRK2 activity (Borgkvist et al. 2014; Dragicevic et al. 2015).
TTCCs seem to have similar bidirectional functions in SN DA neurons as they not only stimulate but also inhibit pacemaker activity via stimulation of potassium channels: Cav3.1 channels can also sensitize Ca2+‐ and NCS‐1 dependent D2‐AR/GIRK2 signaling (Poetschke et al. 2015). Additionally, TTCCs can functionally couple and activate Ca2+‐stimulated A‐type K+ channels (An et al. 2000; Anderson et al. 2010; Jerng and Pfaffinger 2014; Turner and Zamponi 2014; Zamponi et al. 2015) – likely (but not yet demonstrated) also in SN DA neurons. These voltage‐gated A‐type channels are formed by Kv4.3 pore‐forming α‐ and Ca2+‐sensitive KChip3 β‐subunits, and they dynamically inhibit the electrical activity of SN DA neurons, similar as GIRK2 channels (Kimm and Bean 2014; Liss et al. 2001, 2002).
Beyond that, VGCCs can also alter ion channel function and neuronal activity at the level of gene expression, as they are particularly effective in activating Ca2+‐dependent transcription factors, like cAMP response element‐binding protein (CREB), myocyte enhancer factor‐2 (MEF‐2), nuclear factor of activated T cells (NFAT), and downstream regulatory element antagonist modulator (DREAM) (Graef et al. 1999; Mandel and Goodman 1999; Mao et al. 1999; Dolmetsch et al. 2001; Osawa et al. 2001; Dolmetsch 2003; Screaton et al. 2004; Rivas et al. 2011; Putney 2012; Selvakumar et al. 2014). Moreover, the C‐terminus of Cav1.3 and Cav1.2 itself can be cleaved, and translocate to the nucleus in a Ca2+‐dependent fashion, where the LTCC C‐termini act as transcription factors (Dolmetsch 2003; Gomez‐Ospina et al. 2006; Lu et al. 2015). Likewise, the A‐type K+ channel subunit KChip3 (a member of the NCS‐1 like Ca2+ binding protein family) is known as the transcriptional repressor DREAM (Jo et al. 2001; Buxbaum 2004), and can shuttle from the membrane to the nucleus in inverse correlation with cellular Ca2+ levels (Fig. 1) (Li and Adelman 2000; Sours‐Brothers et al. 2009; Mellstrom et al. 2014).
In conclusion, VGCCs modulate the electrical activity of SN DA neurons in both directions: in a positive feed‐forward mechanism by stimulating action potential generation and ATP synthesis, and in a negative feedback mechanism by stimulation of inhibitory K+ channels like GIRK2 or Kv4.3, via calcium‐sensing proteins (e.g. NCS‐1 and KChip3). Moreover, by regulating gene expression, they can modulate SN DA neuron function in opposite manners on different timescales (compare Figs 1 and 2).
Bidirectional roles of metabolically gated K‐ATP channels in SN DA neurons
K‐ATP channels act as metabolic sensors in SN DA neurons
ATP‐sensitive potassium (K‐ATP) channels are heteromultimeric transmembrane proteins, consisting of pore‐forming α‐subunits of the Kir6 type (Kir6.1 or Ki6.2) and regulatory β‐subunits, the sulfonylurea receptors (SUR1 or SUR2A/B) members of the ATP‐binding cassette transporter superfamily (Ashcroft and Gribble 1998; Proks and Ashcroft 2009). Similar as in pancreatic beta‐cells, in mature SN DA neurons K‐ATP channels are formed by Kir6.2 and SUR1 (Liss et al. 2005; Schiemann et al. 2012). However, K‐ATP channels are expressed in a variety of additional neuronal and non‐neuronal cells, and their dysfunction is associated with a variety of diseases (Seino and Miki 2003; McTaggart et al. 2010). Given the crucial role of K‐ATP channel closure for triggering pancreatic insulin secretion in interplay with LTCCs (Ashcroft et al. 1984; Nichols 2006; Clark and Proks 2010; Seino 2012), K‐ATP blockers (sulfonylureas like glibenclamide or tolbutamide) are well‐established drugs for treatment of diabetes type II (T2D), while K‐ATP activators like diazoxide are applied to treat hypertension (Miura and Miki 2003; Ashcroft 2007; Ashcroft and Rorsman 2013; de Wet and Proks 2015).
In view of PD and high vulnerability of SN DA neurons, K‐ATP channels act as so‐called metabolic sensors (Nichols 2006; Liss and Roeper 2010), as their open probability is regulated by ATP‐ and ADP levels, as well as by a variety of other metabolic signals, such as insulin, leptin, ghrelin, long‐chain fatty acids (acyl‐CoA), PIP2, pH, hydrogen peroxide, ROS/RNS, and nitric oxide (as reviewed in (Ashcroft 2007; Dragicevic et al. 2015)). In essence, the open probability of K‐ATP channels is higher in metabolic demand situations, and their activity hyperpolarizes excitable cells, and thus inhibits e.g. energy‐demanding electrical activity of SN DA neurons (Liss and Roeper 2001; Miki and Seino 2005; Nichols 2006; Ashcroft and Rorsman 2013). Thereby, K‐ATP channels protect cells from overexcitability and excitotoxicity – a well‐described mechanism e.g. for ischemic preconditioning and myoprotection (Liss and Roeper 2001; Miki et al. 2001; Yamada and Inagaki 2005; Soundarapandian et al. 2007; Lefer et al. 2009; Flagg et al. 2010).
Considering the differential vulnerability of DA neurons to degeneration, it is important to note that although more resistant VTA DA neurons also express functional K‐ATP channels of the same molecular make‐up, they are not activated in response to metabolic stress that activates K‐ATP channels in SN DA neurons (Liss et al. 1999a, 2005; Schiemann et al. 2012). This differential metabolic sensitivity of SN DA and VTA DA K‐ATP channels is likely caused by differential mitochondrial uncoupling via UCPs that does not compromise ATP production, but lowers ROS and metabolic stress levels (Liss et al. 2005; Guzman et al. 2010).
Concisely, in a feedback mechanism, selective K‐ATP channel activity in SN DA neurons reduces their electrical activity and thus need of energy, particularly in metabolically demanding situations. To avoid, however, a chronic loss of electrical activity and related dopamine release of metabolically challenged SN DA neurons, they are in need of respective feed‐forward control mechanisms.
K‐ATP channels can inhibit but also stimulate activity of SN DA neurons
Indeed, similar as VGCCs, K‐ATP channels have also bidirectional effects in SN DA neurons: in first instance, their activation in response to metabolic stress (e.g. PD‐triggers) reduces SN DA neuron activity as expected (Liss et al. 2005). However, we identified that in vivo, under physiological conditions, within the intact basal ganglia network, K‐ATP channel activity does not inhibit but in contrast stimulate the activity of SN DA neurons by facilitating their switch to NMDA glutamate receptor‐mediated burst activity (Schiemann et al. 2012; da Silva and Costa 2012; Dragicevic et al. 2015). This K‐ATP triggered burst activity of SN DA neurons, associated with a supralinear increase in dopamine release, stimulates novelty‐induced exploration in vivo in mice (Deacon et al. 2006; Schiemann et al. 2012). Burst activity is particularly energy‐demanding, and therefore contributes to metabolic stress; however, its K‐ATP channel‐mediated facilitation in SN DA neurons would allow and maintain goal‐directed behavior in novel situations, particularly under metabolic demand, like food deprivation or fight‐or‐flight situations (Schiemann et al. 2012; Paladini and Roeper 2014; Dragicevic et al. 2015). It is noteworthy, that in subthalamic nucleus neurons, in contrast to SN DA neurons, NMDA‐R activity stimulates K‐ATP channels (in vitro), thereby reducing neuronal burst activity that is pathologically increased in PD (Shen and Johnson 2010).
In conclusion, K‐ATP channels (Kir6.2/SUR1) can modulate the electrical activity of SN DA neurons in both directions: in a negative feedback mechanism by membrane hyperpolarization, and in a positive feed‐forward mechanism, in functional interplay with NMDA glutamate receptors, by stimulation of burst activity (compare Fig. 1 and 2). Yet, a strong K‐ATP activation, for instance in response to PD‐triggers, will at a certain point no longer trigger burst activity of SN DA neurons, but in contrast reduce their activity, or even silence them, as illustrated in Fig. 2.
Bidirectional effects of acute and chronic K‐ATP channel activity for SN DA neuron survival
Considering the preferential degeneration of SN DA neurons in PD, acute and chronic K‐ATP channel activity has also bidirectional – protective but also detrimental – effects on SN DA survival. In line with a well‐described general neuroprotective effect of K‐ATP channel activity in metabolic stress situations, SN DA neurons of K‐ATP channel‐deficient mice [Kir6.2 KO (Minami et al. 2004)] display a significantly higher vulnerability in vivo in acute response to the PD‐toxin MPTP (Liss et al. 2005). However, chronic K‐ATP channel activity in SN DA neurons in vivo in response to sustained metabolic stress is not beneficial anymore, but in contrast seems to trigger degeneration as evident from K‐ATP KO: germline loss of Kir6.2 K‐ATP channels rescued SN DA neurons from selective degeneration in two different PD mouse models (Liss et al. 1999b, 2005), the chronic neurotoxic MPTP/probenecid PD‐model (Jackson‐Lewis and Przedborski 2007; Meredith and Rademacher 2011), as well as the homozygous weaver GIRK2 mutant mouse, a chronic genetic model of PD (Navarro et al. 1996; Oo et al. 1996; Slesinger et al. 1996). In agreement with a neurodegenerative effect of chronic K‐ATP channel activity, pharmacological K‐ATP channel block protects cultured rat SN DA neurons (Toulorge et al. 2010), and PC12 DA cells from degeneration (Nam et al. 2015).
Beyond that, a pathophysiological role of chronic K‐ATP channel activity in human PD is indicated, as remaining SN DA neurons from PD patients express about 2‐fold higher levels of SUR1, the subunit that is responsible for K‐ATP channel trafficking to the plasma membrane (Sharma et al. 1999; Tucker et al. 1997), and about 10‐fold higher levels of the NMDA‐R pore forming subunit NR1 (Schiemann et al. 2012; Dragicevic et al. 2015). Consistent with these human expression data and our functional in vivo mouse data, SN DA neurons of awake PD patients display high levels of burst activity (Zaghloul et al. 2009; Schiemann et al. 2012). Furthermore, there is retrospective epidemiological evidence of a reduced risk for PD in T2D patients that were treated with K‐ATP blockers (Powers et al. 2006; Scigliano et al. 2006; Schernhammer et al. 2011; Wahlqvist et al. 2012; Cereda et al. 2013; Lu et al. 2014; Brauer et al. 2015). Likewise, there is evidence for BBB penetrance of these K‐ATP channel blockers (Ashcroft 2010; Lahmann et al. 2015). This evidence, however, is ambiguous and must be interpreted with caution, as T2D per se seems to affect the risk for PD (Hu et al. 2007; Cereda et al. 2012; Lima et al. 2014; Lu et al. 2014; Zhang and Tian 2014), and both diseases seem to share common pathophysiological pathways (Santiago and Potashkin 2013, 2014), which could skew protective effects of K‐ATP blockers. Nevertheless, pharmacological K‐ATP blockade, selectively in SN DA neurons, could provide another ion channel targeting neuroprotective strategy as a novel PD therapy. However, to the best of our knowledge, currently no clinical studies are further evaluating this avenue.
In summary, mouse model and human data identified that K‐ATP channels in SN DA neurons act as bidirectional metabolic gatekeepers, adapting their activity and thus dopamine release to their metabolic state, based on feed‐forward as well as feedback mechanisms: K‐ATP channel‐mediated feed‐forward burst facilitation, e.g. in metabolic demand situations would enhance dopamine release and exploratory movement. However, it would also boost SN DA energy demand and metabolic stress, and consequently activate further K‐ATP channels. This will, particularly in the presence of additional PD‐stressors, in a feedback mechanism reduce activity and energy consumption of SN DA neurons. In vivo, K‐ATP channel activity in acute response to pathophysiological metabolic stressors is beneficial, but their chronic pathophysiological activity in response to PD‐triggers has opposite effects and triggers SN DA degeneration.
A flexible range of activity pattern and Ca2+ levels is vital for SN DA function and survival
Given the bidirectional functions of Cav1.3 LTCCs as well as of K‐ATP channels for SN DA neuron activity, it is not clear whether the supposed PD protective effect of pharmacological channel inhibition might be caused by a reduction or a stimulation of SN DA activity. Indeed, the relationship between ion channel activity, activity pattern, calcium homeostasis, physiological functions, and vulnerability of SN DA neurons in PD is not a simple one, but is rather complex and context dependent (Fig. 2).
A number of studies indicate that the maintained activity of SN DA neurons is not only crucial for dopamine release and fulfillment of their physiological functions but also for their maintenance and survival during development and adulthood (Michel et al. 2007, 2013; Toulorge et al. 2010, 2011; Dragicevic et al. 2015; Guerreiro et al. 2015; Wang et al. 2015). In accordance with the classical ‘use it or lose it’ principle of neuronal plasticity and neuronal loss (Swaab et al. 2002; Coyle 2003; Coulson et al. 2008; Valenzuela et al. 2012), reduced activity of SN DA neurons seems to facilitate their degeneration in vivo and in vitro (Salthun‐Lassalle et al. 2004; Liss et al. 2005; Janezic et al. 2013). On the other hand, reduced activity has also been shown to be beneficial for SN DA survival, particularly in acute pathophysiological demand situations (Liss et al. 2005; Yamada and Inagaki 2005; Aumann et al. 2008; Virgili et al. 2013). In accordance with classic excitotoxic cell death pathways, elevated activity clearly is detrimental for SN DA survival, as it triggers energy demand, metabolic stress, and pathophysiological Ca2+ overload, and its detrimental consequences (Blandini 2010; Surmeier et al. 2011; Ambrosi et al. 2014; Prentice et al. 2015; Van Laar et al. 2015).
However, Ca2+ apparently also has context‐dependent bidirectional effects on SN DA neuron physiology, viability, and vulnerability. As detailed above, Ca2+ can trigger excitotoxicity and other detrimental pathways, however, it is necessary not only for physiological SN DA functions but also for their survival (Michel et al. 2013). Findings from PINK1 (PARK6) and HtrA2/Omi (PARK13) PD‐gene mice support this view, as their SN DA pacemaker activity is less precise, and burst firing seems to be facilitated in consequence of reduced Ca2+ release from ER and mitochondria, and thus by reduced cytosolic Ca2+ (Bishop et al. 2010).
Given these bidirectional roles of SN DA neuron activity and related Ca2+ signaling, intrinsic or synaptic mechanisms that modulate their activity should have variable, context‐dependent effects on their viability and their vulnerability in PD (Fig. 2). On one hand, mechanisms that maintain or stimulate electrical activity, and thus dopamine release and the ability for voluntary movement – even or particularly under metabolic demand situations (e.g. food deprivation, or fight‐or‐flight situations) – could be beneficial or even life‐saving for the organism. However, as a drawback, ongoing stimulated activity of SN DA neurons and associated metabolic stress might render SN DA neurons more vulnerable to excitotoxicity and PD‐triggers. On the other hand, mechanisms that reduce SN DA activity and calcium‐mediated dopamine release, and thus impair voluntary movement, could be detrimental for the organism, particularly under situations were immediate and/or ongoing motion is required for survival. This would, however, protect SN DA neurons from excitotoxic events.
In conclusion, a context‐dependent, flexible bandwidth of activity patterns and associated Ca2+ levels is necessary to enable respective physiological functions of SN DA neurons. Consequently, as illustrated in Figs 1 and 2, SN DA neurons possess several intrinsic mechanisms to protect and to adapt their activity pattern as well as their calcium homeostasis in both directions within a physiological range.
The physiological bandwidth of activity patterns and Ca2+ levels in SN DA neurons is defined by ion channels, and gets narrowed by PD‐triggers (?)
An intricate network of ion channels that can mediate bidirectional functions on different timelines, and that complement and compensate each other as detailed above, is crucial to enable this physiological flexibility of SN DA neurons. In this sense, in an immediate scenario, LTCC activity and intracellular Ca2+ stabilize and stimulate SN DA activity and their ATP production in a feed‐forward mechanism: the more active the neuron is, the more dopamine gets released, the more ATP is needed and would be produced because of activity‐related VGCC activity and Ca2+ stimulation of TCA, ETC and further enzymes. Furthermore, LTCC activity can homeostatically adapt SN DA function to physiological needs via alterations of Ca2+‐dependent gene expression as already discussed above (Pathak et al. 2015). These short‐ and long‐term physiological functions of VGCCs and Ca2+ in SN DA neurons would ensure their adaptive electrical activity, dopamine release, and context‐specific movement control. The burst‐triggering function of K‐ATP channel activation in SN DA neurons further contributes to this positive safety net of reinforcement mechanisms (Schiemann et al. 2012). However, as detailed above, VGCCs as well as K‐ATP channels not only facilitate and stabilize pacemaker activity, but they can also stimulate inhibitory responses that reduce SN DA activity and related Ca2+ levels in negative feedback loops, e.g. via NCS‐1/D2/GIRK2 or A‐type Kv4.3/KChip3 channel sensitization. Moreover, A‐type channel function in SN DA neurons is also complex and context dependent (Hahn et al. 2003). On one hand, the glia cell line‐derived neurotrophic factor GDNF that promotes maintenance and survival of SN DA neurons (de d'Anglemont Tassigny et al. 2015; Kramer and Liss 2015), can stimulate pacemaker frequency of SN DA neurons by A‐type current inhibition (Yang et al. 2001), and Kv4‐blockers infused in vivo into the striatum reduce PD‐symptoms in mice (Aidi‐Knani et al. 2015). However, on the other hand, over‐expression of the mutated PARK1‐gene product A53T α‐synuclein, which triggers SN DA degeneration, has GDNF‐like effects on A‐type currents in vivo in mouse SN DA neurons (Subramaniam et al. 2014). Moreover, remaining human SN DA neurons from PD patients display not only elevated burst activity, but also massively elevated levels of Kv4.3 mRNA – possibly as a compensatory protective feedback mechanism (Fig. 2) (Schiemann et al. 2012; Schlaudraff et al. 2014; Dragicevic et al. 2015). Beyond this already complex scenario, VGCCs, A‐type Kv4.3 channels, GIRK2, and K‐ATP channels modulate each other's activity: elevated Ca2+ levels and associated metabolic stress because of VGCC activation (in particular Cav1.3) can sensitize D2/GIRK2 responses, and increase the open probability of Ca2+‐sensitive A‐type Kv4.3 channels as well as of metabolically sensitive K‐ATP channels. Membrane hyperpolarization and reduced SN DA activity resulting from these K+ channel activities in turn could increase VGCC currents, in particular of LVA Cav1.3 LTCCs and Cav3.1 TTCCs.
It should be emphasized again that less vulnerable VTA DA neurons, which are not essential for voluntary movement, do neither display VGCC‐mediated intracellular Ca2+ oscillations during pacemaking, nor A‐type K+ channel‐mediated frequency control, or K‐ATP channel‐mediated high metabolic sensitivity, as detailed above. However, other neurons that show high vulnerability to PD‐triggers, and that are important for fight‐or‐flight responses, like the noradrenergic LC neurons, possess ion channel activities similar to those of SN DA neurons (Koyama et al. 1999; Samuels and Szabadi 2008; de Oliveira et al. 2010; Tovar et al. 2013; Sanchez‐Padilla et al. 2014; Hopp et al. 2015; Matschke et al. 2015).
In conclusion, despite their complex homeostatic bidirectional regulatory mechanisms, the ‘high calcium, high activity, high metabolism’ phenotype of SN DA neurons means that they are energetically ‘living on the edge’. Their massive axonal fields and lower number of mitochondria might further contribute to their fragile homeostasis (Liang et al. 2007; Surmeier et al. 2010; Bolam and Pissadaki 2012; Ciron et al. 2015; Hunn et al. 2015). Hence, any factor that perturbs their delicate metabolic balance (e.g. PD‐triggers) might ‘tip them over the edge’. Meaning that their feedback and feed‐forward control mechanisms are no longer sufficient to keep SN DA activity and calcium homeostasis within a desired physiological range, and consequently detrimental pathways can trigger degeneration (Pissadaki and Bolam 2013; Surmeier and Schumacker 2013; Dragicevic et al. 2015). In other words, as illustrated in figure 2, PD‐trigger factors (e.g. environmental factors or PARK‐genes, impairing mitochondrial and/or lysosomal function) would narrow the physiological bandwidth of flexible SN DA activity and calcium signaling in both directions. Consequently, reduced as well as elevated activity‐ and calcium‐levels could tip SN DA neurons more easily ‘over their physiological edge’ (Pissadaki and Bolam 2013). In this scenario, the same SN DA activity or oscillatory calcium signal that enables their physiological function, could – in the presence of PD‐triggers – stimulate their degeneration, by e.g. inducing excitotoxicitiy or apoptosis as described above.
Summary
Taken together, the bidirectional effects of SN DA activity, calcium homeostasis and related ion channel activity on their viability and vulnerability illustrate the general dilemma of these neurons that seem to be committed to maintain their flexible physiological activity. On one hand, they need stimulatory intrinsic (and extrinsic) feed‐forward mechanisms to ensure their homeostatic activity, calcium signaling, and dopamine release within the basal ganglia network, even under metabolically demanding situations, as well as to prevent cell death in a ‘use it or lose it’ context. On the other hand, SN DA neurons need respective inhibitory feedback mechanisms to prevent overexcitability, Ca2+ overload, excitotoxicity, and its related cell death mechanisms (compare Fig. 1 and 2).
These context‐dependent stimulatory and inhibitory mechanisms are enabled by a network of ion channels (Fig. 1) that can mediate bidirectional functions, and enable a flexible bandwidth of SN DA activity patterns. PD‐trigger factors could narrow this physiological bandwidth at both ends, and thus facilitate pathophysiological and degenerative pathways (Fig. 2). To make things worse, once the intricate steady‐state of SN DA neurons gets out of balance, the different players that enable and maintain their physiological flexibility, could now – not least because of their complex interactions – augment detrimental pathophysiological changes of SN DA activity pattern and/or calcium load, leading to a vicious self‐energizing spiral that becomes independent from its initial source (e.g. PD‐triggers), and progressively fortify SN DA degeneration.
Voltage‐gated calcium channels (particularly the Cav1.3 L‐type and the Cav3.1 T‐type) as well as metabolically gated K‐ATP channels (particularly the Kir6.2/SUR1 type) seem to have central roles in this scenario: the activity of both channel types is crucial for the specific movement‐related physiological functions of SN DA neurons, and thus for the viability of the organism. However, their systemic block or general loss reduces the particularly high vulnerability of SN DA neurons to metabolic stress, degeneration and to PD. The functional expression of Cav1.3 LTCCs and of K‐ATP channels and related signaling networks in SN DA but not VTA DA neurons might thus reflect a kind of antagonistic pleiotropy: being beneficial and conferring a fitness advantage in youth and young adult life, but might have detrimental effects on SN DA survival later in life during aging, and in PD (Kirkwood and Austad 2000; Lambeth 2007; Parsons 2007; Surmeier et al. 2012).
Given the promising results from mouse models and epidemiological studies, pharmacological inhibition of these channels consequently offers novel therapeutic avenues for neuroprotective PD‐therapies – which need to start before PD‐motor symptoms manifest and most SN DA neurons are already lost. The use of well‐established LTCC blockers (phase III isradipine study in early PD, clinicaltrials.gov identifier: NCT02168842), and the development of more specific Cav1.3 LTCC blockers (Kang et al. 2012), are the most advanced strategies. Noteworthy in a therapeutic context is that LTCCs, as well as K‐ATP and other here discussed ion channels, are not only expressed in SN DA neurons, but in a variety of other neuronal and non‐neuronal cells. Given the emerging complex, interdependent, and bidirectional functions of these ion channels that are not yet fully understood, and their intricate signaling network in SN DA neurons (that might well be even much more complex than outlined here), the pharmacological modulation of just one parameter (i.e. block of one ion channel type) might also have a variety of – unpredictable – consequences on the viability and/or the physiological function of SN DA neurons, and thus unwanted side‐effects besides the desired neuroprotection. First safety studies indicate that this seems not to be the case for isradipine (Simuni et al. 2010). However, further research is needed to develop specific pharmacological ion channel blockers (or activators), and to further dissect their complex acute and chronic effects – on SN DA neurons as well as on the whole human body – in health and in PD.
Acknowledgments and conflict of interest disclosure
The authors declare no sources of conflicts of interest, and apologize to all colleagues whose work was not cited due to space limitations. BL is supported by grants from the DFG (LI1745/1), the FWF (SFB F4412), and the Alfried Krupp Foundation. We are particularly grateful to Jim Surmeier for critically reading the manuscript and for ongoing inspiring discussions. In the same view, we thank Paul Dietl, Edgar Kramer and Joerg Striessnig for discussions and for reading parts of the manuscript.
This article is part of a special issue on Parkinson disease .
References
- Adams P. J. and Snutch T. P. (2007) Calcium channelopathies: voltage‐gated calcium channels. Subcell. Biochem. 45, 215–251. [DOI] [PubMed] [Google Scholar]
- Agid Y., Ruberg M., Javoy‐Agid F. et al (1993) Are dopaminergic neurons selectively vulnerable to Parkinson's disease? Adv. Neurol. 60, 148–164. [PubMed] [Google Scholar]
- Aidi‐Knani S., Regaya I., Amalric M. and Mourre C. (2015) Kv4 channel blockade reduces motor and neuropsychiatric symptoms in rodent models of Parkinson's disease. Behav. Pharmacol. 26, 91–100. [DOI] [PubMed] [Google Scholar]
- Albin R. L. and Dauer W. T. (2014) Magic shotgun for Parkinson's disease? Brain 137, 1274–1275. [DOI] [PubMed] [Google Scholar]
- Alexeyev M., Shokolenko I., Wilson G. and LeDoux S. (2013) The maintenance of mitochondrial DNA integrity–critical analysis and update. Cold Spring Harb Perspect. Biol. 5, a012641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosi G., Cerri S. and Blandini F. (2014) A further update on the role of excitotoxicity in the pathogenesis of Parkinson's disease. J. Neural. Transm. (Vienna) 121, 849–859. [DOI] [PubMed] [Google Scholar]
- Ambrosi G., Ghezzi C., Zangaglia R., Levandis G., Pacchetti C. and Blandini F. (2015) Ambroxol‐induced rescue of defective glucocerebrosidase is associated with increased LIMP‐2 and saposin C levels in GBA1 mutant Parkinson's disease cells. Neurobiol. Dis. 82, 235–242. [DOI] [PubMed] [Google Scholar]
- An W. F., Bowlby M. R., Betty M. et al (2000) Modulation of A‐type potassium channels by a family of calcium sensors. Nature 403, 553–556. [DOI] [PubMed] [Google Scholar]
- Anderson D., Mehaffey W. H., Iftinca M., Rehak R., Engbers J. D., Hameed S., Zamponi G. W. and Turner R. W. (2010) Regulation of neuronal activity by Cav3‐Kv4 channel signaling complexes. Nat. Neurosci. 13, 333–337. [DOI] [PubMed] [Google Scholar]
- de d'Anglemont Tassigny X., Pascual A. and Lopez‐Barneo J. (2015) GDNF‐based therapies, GDNF‐producing interneurons, and trophic support of the dopaminergic nigrostriatal pathway. Implications for Parkinson's disease. Front. Neuroanat., 9, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong C. M. and Matteson D. R. (1985) Two distinct populations of calcium channels in a clonal line of pituitary cells. Science 227, 65–67. [DOI] [PubMed] [Google Scholar]
- Ashcroft F. M. (2007) The Walter B. Cannon Physiology in Perspective Lecture, 2007. ATP‐sensitive K+ channels and disease: from molecule to malady. Am. J. Physiol. Endocrinol. Metab. 293, E880–E889. [DOI] [PubMed] [Google Scholar]
- Ashcroft F. M. (2010) New uses for old drugs: neonatal diabetes and sulphonylureas. Cell Metab. 11, 179–181. [DOI] [PubMed] [Google Scholar]
- Ashcroft F. M. and Gribble F. M. (1998) Correlating structure and function in ATP‐sensitive K+ channels. Trends Neurosci. 21, 288–294. [DOI] [PubMed] [Google Scholar]
- Ashcroft F. M. and Rorsman P. (2013) K(ATP) channels and islet hormone secretion: new insights and controversies. Nat. Rev. Endocrinol. 9, 660–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft F. M., Harrison D. E. and Ashcroft S. J. (1984) Glucose induces closure of single potassium channels in isolated rat pancreatic beta‐cells. Nature 312, 446–448. [DOI] [PubMed] [Google Scholar]
- Asselta R., Rimoldi V., Siri C. et al (2014) Glucocerebrosidase mutations in primary parkinsonism. Parkinsonism Relat. Disord. 20, 1215–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aumann T. D., Gantois I., Egan K., Vais A., Tomas D., Drago J. and Horne M. K. (2008) SK channel function regulates the dopamine phenotype of neurons in the Substantia nigra pars compacta. Exp. Neurol. 213, 419–430. [DOI] [PubMed] [Google Scholar]
- Bakowski D., Nelson C. and Parekh A. B. (2012) Endoplasmic reticulum‐mitochondria coupling: local Ca(2)(+) signalling with functional consequences. Pflugers Arch. 464, 27–32. [DOI] [PubMed] [Google Scholar]
- Baughman J. M., Perocchi F., Girgis H. S. et al (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean B. P. (2007a) The action potential in mammalian central neurons. Nat. Rev. Neurosci. 8, 451–465. [DOI] [PubMed] [Google Scholar]
- Bean B. P. (2007b) Neurophysiology: stressful pacemaking. Nature 447, 1059–1060. [DOI] [PubMed] [Google Scholar]
- Beaulieu J. M., Espinoza S. and Gainetdinov R. R. (2015) Dopamine receptors ‐ IUPHAR Review 13. Br. J. Pharmacol. 172, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker C., Jick S. S. and Meier C. R. (2008) Use of antihypertensives and the risk of Parkinson disease. Neurology 70, 1438–1444. [DOI] [PubMed] [Google Scholar]
- Beier K. T., Steinberg E. E., DeLoach K. E. et al (2015) Circuit architecture of VTA dopamine neurons revealed by systematic Input‐output mapping. Cell 162, 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilina A. and Cookson M. R. (2015) Genes associated with Parkinson's disease: regulation of autophagy and beyond. J. Neurochem. 139 (Suppl. 1), 91–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A., Krishnan K. J., Morris C. M. et al (2006) High levels of mitochondrial DNA deletions in Substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 38, 515–517. [DOI] [PubMed] [Google Scholar]
- Bendikov‐Bar I., Maor G., Filocamo M. and Horowitz M. (2013) Ambroxol as a pharmacological chaperone for mutant glucocerebrosidase. Blood Cells Mol. Dis. 50, 141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman S. B. and Hastings T. G. (1999) Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: implications for Parkinson's disease. J. Neurochem. 73, 1127–1137. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I. and Mattson M. P. (2008) Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 31, 454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop M. W., Chakraborty S., Matthews G. A., Dougalis A., Wood N. W., Festenstein R. and Ungless M. A. (2010) Hyperexcitable Substantia nigra dopamine neurons in PINK1‐ and HtrA2/Omi‐deficient mice. J. Neurophysiol. 104, 3009–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorklund A. and Dunnett S. B. (2007) Dopamine neuron systems in the brain: an update. Trends Neurosci. 30, 194–202. [DOI] [PubMed] [Google Scholar]
- Blandini F. (2010) An update on the potential role of excitotoxicity in the pathogenesis of Parkinson's disease. Funct. Neurol. 25, 65–71. [PubMed] [Google Scholar]
- Blesa J. and Przedborski S. (2014) Parkinson's disease: animal models and dopaminergic cell vulnerability. Front. Neuroanat. 8, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolam J. P. and Pissadaki E. K. (2012) Living on the edge with too many mouths to feed: why dopamine neurons die. Mov. Disord. 27, 1478–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond C. T., Maylie J. and Adelman J. P. (2005) SK channels in excitability, pacemaking and synaptic integration. Curr. Opin. Neurobiol. 15, 305–311. [DOI] [PubMed] [Google Scholar]
- Bonifati V., Rizzu P., van Baren M. J. et al (2003) Mutations in the DJ‐1 gene associated with autosomal recessive early‐onset parkinsonism. Science 299, 256–259. [DOI] [PubMed] [Google Scholar]
- Borgkvist A., Mosharov E. V. and Sulzer D. (2014) Calcium currents regulate dopamine autoreceptors. Brain 137, 2113–2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdenx M., Bezard E. and Dehay B. (2014) Lysosomes and alpha‐synuclein form a dangerous duet leading to neuronal cell death. Front. Neuroanat. 8, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdy R. and Barrot M. (2012) A new control center for dopaminergic systems: pulling the VTA by the tail. Trends Neurosci. 35, 681–690. [DOI] [PubMed] [Google Scholar]
- Bozzi Y. and Borrelli E. (2006) Dopamine in neurotoxicity and neuroprotection: what do D2 receptors have to do with it? Trends Neurosci. 29, 167–174. [DOI] [PubMed] [Google Scholar]
- Braak H., Ghebremedhin E., Rub U., Bratzke H. and Del Tredici K. (2004) Stages in the development of Parkinson's disease‐related pathology. Cell Tissue Res. 318, 121–134. [DOI] [PubMed] [Google Scholar]
- Branch S. Y., Sharma R. and Beckstead M. J. (2014) Aging decreases L‐type calcium channel currents and pacemaker firing fidelity in Substantia nigra dopamine neurons. J. Neurosci. 34, 9310–9318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauer R., Bhaskaran K., Chaturvedi N., Dexter D. T., Smeeth L. and Douglas I. (2015) Glitazone Treatment and Incidence of Parkinson's Disease among People with Diabetes: A Retrospective Cohort Study. PLoS Med. 12(7), e1001854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brichta L. and Greengard P. (2014) Molecular determinants of selective dopaminergic vulnerability in Parkinson's disease: an update. Front. Neuroanat. 8, 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brimblecombe K. R., Gracie C. J., Platt N. J. and Cragg S. J. (2015) Gating of dopamine transmission by calcium and axonal N‐, Q‐, T‐ and L‐type voltage‐gated calcium channels differs between striatal domains. J. Physiol. 593, 929–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brini M. (2003) Ca(2 + ) signalling in mitochondria: mechanism and role in physiology and pathology. Cell Calcium 34, 399–405. [DOI] [PubMed] [Google Scholar]
- Brini M., Cali T., Ottolini D. and Carafoli E. (2014) Neuronal calcium signaling: function and dysfunction. Cell. Mol. Life Sci. 71, 2787–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brisch R., Saniotis A., Wolf R. et al (2014) The role of dopamine in schizophrenia from a neurobiological and evolutionary perspective: old fashioned, but still in vogue. Front. Psychiatry. 5, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg‐Martin E. S., Matsumoto M. and Hikosaka O. (2010) Dopamine in motivational control: rewarding, aversive, and alerting. Neuron 68, 815–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- der van Brug M. P., Singleton A., Gasser T. and Lewis P. A. (2015) Parkinson's disease: from human genetics to clinical trials. Sci. Transl. Med. 7, 205, ps220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne R. D. and Haynes L. P. (2014) Sense and specificity in neuronal calcium signalling. Biochim. Biophys. Acta 1853, 1921–1932. doi:10.1016/j.bbamcr.2014.1010.1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum J. D. (2004) A role for calsenilin and related proteins in multiple aspects of neuronal function. Biochem. Biophys. Res. Commun. 322, 1140–1144. [DOI] [PubMed] [Google Scholar]
- Cali T., Ottolini D., Negro A. and Brini M. (2012) alpha‐Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum‐mitochondria interactions. J. Biol. Chem. 287, 17914–17929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cali T., Ottolini D. and Brini M. (2014) Calcium signaling in Parkinson's disease. Cell Tissue Res. 357, 439–454. [DOI] [PubMed] [Google Scholar]
- Catterall W. A. and Zheng N. (2015) Deciphering voltage‐gated Na(+) and Ca(2 + ) channels by studying prokaryotic ancestors. Trends Biochem. Sci. 40, 526–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall W. A., Perez‐Reyes E., Snutch T. P. and Striessnig J. (2005) International Union of Pharmacology. XLVIII. Nomenclature and structure‐function relationships of voltage‐gated calcium channels. Pharmacol. Rev. 57, 411–425. [DOI] [PubMed] [Google Scholar]
- Catterall W. A., Leal K. and Nanou E. (2013) Calcium channels and short‐term synaptic plasticity. J. Biol. Chem. 288, 10742–10749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cens T., Rousset M., Leyris J. P., Fesquet P. and Charnet P. (2006) Voltage‐ and calcium‐dependent inactivation in high voltage‐gated Ca(2 + ) channels. Prog. Biophys. Mol. Biol. 90, 104–117. [DOI] [PubMed] [Google Scholar]
- Cereda E., Barichella M., Cassani E., Caccialanza R. and Pezzoli G. (2012) Clinical features of Parkinson disease when onset of diabetes came first: A case‐control study. Neurology. 78(19), 1507–1511. [DOI] [PubMed] [Google Scholar]
- Cereda E., Barichella M., Pedrolli C., Klersy C., Cassani E., Caccialanza R. and Pezzoli G. (2013) Diabetes and risk of Parkinson's disease. Mov. Disord. 28, 257. [DOI] [PubMed] [Google Scholar]
- Chan C. S., Guzman J. N., Ilijic E., Mercer J. N., Rick C., Tkatch T., Meredith G. E. and Surmeier D. J. (2007) ‘Rejuvenation’ protects neurons in mouse models of Parkinson's disease. Nature 447, 1081–1086. [DOI] [PubMed] [Google Scholar]
- Christel C. and Lee A. (2012) Ca2 + ‐dependent modulation of voltage‐gated Ca2 + channels. Biochim. Biophys. Acta 1820, 1243–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen K. A., Myers J. T. and Swanson J. A. (2002) pH‐dependent regulation of lysosomal calcium in macrophages. J. Cell Sci. 115, 599–607. [DOI] [PubMed] [Google Scholar]
- Ciron C., Zheng L., Bobela W., Knott G. W., Leone T. C., Kelly D. P. and Schneider B. L. (2015) PGC‐1alpha activity in nigral dopamine neurons determines vulnerability to alpha‐synuclein. Acta Neuropathol. Commun. 3, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark R. and Proks P. (2010) ATP‐sensitive potassium channels in health and disease. Adv. Exp. Med. Biol. 654, 165–192. [DOI] [PubMed] [Google Scholar]
- Coca A., Mazon P., Aranda P. et al (2013) Role of dihydropyridinic calcium channel blockers in the management of hypertension. Expert Rev. Cardiovasc. Ther. 11, 91–105. [DOI] [PubMed] [Google Scholar]
- Collier T. J., Kanaan N. M. and Kordower J. H. (2011) Ageing as a primary risk factor for Parkinson's disease: evidence from studies of non‐human primates. Nat. Rev. Neurosci. 12, 359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun P., Wyrembak J., Schriner S. E., Chen H. W., Marciniack C., Laferla F. and Wallace D. C. (2012) A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim. Biophys. Acta 1820, 553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa R. M. (2011) A selectionist account of de novo action learning. Curr. Opin. Neurobiol. 21, 579–586. [DOI] [PubMed] [Google Scholar]
- Coulson E. J., May L. M., Osborne S. L., Reid K., Underwood C. K., Meunier F. A., Bartlett P. F. and Sah P. (2008) p75 neurotrophin receptor mediates neuronal cell death by activating GIRK channels through phosphatidylinositol 4,5‐bisphosphate. J. Neurosci. 28, 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle J. T. (2003) Use it or lose it–do effortful mental activities protect against dementia? N. Engl. J. Med. 348, 2489–2490. [DOI] [PubMed] [Google Scholar]
- Crocker S. J., Smith P. D., Jackson‐Lewis V. et al (2003) Inhibition of calpains prevents neuronal and behavioral deficits in an MPTP mouse model of Parkinson's disease. J. Neurosci. 23, 4081–4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlstrom A. and Fuxe K. (1964) Localization of monoamines in the lower brain stem. Experientia 20, 398–399. [DOI] [PubMed] [Google Scholar]
- Damier P., Hirsch E. C., Agid Y. and Graybiel A. M. (1999a) The Substantia nigra of the human brain. I. Nigrosomes and the nigral matrix, a compartmental organization based on calbindin D(28K) immunohistochemistry. Brain, 122 (Pt 8), 1421–1436. [DOI] [PubMed] [Google Scholar]
- Damier P., Hirsch E. C., Agid Y. and Graybiel A. M. (1999b) The Substantia nigra of the human brain. II. Patterns of loss of dopamine‐containing neurons in Parkinson's disease. Brain, 122 (Pt 8), 1437–1448. [DOI] [PubMed] [Google Scholar]
- D'Ardenne K., Eshel N., Luka J., Lenartowicz A., Nystrom L. E. and Cohen J. D. (2012) Role of prefrontal cortex and the midbrain dopamine system in working memory updating. Proc. Natl Acad. Sci. USA 109, 19900–19909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Mei C., Ramos M., Iitaka C. and Borrelli E. (2009) Getting specialized: presynaptic and postsynaptic dopamine D2 receptors. Curr. Opin. Pharmacol. 9, 53–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D., Raffaello A., Teardo E., Szabo I. and Rizzuto R. (2011) A forty‐kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon R. M., Brook R. C., Meyer D., Haeckel O., Ashcroft F. M., Miki T., Seino S. and Liss B. (2006) Behavioral phenotyping of mice lacking the K ATP channel subunit Kir6.2. Physiol. Behav. 87, 723–733. [DOI] [PubMed] [Google Scholar]
- Dehay B., Martinez‐Vicente M., Caldwell G. A., Caldwell K. A., Yue Z., Cookson M. R., Klein C., Vila M. and Bezard E. (2013) Lysosomal impairment in Parkinson's disease. Mov. Disord. 28, 725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deister C. A., Teagarden M. A., Wilson C. J. and Paladini C. A. (2009) An intrinsic neuronal oscillator underlies dopaminergic neuron bursting. J. Neurosci. 29, 15888–15897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Tredici K. and Braak H. (2013) Dysfunction of the locus coeruleus‐norepinephrine system and related circuitry in Parkinson's disease‐related dementia. J. Neurol. Neurosurg. Psychiatry 84, 774–783. [DOI] [PubMed] [Google Scholar]
- Deleidi M. and Gasser T. (2013) The role of inflammation in sporadic and familial Parkinson's disease. Cell. Mol. Life Sci. 70, 4259–4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De‐Miguel F. F. and Nicholls J. G. (2015) Release of chemical transmitters from cell bodies and dendrites of nerve cells. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370(1672), pii: 20140181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fonzo A., Dekker M. C., Montagna P. et al (2009) FBXO7 mutations cause autosomal recessive, early‐onset parkinsonian‐pyramidal syndrome. Neurology 72, 240–245. [DOI] [PubMed] [Google Scholar]
- Diepenbroek M., Casadei N., Esmer H. et al (2014) Overexpression of the calpain‐specific inhibitor calpastatin reduces human alpha‐Synuclein processing, aggregation and synaptic impairment in [A30P]alphaSyn transgenic mice. Hum. Mol. Genet. 23, 3975–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich A., Fahlbusch M. and Gudermann T. (2014) Classical transient receptor potential 1 (TRPC1): channel or channel regulator? Cells 3, 939–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch R. (2003) Excitation‐transcription coupling: signaling by ion channels to the nucleus. Sci STKE, 2003, 166, PE4. [DOI] [PubMed] [Google Scholar]
- Dolmetsch R. E., Pajvani U., Fife K., Spotts J. M. and Greenberg M. E. (2001) Signaling to the nucleus by an L‐type calcium channel ‐ Calmodulin complex through the MAP kinase pathway. Science 294, 333–339. [DOI] [PubMed] [Google Scholar]
- Dopeso‐Reyes I. G., Rico A. J., Roda E., Sierra S., Pignataro D., Lanz M., Sucunza D., Chang‐Azancot L. and Lanciego J. L. (2014) Calbindin content and differential vulnerability of midbrain efferent dopaminergic neurons in macaques. Front. Neuroanat. 8, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragicevic E., Poetschke C., Duda J. et al (2014) Cav1.3 channels control D2‐autoreceptor responses via NCS‐1 in Substantia nigra dopamine neurons. Brain 137(8), 2287–2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragicevic E., Schiemann J. and Liss B. (2015) Dopamine midbrain neurons in health and Parkinson's disease: emerging roles of voltage‐gated calcium channels and ATP‐sensitive potassium channels. Neuroscience 284C, 798–814. [DOI] [PubMed] [Google Scholar]
- Drago I., Pizzo P. and Pozzan T. (2011) After half a century mitochondrial calcium in‐ and efflux machineries reveal themselves. EMBO J. 30, 4119–4125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drion G., Massotte L., Sepulchre R. and Seutin V. (2011) How modeling can reconcile apparently discrepant experimental results: the case of pacemaking in dopaminergic neurons. PLoS Comput. Biol. 7, e1002050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryanovski D. I., Guzman J. N., Xie Z., Galteri D. J., Volpicelli‐Daley L. A., Lee V. M., Miller R. J., Schumacker P. T. and Surmeier D. J. (2013) Calcium entry and alpha‐synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J. Neurosci. 33, 10154–10164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufty B. M., Warner L. R., Hou S. T. et al (2007) Calpain‐cleavage of alpha‐synuclein: connecting proteolytic processing to disease‐linked aggregation. Am. J. Pathol. 170, 1725–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durante P., Cardenas C. G., Whittaker J. A., Kitai S. T. and Scroggs R. S. (2004) Low‐threshold L‐type calcium channels in rat dopamine neurons. J Neurophysiol. 91, 1450–1454. [DOI] [PubMed] [Google Scholar]
- Epstein B. J., Vogel K. and Palmer B. F. (2007) Dihydropyridine calcium channel antagonists in the management of hypertension. Drugs 67, 1309–1327. [DOI] [PubMed] [Google Scholar]
- Fearnley J. M. and Lees A. J. (1991) Ageing and Parkinson's disease: Substantia nigra regional selectivity. Brain 114(Pt 5), 2283–2301. [DOI] [PubMed] [Google Scholar]
- Flagg T. P., Enkvetchakul D., Koster J. C. and Nichols C. G. (2010) Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiol. Rev. 90, 799–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flockerzi V., Bosse E., Biel M., Hullin R. and Hofmann F. (1991) High voltage activated calcium channels: molecular composition and function. Eur. Heart J. 12, 95–98. [DOI] [PubMed] [Google Scholar]
- Fois G., Hobi N., Felder E., Ziegler A., Miklavc P., Walther P., Radermacher P., Haller T. and Dietl P. (2015) A new role for an old drug: ambroxol triggers lysosomal exocytosis via pH‐dependent Ca(2 + ) release from acidic Ca(2 + ) stores. Cell Calcium 58, 628–637. [DOI] [PubMed] [Google Scholar]
- Ford C. P. (2014) The role of D2‐autoreceptors in regulating dopamine neuron activity and transmission. Neuroscience 282, 13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford C. P., Gantz S. C., Phillips P. E. and Williams J. T. (2010) Control of extracellular dopamine at dendrite and axon terminals. J. Neurosci. 30, 6975–6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz O., Liss B., Neu A. and Roeper J. (2000) Single‐cell mRNA expression of HCN1 correlates with a fast gating phenotype of hyperpolarization‐activated cyclic nucleotide‐gated ion channels (Ih) in central neurons. Eur. J. Neurosci. 12, 2685–2693. [DOI] [PubMed] [Google Scholar]
- Funayama M., Hasegawa K., Kowa H., Saito M., Tsuji S. and Obata F. (2002) A new locus for Parkinson's disease (PARK8) maps to chromosome 12p11.2‐q13.1. Ann. Neurol. 51, 296–301. [DOI] [PubMed] [Google Scholar]
- Games D., Valera E., Spencer B. et al (2014) Reducing C‐terminal‐truncated alpha‐synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson's disease‐like models. J. Neurosci. 34, 9441–9454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan‐Or Z., Dion P. A. and Rouleau G. A. (2015) Genetic perspective on the role of the autophagy‐lysosome pathway in Parkinson disease. Autophagy 11, 1443–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasser T., Hardy J. and Mizuno Y. (2011) Milestones in PD genetics. Mov. Disord. 26, 1042–1048. [DOI] [PubMed] [Google Scholar]
- Gerfen C. R. and Surmeier D. J. (2011) Modulation of striatal projection systems by dopamine. Annu. Rev. Neurosci. 34, 441–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleichmann M. and Mattson M. P. (2011) Neuronal calcium homeostasis and dysregulation. Antioxid. Redox Signal. 14, 1261–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M., Spillantini M. G., Del Tredici K. and Braak H. (2013) 100 years of Lewy pathology. Nat. Rev. Neurol. 9, 13–24. [DOI] [PubMed] [Google Scholar]
- Goldberg M. S., Pisani A., Haburcak M. et al (2005) Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism‐linked gene DJ‐1. Neuron 45, 489–496. [DOI] [PubMed] [Google Scholar]
- Gomez‐Ospina N., Tsuruta F., Barreto‐Chang O., Hu L. and Dolmetsch R. (2006) The C terminus of the L‐type voltage‐gated calcium channel Ca(V)1.2 encodes a transcription factor. Cell 127, 591–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Suaga P., Churchill G. C., Patel S. and Hilfiker S. (2012) A link between LRRK2, autophagy and NAADP‐mediated endolysosomal calcium signalling. Biochem. Soc. Trans. 40, 1140–1146. [DOI] [PubMed] [Google Scholar]
- Gonzalez‐Hernandez T., Cruz‐Muros I., Afonso‐Oramas D., Salas‐Hernandez J. and Castro‐Hernandez J. (2010) Vulnerability of mesostriatal dopaminergic neurons in Parkinson's disease. Front. Neuroanat. 4, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace A. A. and Bunney B. S. (1984a) The control of firing pattern in nigral dopamine neurons: burst firing. J. Neurosci. 4, 2877–2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace A. A. and Bunney B. S. (1984b) The control of firing pattern in nigral dopamine neurons: single spike firing. J. Neurosci. 4, 2866–2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace A. A. and Onn S. P. (1989) Morphology and electrophysiological properties of immunocytochemically identified rat dopamine neurons recorded in vitro. J. Neurosci. 9, 3463–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graef I. A., Mermelstein P. G., Stankunas K., Neilson J. R., Deisseroth K., Tsien R. W. and Crabtree G. R. (1999) L‐type calcium channels and GSK‐3 regulate the activity of NF‐ATc4 in hippocampal neurons. Nature 401, 703–708. [DOI] [PubMed] [Google Scholar]
- Greene J. G. (2014) Causes and consequences of degeneration of the dorsal motor nucleus of the vagus nerve in Parkinson's disease. Antioxid. Redox Signal. 21, 649–667. [DOI] [PubMed] [Google Scholar]
- Grillner P. and Mercuri N. B. (2002) Intrinsic membrane properties and synaptic inputs regulating the firing activity of the dopamine neurons. Behav. Brain Res. 130, 149–169. [DOI] [PubMed] [Google Scholar]
- Guardia‐Laguarta C., Area‐Gomez E., Schon E. A. and Przedborski S. (2015) A new role for alpha‐synuclein in Parkinson's disease: alteration of ER‐mitochondrial communication. Mov. Disord. 30, 1026–1033. [DOI] [PubMed] [Google Scholar]
- Gudala K., Kanukula R. and Bansal D. (2015) Reduced Risk of Parkinson's Disease in Users of Calcium Channel Blockers: A Meta‐Analysis. Int J Chronic Dis 2015, 697404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueguinou M., Chantome A., Fromont G., Bougnoux P., Vandier C. and Potier‐Cartereau M. (2014) KCa and Ca(2 + ) channels: the complex thought. Biochim. Biophys. Acta 1843, 2322–2333. [DOI] [PubMed] [Google Scholar]
- Guerreiro S., Florence C., Rousseau E., Hamadat S., Hirsch E. C. and Michel P. P. (2015) The sleep‐modulating peptide orexin‐B protects midbrain dopamine neurons from degeneration, alone or in cooperation with nicotine. Mol. Pharmacol. 87, 525–532. [DOI] [PubMed] [Google Scholar]
- Guzman J. N., Sanchez‐Padilla J., Chan C. S. and Surmeier D. J. (2009) Robust pacemaking in Substantia nigra dopaminergic neurons. J. Neurosci. 29, 11011–11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman J. N., Sanchez‐Padilla J., Wokosin D., Kondapalli J., Ilijic E., Schumacker P. T. and Surmeier D. J. (2010) Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ‐1. Nature 468, 696–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hage T. A. and Khaliq Z. M. (2015) Tonic firing rate controls dendritic Ca2 + signaling and synaptic gain in Substantia nigra dopamine neurons. J. Neurosci. 35, 5823–5836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn J., Tse T. E. and Levitan E. S. (2003) Long‐term K+ channel‐mediated dampening of dopamine neuron excitability by the antipsychotic drug haloperidol. J Neurosci. 23, 10859–10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnoczky G., Booth D., Csordas G. et al (2014) Reliance of ER‐mitochondrial calcium signaling on mitochondrial EF‐hand Ca binding proteins: miros, MICUs, LETM1 and solute carriers. Curr. Opin. Cell Biol. 29C, 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao L. Y., Giasson B. I. and Bonini N. M. (2010) DJ‐1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc. Natl Acad. Sci. USA 107, 9747–9752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J. (2010) Genetic analysis of pathways to Parkinson disease. Neuron 68, 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzen E. L., Arzimanoglou A., Brashear A. et al (2014) Distinct neurological disorders with ATP1A3 mutations. Lancet. Neurol. 13, 503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henchcliffe C. and Beal M. F. (2008) Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat. Clin. Pract. Neurol. 4, 600–609. [DOI] [PubMed] [Google Scholar]
- Henny P., Brown M. T., Northrop A., Faunes M., Ungless M. A., Magill P. J. and Bolam J. P. (2012) Structural correlates of heterogeneous in vivo activity of midbrain dopaminergic neurons. Nat. Neurosci. 15, 613–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindle J. V. (2010) Ageing, neurodegeneration and Parkinson's disease. Age Ageing 39, 156–161. [DOI] [PubMed] [Google Scholar]
- Hirsch E. C. and Hunot S. (2009) Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurol. 8, 382–397. [DOI] [PubMed] [Google Scholar]
- Hirsch E., Graybiel A. M. and Agid Y. A. (1988) Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson's disease. Nature 334, 345–348. [DOI] [PubMed] [Google Scholar]
- Hofmann F., Flockerzi V., Kahl S. and Wegener J. W. (2014) L‐type CaV1.2 calcium channels: from in vitro findings to in vivo function. Physiol. Rev. 94, 303–326. [DOI] [PubMed] [Google Scholar]
- Hoglinger G. U., Alvarez‐Fischer D., Arias‐Carrion O. et al (2015) A new dopaminergic nigro‐olfactory projection. Acta Neuropathol. 130, 333–348. [DOI] [PubMed] [Google Scholar]
- Hopp S. C., Royer S. E., D'Angelo H. M., Kaercher R. M., Fisher D. A. and Wenk G. L. (2015) Differential neuroprotective and anti‐inflammatory effects of L‐type voltage dependent calcium channel and ryanodine receptor antagonists in the Substantia nigra and locus coeruleus. J. Neuroimmune Pharmacol. 10, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppins S. and Nunnari J. (2012) Cell Biology. Mitochondrial dynamics and apoptosis–the ER connection. Science 337, 1052–1054. [DOI] [PubMed] [Google Scholar]
- Howarth C., Gleeson P. and Attwell D. (2012) Updated energy budgets for neural computation in the neocortex and cerebellum. J. Cereb. Blood Flow Metab. 32, 1222–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu G., Jousilahti P., Bidel S., Antikainen R. and Tuomilehto J. (2007) Type 2 diabetes and the risk of Parkinson's disease. Diabetes Care 30, 842–847. [DOI] [PubMed] [Google Scholar]
- Huang H., Ng C. Y., Yu D., Zhai J., Lam Y. and Soong T. W. (2014) Modest CaV1.342‐selective inhibition by compound 8 is beta‐subunit dependent. Nat. Commun., 5, 4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunn B. H., Cragg S. J., Bolam J. P., Spillantini M. G. and Wade‐Martins R. (2015) Impaired intracellular trafficking defines early Parkinson's disease. Trends Neurosci. 38, 178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson‐Lewis V. and Przedborski S. (2007) Protocol for the MPTP mouse model of Parkinson's disease. Nat. Protoc. 2, 141–151. [DOI] [PubMed] [Google Scholar]
- Janezic S., Threlfell S., Dodson P. D. et al (2013) Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc. Natl Acad. Sci. USA 110, E4016–E4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang M., Jang J. Y., Kim S. H., Uhm K. B., Kang Y. K., Kim H. J., Chung S. and Park M. K. (2011) Functional organization of dendritic Ca2 + signals in midbrain dopamine neurons. Cell Calcium 50, 370–380. [DOI] [PubMed] [Google Scholar]
- Jellinger K. A. (1991) Pathology of Parkinson's disease. Changes other than the nigrostriatal pathway. Mol. Chem. Neuropathol. 14, 153–197. [DOI] [PubMed] [Google Scholar]
- Jerng H. H. and Pfaffinger P. J. (2014) Modulatory mechanisms and multiple functions of somatodendritic A‐type K (+) channel auxiliary subunits. Front Cell Neurosci. 8, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhou T. C., Good C. H., Rowley C. S., Xu S. P., Wang H., Burnham N. W., Hoffman A. F., Lupica C. R. and Ikemoto S. (2013) Cocaine drives aversive conditioning via delayed activation of dopamine‐responsive habenular and midbrain pathways. J. Neurosci. 33, 7501–7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X. and Costa R. M. (2010) Start/stop signals emerge in nigrostriatal circuits during sequence learning. Nature 466, 457–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo D. G., Kim M. J., Choi Y. H., Kim I. K., Song Y. H., Woo H. N., Chung C. W. and Jung Y. K. (2001) Pro‐apoptotic function of calsenilin/DREAM/KChIP3. FASEB J. 15, 589–591. [DOI] [PubMed] [Google Scholar]
- Johar K., Priya A. and Wong‐Riley M. T. (2014) Regulation of Na(+)/K(+)‐ATPase by neuron‐specific transcription factor Sp4: implication in the tight coupling of energy production, neuronal activity and energy consumption in neurons. Eur. J. Neurosci. 39, 566–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S. W. and Wu Y. N. (2004) Multiple mechanisms underlie burst firing in rat midbrain dopamine neurons in vitro. Brain Res. 1019, 293–296. [DOI] [PubMed] [Google Scholar]
- Johnson S. W., Seutin V. and North R. A. (1992) Burst firing in dopamine neurons induced by N‐methyl‐D‐aspartate: role of electrogenic sodium pump. Science 258, 665–667. [DOI] [PubMed] [Google Scholar]
- Kabbani N., Woll M. P., Nordman J. C. and Levenson R. (2012) Dopamine receptor interacting proteins: targeting neuronal calcium sensor‐1/D2 dopamine receptor interaction for antipsychotic drug development. Curr. Drug Targets 13, 72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia L. V. and Lang A. E. (2015) Parkinson's disease. Lancet 386, 896–912. [DOI] [PubMed] [Google Scholar]
- Kang S., Cooper G., Dunne S. F., Dusel B., Luan C. H., Surmeier D. J. and Silverman R. B. (2012) CaV1.3‐selective L‐type calcium channel antagonists as potential new therapeutics for Parkinson's disease. Nat. Commun., 3, 1146. [DOI] [PubMed] [Google Scholar]
- Kang S., Cooper G., Dunne S. F., Luan C. H., Surmeier D. J. and Silverman R. B. (2013) Structure‐activity relationship of N, N’‐disubstituted pyrimidinetriones as Ca(V)1.3 calcium channel‐selective antagonists for Parkinson's disease. J. Med. Chem. 56, 4786–4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaliq Z. M. and Bean B. P. (2010) Pacemaking in dopaminergic ventral tegmental area neurons: depolarizing drive from background and voltage‐dependent sodium conductances. J. Neurosci. 30, 7401–7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan N. L., Jain S., Lynch J. M. et al (2005) Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson's disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain 128, 2786–2796. [DOI] [PubMed] [Google Scholar]
- Kieburtz K. and Wunderle K. B. (2013) Parkinson's disease: evidence for environmental risk factors. Mov. Disord. 28, 8–13. [DOI] [PubMed] [Google Scholar]
- Kilpatrick B. S., Magalhaes J., Beavan M. S. et al (2015) Endoplasmic reticulum and lysosomal Ca2 + stores are remodelled in GBA1–linked Parkinson disease patient fibroblasts. Cell Calcium 1, 12–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimm T. and Bean B. P. (2014) Inhibition of A‐type potassium current by the peptide toxin SNX‐482. J Neurosci. 34, 9182–9189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimm T., Khaliq Z. M. and Bean B. P. (2015) Differential regulation of action potential shape and burst‐frequency firing by BK and Kv2 channels in Substantia nigra dopaminergic neurons. J. Neurosci. 35, 16404–16417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood T. B. and Austad S. N. (2000) Why do we age? Nature 408, 233–238. [DOI] [PubMed] [Google Scholar]
- Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y. and Shimizu N. (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392, 605–608. [DOI] [PubMed] [Google Scholar]
- Kordower J. H., Olanow C. W., Dodiya H. B., Chu Y., Beach T. G., Adler C. H., Halliday G. M. and Bartus R. T. (2013) Disease duration and the integrity of the nigrostriatal system in Parkinson's disease. Brain 136, 2419–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschak A., Reimer D., Huber I., Grabner M., Glossmann H., Engel J. and Striessnig J. (2001) alpha 1D (CaV1.3) subunits can form l‐type Ca2 + channels activating at negative voltages. J. Biol. Chem. 276, 22100–22106. [DOI] [PubMed] [Google Scholar]
- Koyama S., Jin Y. H. and Akaike N. (1999) ATP‐sensitive and Ca2 + ‐activated K+ channel activities in the rat locus coeruleus neurons during metabolic inhibition. Brain Res. 828, 189–192. [DOI] [PubMed] [Google Scholar]
- Kramer E. R. and Liss B. (2015) Mechanisms of development and maintenance of dopaminergic neurons; in press. FEBS Lett. 589, 3760–3772. [DOI] [PubMed] [Google Scholar]
- Kraytsberg Y., Kudryavtseva E., McKee A. C., Geula C., Kowall N. W. and Khrapko K. (2006) Mitochondrial DNA deletions are abundant and cause functional impairment in aged human Substantia nigra neurons. Nat. Genet. 38, 518–520. [DOI] [PubMed] [Google Scholar]
- Kruger R., Sharma M., Riess O. et al (2011) A large‐scale genetic association study to evaluate the contribution of Omi/HtrA2 (PARK13) to Parkinson's disease. Neurobiol. Aging 32, 548, e549–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laguna A., Schintu N., Nobre A. et al (2015) Dopaminergic control of autophagic‐lysosomal function implicates Lmx1b in Parkinson's disease. Nat. Neurosci. 18, 826–835. [DOI] [PubMed] [Google Scholar]
- Lahmann C., Kramer H. B. and Ashcroft F. M. (2015) Systemic administration of glibenclamide fails to achieve therapeutic levels in the brain and cerebrospinal fluid of rodents. PLoS ONE 10, e0134476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambeth J. D. (2007) Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radic. Biol. Med. 43, 332–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammel S., Hetzel A., Hackel O., Jones I., Liss B. and Roeper J. (2008) Unique properties of mesoprefrontal neurons within a dual mesocorticolimbic dopamine system. Neuron 57, 760–773. [DOI] [PubMed] [Google Scholar]
- Lammel S., Lim B. K., Ran C., Huang K. W., Betley M. J., Tye K. M., Deisseroth K. and Malenka R. C. (2012) Input‐specific control of reward and aversion in the ventral tegmental area. Nature 491, 212–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang Y., Gong D., Fan Y. (2015) Pharmacoepidemiol Drug Saf. Calcium channel blocker use and risk of Parkinson's disease: a meta‐analysis. 4, 559–566. [DOI] [PubMed] [Google Scholar]
- Langston J. W. and Ballard P. A., Jr (1983) Parkinson's disease in a chemist working with 1‐methyl‐4‐phenyl‐1,2,5,6‐tetrahydropyridine. N. Engl. J. Med. 309, 310. [DOI] [PubMed] [Google Scholar]
- Laughlin S. B., van de Ruyter Steveninck R. R. and Anderson J. C. (1998) The metabolic cost of neural information. Nat. Neurosci. 1, 36–41. [DOI] [PubMed] [Google Scholar]
- Lee C. R. and Tepper J. M. (2009) Basal ganglia control of Substantia nigra dopaminergic neurons. J. Neural. Transm. Suppl. 73, 71–90. [DOI] [PubMed] [Google Scholar]
- Lefer D. J., Nichols C. G. and Coetzee W. A. (2009) Sulfonylurea receptor 1 subunits of ATP‐sensitive potassium channels and myocardial ischemia/reperfusion injury. Trends Cardiovasc. Med. 19, 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehericy S., Bardinet E., Poupon C., Vidailhet M. and Francois C. (2014) 7 Tesla magnetic resonance imaging: a closer look at Substantia nigra anatomy in Parkinson's disease. Mov. Disord. 29, 1574–1581. [DOI] [PubMed] [Google Scholar]
- Lerner T. N., Shilyansky C., Davidson T. J. et al (2015) Intact‐brain analyses reveal distinct information carried by SNc dopamine subcircuits. Cell 162, 635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M. and Adelman J. P. (2000) ChIPping away at potassium channel regulation. Nat. Neurosci. 3, 202–204. [DOI] [PubMed] [Google Scholar]
- Liang C. L., Wang T. T., Luby‐Phelps K. and German D. C. (2007) Mitochondria mass is low in mouse Substantia nigra dopamine neurons: implications for Parkinson's disease. Exp. Neurol. 203, 370–380. [DOI] [PubMed] [Google Scholar]
- Lieb A., Ortner N. and Striessnig J. (2014) C‐terminal modulatory domain controls coupling of voltage‐sensing to pore opening in Cav1.3 L‐type Ca(2 + ) channels. Biophys. J . 106, 1467–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima M. M., Targa A. D., Noseda A. C., Rodrigues L. S., Delattre A. M., dos Santos F. V., Fortes M. H., Maturana M. J. and Ferraz A. C. (2014) Does Parkinson's disease and type‐2 diabetes mellitus present common pathophysiological mechanisms and treatments? CNS Neurol. Disord. Drug Targets 13, 418–428. [DOI] [PubMed] [Google Scholar]
- Lipscombe D., Helton T. D. and Xu W. (2004) L‐type calcium channels: the low down. J. Neurophysiol. 92, 2633–2641. [DOI] [PubMed] [Google Scholar]
- Lipscombe D., Allen S. E. and Toro C. P. (2013) Control of neuronal voltage‐gated calcium ion channels from RNA to protein. Trends Neurosci. 36, 598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liss B. and Roeper J. (2001) Molecular physiology of neuronal K‐ATP channels (review). Mol. Membr. Biol. 18, 117–127. [PubMed] [Google Scholar]
- Liss B. and Roeper J. (2010) Ion channels and regulation of dopamine neuron activity in Dopamine handbook (Leslie S. D. I., Iversen L., Dunnett S. B. and Björklund A. eds), pp. 118–138. Oxford, University press. [Google Scholar]
- Liss B., Bruns R. and Roeper J. (1999a) Alternative sulfonylurea receptor expression defines metabolic sensitivity of K‐ATP channels in dopaminergic midbrain neurons. EMBO J. 18, 833–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liss B., Neu A. and Roeper J. (1999b) The weaver mouse gain‐of‐function phenotype of dopaminergic midbrain neurons is determined by coactivation of wvGirk2 and K‐ATP channels. J. Neurosci. 19, 8839–8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liss B., Franz O., Sewing S., Bruns R., Neuhoff H. and Roeper J. (2001) Tuning pacemaker frequency of individual dopaminergic neurons by Kv4.3L and KChip3.1 transcription. EMBO J. 20, 5715–5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liss B., An W. F. and Roeper J. (2002) Differential co‐expression of KChip subunits defines inactivation kinetics of A‐type potassium channels in dopaminergic VTA neurons. 32th annual SFN meeing, Abstr. 438.17.
- Liss B., Haeckel O., Wildmann J., Miki T., Seino S. and Roeper J. (2005) K‐ATP channels promote the differential degeneration of dopaminergic midbrain neurons. Nat. Neurosci. 8, 1742–1751. [DOI] [PubMed] [Google Scholar]
- Lu L., Fu D. L., Li H. Q., Liu A. J., Li J. H. and Zheng G. Q. (2014) Diabetes and risk of Parkinson's disease: an updated meta‐analysis of case‐control studies. PLoS ONE 9, e85781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L., Sirish P., Zhang Z. et al (2015) Regulation of gene transcription by voltage‐gated L‐type calcium channel, Cav1.3. J. Biol. Chem. 290, 4663–4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher C. and Slesinger P. A. (2010) Emerging roles for G protein‐gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat. Rev. Neurosci. 11, 301–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maegawa G. H., Tropak M. B., Buttner J. D. et al (2009) Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J. Biol. Chem. 284, 23502–23516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majewski L. and Kuznicki J. (2015) SOCE in neurons: Signaling or just refilling? Biochim. Biophys. Acta 1853, 1940–1952. [DOI] [PubMed] [Google Scholar]
- Malli R., Frieden M., Osibow K., Zoratti C., Mayer M., Demaurex N. and Graier W. F. (2003) Sustained Ca2 + transfer across mitochondria is Essential for mitochondrial Ca2 + buffering, sore‐operated Ca2 + entry, and Ca2 + store refilling. J. Biol. Chem. 278, 44769–44779. [DOI] [PubMed] [Google Scholar]
- Mandel G. and Goodman R. H. (1999) Cell signalling. DREAM on without calcium. Nature 398, 29–30. [DOI] [PubMed] [Google Scholar]
- Mao Z., Bonni A., Xia F., Nadal‐Vicens M. and Greenberg M. E. (1999) Neuronal activity‐dependent cell survival mediated by transcription factor MEF2. Science 286, 785–790. [DOI] [PubMed] [Google Scholar]
- Margolis E. B., Coker A. R., Driscoll J. R., Lemaitre A. I. and Fields H. L. (2010) Reliability in the identification of midbrain dopamine neurons. PLoS ONE 5, e15222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marras C., Gruneir A., Rochon P., Wang X., Anderson G., Brotchie J., Bell C. M., Fox S. and Austin P. C. (2012) Dihydropyridine calcium channel blockers and the progression of parkinsonism. Ann. Neurol. 71, 362–369. [DOI] [PubMed] [Google Scholar]
- Martin L. J., Semenkow S., Hanaford A. and Wong M. (2014) Mitochondrial permeability transition pore regulates Parkinson's disease development in mutant alpha‐synuclein transgenic mice. Neurobiol. Aging 35, 1132–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matschke L. A., Bertoune M., Roeper J., Snutch T. P., Oertel W. H., Rinne S. and Decher N. (2015) A concerted action of L‐ and T‐type Ca(2 + ) channels regulates locus coeruleus pacemaking. Mol. Cell Neurosci. 68, 293–302. [DOI] [PubMed] [Google Scholar]
- Mazzulli J. R., Xu Y. H., Sun Y., Knight A. L., McLean P. J., Caldwell G. A., Sidransky E., Grabowski G. A. and Krainc D. (2011) Gaucher disease glucocerebrosidase and alpha‐synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146, 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill A., Magalhaes J., Shen C. et al (2014) Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation‐linked Parkinson disease cells. Brain 137, 1481–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTaggart J. S., Clark R. H. and Ashcroft F. M. (2010) The role of the KATP channel in glucose homeostasis in health and disease: more than meets the islet. J. Physiol. 588, 3201–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellstrom B., Sahun I., Ruiz‐Nuno A. et al (2014) DREAM controls the on/off switch of specific activity‐dependent transcription pathways. Mol. Cell. Biol. 34, 877–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercuri N. B., Bonci A., Calabresi P., Stratta F., Stefani A. and Bernardi G. (1994) Effects of dihydropyridine calcium antagonists on rat midbrain dopaminergic neurones. Br. J. Pharmacol. 113, 831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith G. E. and Rademacher D. J. (2011) MPTP mouse models of Parkinson's disease: an update. J. Parkinsons Dis. 1, 19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel P. P., Alvarez‐Fischer D., Guerreiro S., Hild A., Hartmann A. and Hirsch E. C. (2007) Role of activity‐dependent mechanisms in the control of dopaminergic neuron survival. J. Neurochem. 101, 289–297. [DOI] [PubMed] [Google Scholar]
- Michel P. P., Toulorge D., Guerreiro S. and Hirsch E. C. (2013) Specific needs of dopamine neurons for stimulation in order to survive: implication for Parkinson disease. FASEB J. 27, 3414–3423. [DOI] [PubMed] [Google Scholar]
- Miki T. and Seino S. (2005) Roles of KATP channels as metabolic sensors in acute metabolic changes. J. Mol. Cell. Cardiol. 38, 917–925. [DOI] [PubMed] [Google Scholar]
- Miki T., Liss B., Minami K. et al (2001) ATP‐sensitive K+ channels in the hypothalamus are essential for the maintenance of glucose homeostasis. Nat. Neurosci. 4, 507–512. [DOI] [PubMed] [Google Scholar]
- Minami K., Miki T., Kadowaki T. and Seino S. (2004) Roles of ATP‐sensitive K+ channels as metabolic sensors: studies of Kir6.x null mice. Diabetes, 53 Suppl 3, S176–S180. [DOI] [PubMed] [Google Scholar]
- Miura T. and Miki T. (2003) ATP‐sensitive K+ channel openers: old drugs with new clinical benefits for the heart. Curr. Vasc. Pharmacol. 1, 251–258. [DOI] [PubMed] [Google Scholar]
- Murphy M. P. (2009) How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam Y. J., da Lee H., Lee M. S. and Lee C. S. (2015) K(ATP) channel block prevents proteasome inhibitor‐induced apoptosis in differentiated PC12 cells. Eur. J. Pharmacol. 764, 582–591. [DOI] [PubMed] [Google Scholar]
- Navarro B., Kennedy M. E., Velimirovic B., Bhat D., Peterson A. S. and Clapham D. E. (1996) Nonselective and G betagamma‐insensitive weaver K+ channels. Science 272, 1950–1953. [DOI] [PubMed] [Google Scholar]
- Nedergaard S., Flatman J. A. and Engberg I. (1993) Nifedipine‐ and omega‐conotoxin‐sensitive Ca2 + conductances in guinea‐pig Substantia nigra pars compacta neurones. J. Physiol. 466, 727–747. [PMC free article] [PubMed] [Google Scholar]
- Neuhoff H., Neu A., Liss B. and Roeper J. (2002) I(h) channels contribute to the different functional properties of identified dopaminergic subpopulations in the midbrain. J. Neurosci. 22, 1290–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng E., Varaschin R. K., Su P., Browne C. J., Hermainski J., Le Foll B., Pongs O., Liu F., Trudeau L. E., Roder J.C.. and Wong A. H. (2016) Neuronal calcium sensor‐1 deletion in the mouse decreases motivation and dopamine release in the nucleus accumbens. Behav Brain Res. 301, 213–225. [DOI] [PubMed] [Google Scholar]
- Nichols C. G. (2006) KATP channels as molecular sensors of cellular metabolism. Nature 440, 470–476. [DOI] [PubMed] [Google Scholar]
- Obeso J. A. and Lanciego J. L. (2011) Past, present, and future of the pathophysiological model of the Basal Ganglia. Front. Neuroanat. 5, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira R. B., Howlett M. C., Gravina F. S., Imtiaz M. S., Callister R. J., Brichta A. M. and van Helden D. F. (2010) Pacemaker currents in mouse locus coeruleus neurons. Neuroscience 170, 166–177. [DOI] [PubMed] [Google Scholar]
- Olson P. A., Tkatch T., Hernandez‐Lopez S., Ulrich S., Ilijic E., Mugnaini E., Zhang H., Bezprozvanny I. and Surmeier D. J. (2005) G‐protein‐coupled receptor modulation of striatal CaV1.3 L‐type Ca2 + channels is dependent on a Shank‐binding domain. J. Neurosci. 25, 1050–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oo T. F., Blazeski R., Harrison S. M., Henchcliffe C., Mason C. A., Roffler‐Tarlov S. K. and Burke R. E. (1996) Neuron death in the Substantia nigra of weaver mouse occurs late in development and is not apoptotic. J. Neurosci. 16, 6134–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortner N. J., Bock G., Vandael D. H., Mauersberger R., Draheim H. J., Gust R., Carbone E., Tuluc P. and Striessnig J. (2014) Pyrimidine‐2,4,6‐triones are a new class of voltage‐gated L‐type Ca2 + channel activators. Nat. Commun. 5, 3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osawa M., Tong K. I., Lilliehook C., Wasco W., Buxbaum J. D., Cheng H. Y., Penninger J. M., Ikura M. and Ames J. B. (2001) Calcium‐regulated DNA binding and oligomerization of the neuronal calcium‐sensing protein, calsenilin/DREAM/KChIP3. J. Biol. Chem. 276, 41005–41013. [DOI] [PubMed] [Google Scholar]
- Osborne P. B., Halliday G. M., Cooper H. M. and Keast J. R. (2005) Localization of immunoreactivity for deleted in colorectal cancer (DCC), the receptor for the guidance factor netrin‐1, in ventral tier dopamine projection pathways in adult rodents. Neuroscience 131, 671–681. [DOI] [PubMed] [Google Scholar]
- Osellame L. D. and Duchen M. R. (2014) Quality control gone wrong: mitochondria, lysosomal storage disorders and neurodegeneration. Br. J. Pharmacol. 171, 1958–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacelli C., Giguere N., Bourque M. J., Levesque M., Slack R. S. and Trudeau L. E. (2015) Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Curr. Biol. 25, 2349–2360. [DOI] [PubMed] [Google Scholar]
- Paladini C. A. and Roeper J. (2014) Generating bursts (and pauses) in the dopamine midbrain neurons. Neuroscience 282C, 109–121. [DOI] [PubMed] [Google Scholar]
- Parkinson Study, G (2013) Phase II safety, tolerability, and dose selection study of isradipine as a potential disease‐modifying intervention in early Parkinson's disease (STEADY‐PD). Mov. Disord. 28, 1823–1831. [DOI] [PubMed] [Google Scholar]
- Parsons P. A. (2007) Antagonistic pleiotropy and the stress theory of aging. Biogerontology 8, 613–617. [DOI] [PubMed] [Google Scholar]
- Pasternak B., Svanstrom H., Nielsen N. M., Fugger L., Melbye M. and Hviid A. (2012) Use of calcium channel blockers and Parkinson's disease. Am. J. Epidemiol. 175, 627–635. [DOI] [PubMed] [Google Scholar]
- Pathak T., Agrawal T., Richhariya S., Sadaf S. and Hasan G. (2015) Store‐operated calcium entry through orai is required for transcriptional maturation of the flight circuit in drosophila. J. Neurosci. 35, 13784–13799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Reyes E. (2003) Molecular physiology of low‐voltage‐activated t‐type calcium channels. Physiol. Rev. 83, 117–161. [DOI] [PubMed] [Google Scholar]
- Perier C., Bove J. and Vila M. (2012) Mitochondria and programmed cell death in Parkinson's disease: apoptosis and beyond. Antioxid. Redox Signal. 16, 883–895. [DOI] [PubMed] [Google Scholar]
- Pignatelli M. and Bonci A. (2015) Role of dopamine neurons in reward and aversion: a synaptic plasticity perspective. Neuron 86, 1145–1157. [DOI] [PubMed] [Google Scholar]
- Pilsl A. and Winklhofer K. F. (2012) Parkin, PINK1 and mitochondrial integrity: emerging concepts of mitochondrial dysfunction in Parkinson's disease. Acta Neuropathol. 123, 173–188. [DOI] [PubMed] [Google Scholar]
- Pissadaki E. K. and Bolam J. P. (2013) The energy cost of action potential propagation in dopamine neurons: clues to susceptibility in Parkinson's disease. Front Comput. Neurosci. 7, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platzer J., Engel J., Schrott‐Fischer A., Stephan K., Bova S., Chen H., Zheng H. and Striessnig J. (2000) Congenital deafness and sinoatrial node dysfunction in mice lacking class D L‐type Ca2 + channels. Cell 102, 89–97. [DOI] [PubMed] [Google Scholar]
- Plun‐Favreau H., Klupsch K., Moisoi N. et al (2007) The mitochondrial protease HtrA2 is regulated by Parkinson's disease‐associated kinase PINK1. Nat. Cell Biol. 9, 1243–1252. [DOI] [PubMed] [Google Scholar]
- Poetschke C., Dragicevic E., Duda J., Benkert J., Dougalis A., DeZio R., Snutch T. P., Striessnig J. and Liss B. (2015) Compensatory T‐type Ca channel activity alters D2‐autoreceptor responses of Substantia nigra dopamine neurons from Cav1.3 L‐type Ca channel KO mice. Sci. Rep., 5, 13688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin J. F., Zou J., Drouin‐Ouellet J., Kim K. Y., Cicchetti F. and Awatramani R. B. (2014) Defining midbrain dopaminergic neuron diversity by single‐cell gene expression profiling. Cell Rep. 9, 930–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers K. M., Smith‐Weller T., Franklin G. M., Longstreth W. T., Jr , Swanson P. D. and Checkoway H. (2006) Diabetes, smoking, and other medical conditions in relation to Parkinson's disease risk. Parkinsonism Relat. Disord. 12, 185–189. [DOI] [PubMed] [Google Scholar]
- Prentice H., Modi J. P. and Wu J. Y. (2015) Mechanisms of Neuronal Protection against Excitotoxicity, Endoplasmic Reticulum Stress, and Mitochondrial Dysfunction in Stroke and Neurodegenerative Diseases. Oxid. Med. Cell Longev. 2015, 964518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proks P. and Ashcroft F. M. (2009) Modeling K(ATP) channel gating and its regulation. Prog. Biophys. Mol. Biol. 99, 7–19. [DOI] [PubMed] [Google Scholar]
- Puopolo M., Raviola E. and Bean B. P. (2007) Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J. Neurosci. 27, 645–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putney J. W. (2012) Calcium signaling: deciphering the calcium‐NFAT pathway. Curr. Biol. 22, R87–R89. [DOI] [PubMed] [Google Scholar]
- Putzier I., Kullmann P. H., Horn J. P. and Levitan E. S. (2009) CaV1.3 channel voltage dependence, not Ca2 + selectivity, drives pacemaker activity and amplifies bursts in nigral dopamine neurons. J. Neurosci. 29, 15414–15419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanan V. K. and Saykin A. J. (2013) Pathways to neurodegeneration: mechanistic insights from GWAS in Alzheimer's disease, Parkinson's disease, and related disorders. Am. J. Neurodegener. Dis. 2, 145–175. [PMC free article] [PubMed] [Google Scholar]
- Ramirez A., Heimbach A., Grundemann J. et al (2006) Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P‐type ATPase. Nat. Genet. 38, 1184–1191. [DOI] [PubMed] [Google Scholar]
- Reeve A. K., Krishnan K. J., Elson J. L., Morris C. M., Bender A., Lightowlers R. N. and Turnbull D. M. (2008) Nature of mitochondrial DNA deletions in Substantia nigra neurons. Am. J. Hum. Genet. 82, 228–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve A., Simcox E. and Turnbull D. (2014) Ageing and Parkinson's disease: why is advancing age the biggest risk factor? Ageing Res. Rev. 14, 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice M. E. and Patel J. C. (2015) Somatodendritic dopamine release: recent mechanistic insights. Philos. Trans. R. Soc. Lond. B Biol. Sci. 370, pii 20140185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice M. E., Patel J. C. and Cragg S. J. (2011) Dopamine release in the basal ganglia. Neuroscience 198, 112–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz B., Rhodes S. L., Qian L., Schernhammer E., Olsen J. H. and Friis S. (2010) L‐type calcium channel blockers and Parkinson disease in Denmark. Ann. Neurol. 67, 600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas M., Villar D., Gonzalez P., Dopazo X. M., Mellstrom B. and Naranjo J. R. (2011) Building the DREAM interactome. Sci. China Life Sci. 54, 786–792. [DOI] [PubMed] [Google Scholar]
- Rizzuto R., Marchi S., Bonora M. et al (2009) Ca(2 + ) transfer from the ER to mitochondria: when, how and why. Biochim. Biophys. Acta 1787, 1342–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M., Rodriguez‐Sabate C., Morales I., Sanchez A. and Sabate M. (2015) Parkinson's disease as a result of aging. Aging Cell 14, 293–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeper J. (2013) Dissecting the diversity of midbrain dopamine neurons. Trends Neurosci. 36, 336–342. [DOI] [PubMed] [Google Scholar]
- Roselli F. and Caroni P. (2015) From intrinsic firing properties to selective neuronal vulnerability in neurodegenerative diseases. Neuron 85, 901–910. [DOI] [PubMed] [Google Scholar]
- Ryan B. J., Hoek S., Fon E. A. and Wade‐Martins R. (2015) Mitochondrial dysfunction and mitophagy in Parkinson's: from familial to sporadic disease. Trends Biochem. Sci. 40, 200–210. [DOI] [PubMed] [Google Scholar]
- Salthun‐Lassalle B., Hirsch E. C., Wolfart J., Ruberg M. and Michel P. P. (2004) Rescue of mesencephalic dopaminergic neurons in culture by low‐level stimulation of voltage‐gated sodium channels. J. Neurosci. 24, 5922–5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels E. R. and Szabadi E. (2008) Functional neuroanatomy of the noradrenergic locus coeruleus: its roles in the regulation of arousal and autonomic function part I: principles of functional organisation. Curr. Neuropharmacol. 6, 235–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Padilla J., Guzman J. N., Ilijic E. et al (2014) Mitochondrial oxidant stress in locus coeruleus is regulated by activity and nitric oxide synthase. Nat. Neurosci. 17, 832–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago J. A. and Potashkin J. A. (2013) Shared dysregulated pathways lead to Parkinson's disease and diabetes. Trends Mol. Med. 19, 176–186. [DOI] [PubMed] [Google Scholar]
- Santiago J. A. and Potashkin J. A. (2014) System‐based approaches to decode the molecular links in Parkinson's disease and diabetes. Neurobiol. Dis. 72 Pt A, 84–91. [DOI] [PubMed] [Google Scholar]
- Schapira A. H. (2008) Mitochondria in the aetiology and pathogenesis of Parkinson's disease. Lancet Neurol. 7, 97–109. [DOI] [PubMed] [Google Scholar]
- Schapira A. H. (2010) Complex I: inhibitors, inhibition and neurodegeneration. Exp. Neurol. 224, 331–335. [DOI] [PubMed] [Google Scholar]
- Schapira A. H. (2015) Glucocerebrosidase and Parkinson disease: recent advances. Mol. Cell Neurosci. 66, 37–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira A. H. and Gegg M. E. (2013) Glucocerebrosidase in the pathogenesis and treatment of Parkinson disease. Proc. Natl Acad. Sci. USA 110, 3214–3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharinger A., Eckrich S., Vandael D. H. et al (2015) Cell‐type‐specific tuning of Cav1.3 Ca(2 + )‐channels by a C‐terminal automodulatory domain. Front Cell Neurosci. 9, 309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schernhammer E., Hansen J., Rugbjerg K., Wermuth L. and Ritz B. (2011) Diabetes and the risk of developing Parkinson's disease in Denmark. Diabetes Care 34, 1102–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiemann J., Schlaudraff F., Klose V. et al (2012) K‐ATP channels in dopamine Substantia nigra neurons control bursting and novelty‐induced exploration. Nat. Neurosci. 15, 1272–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaudraff F., Grundemann J., Fauler M., Dragicevic E., Hardy J. and Liss B. (2014) Orchestrated increase of dopamine and PARK mRNAs but not miR‐133b in dopamine neurons in Parkinson's disease. Neurobiol. Aging 35, 2302–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schondorf D. C., Aureli M., McAllister F. E. et al (2014) iPSC‐derived neurons from GBA1‐associated Parkinson's disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 5, 4028. [DOI] [PubMed] [Google Scholar]
- Schultz W., Carelli R. M. and Wightman R. M. (2015) Phasic dopamine signals: from subjective reward value to formal economic utility. Curr. Opin. Behav. Sci. 5, 147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwaller B. (2010) Cytosolic Ca2 + buffers. Cold Spring Harb. Perspect. Biol. 2, a004051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scigliano G., Musicco M., Soliveri P., Piccolo I., Ronchetti G. and Girotti F. (2006) Reduced risk factors for vascular disorders in Parkinson disease patients: a case‐control study. Stroke 37, 1184–1188. [DOI] [PubMed] [Google Scholar]
- Screaton R. A., Conkright M. D., Katoh Y. et al (2004) The CREB coactivator TORC2 functions as a calcium‐ and cAMP‐sensitive coincidence detector. Cell 119, 61–74. [DOI] [PubMed] [Google Scholar]
- Seino S. (2012) Cell signalling in insulin secretion: the molecular targets of ATP, cAMP and sulfonylurea. Diabetologia 55, 2096–2108. [DOI] [PubMed] [Google Scholar]
- Seino S. and Miki T. (2003) Physiological and pathophysiological roles of ATP‐sensitive K+ channels. Prog. Biophys. Mol. Biol. 81, 133–176. [DOI] [PubMed] [Google Scholar]
- Selvakumar A., Antony C., Singhal J., Tiwari B. K., Singh Y. and Natarajan K. (2014) Reciprocal regulation of reactive oxygen species and phospho‐CREB regulates voltage gated calcium channel expression during mycobacterium tuberculosis infection. PLoS ONE 9, e96427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj S., Sun Y., Watt J. A., Wang S., Lei S., Birnbaumer L. and Singh B. B. (2012) Neurotoxin‐induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J. Clin. Invest. 122, 1354–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N., Crane A., Clement J. P., Gonzalez G., Babenko A. P., Bryan J., Aguilar‐Bryan L. (1999) The C terminus of SUR1 is required for trafficking of KATP channels. J. Biol. Chem. 274, 20628–20632. [DOI] [PubMed] [Google Scholar]
- Shen K. Z. and Johnson S. W. (2010) Ca2 + influx through NMDA‐gated channels activates ATP‐sensitive K+ currents through a nitric oxide‐cGMP pathway in subthalamic neurons. J. Neurosci. 30, 1882–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebert M., Sidransky E. and Westbroek W. (2014) Glucocerebrosidase is shaking up the synucleinopathies. Brain 137, 1304–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva J. A. and Costa R. M. (2012) Bursting for exploration. Nat. Neurosci. 15, 1178–1179. [DOI] [PubMed] [Google Scholar]
- Simms B. A. and Zamponi G. W. (2014) Neuronal voltage‐gated calcium channels: structure, function, and dysfunction. Neuron 82, 24–45. [DOI] [PubMed] [Google Scholar]
- Simuni T., Borushko E., Avram M. J. et al (2010) Tolerability of isradipine in early Parkinson's disease: a pilot dose escalation study. Mov. Disord. 25, 2863–2866. [DOI] [PubMed] [Google Scholar]
- Singleton A. B., Farrer M. J. and Bonifati V. (2013) The genetics of Parkinson's disease: progress and therapeutic implications. Mov. Disord. 28, 14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnegger‐Brauns M. J., Hetzenauer A., Huber I. G. et al (2004) Isoform‐specific regulation of mood behavior and pancreatic beta cell and cardiovascular function by L‐type Ca 2 + channels. J. Clin. Invest. 113, 1430–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slesinger P. A., Patil N., Liao Y. J., Jan Y. N., Jan L. Y. and Cox D. R. (1996) Functional effects of the mouse weaver mutation on G protein‐gated inwardly rectifying K+ channels. Neuron 16, 321–331. [DOI] [PubMed] [Google Scholar]
- Snutch T. P., Sutton K. G. and Zamponi G. W. (2001) Voltage‐dependent calcium channels–beyond dihydropyridine antagonists. Curr. Opin. Pharmacol. 1, 11–16. [DOI] [PubMed] [Google Scholar]
- Sommer W. H., Costa R. M. and Hansson A. C. (2014) Dopamine systems adaptation during acquisition and consolidation of a skill. Front Integr Neurosci. 8, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soundarapandian M. M., Zhong X., Peng L., Wu D. and Lu Y. (2007) Role of K(ATP) channels in protection against neuronal excitatory insults. J. Neurochem. 103, 1721–1729. [DOI] [PubMed] [Google Scholar]
- Sours‐Brothers S., Ma R. and Koulen P. (2009) Ca2 + ‐sensitive transcriptional regulation: direct DNA interaction by DREAM. Front Biosci (Landmark Ed) 14, 1851–1856. [DOI] [PubMed] [Google Scholar]
- Stocker M. (2004) Ca(2 + )‐activated K+ channels: molecular determinants and function of the SK family. Nat. Rev. Neurosci. 5, 758–770. [DOI] [PubMed] [Google Scholar]
- Striessnig J. and Koschak A. (2008) Exploring the function and pharmacotherapeutic potential of voltage‐gated Ca2 + channels with gene knockout models. Channels (Austin) 2, 233–251. [DOI] [PubMed] [Google Scholar]
- Striessnig J., Grabner M., Mitterdorfer J., Hering S., Sinnegger M. J. and Glossmann H. (1998) Structural basis of drug binding to L Ca2 + channels. Trends Pharmacol. Sci. 19, 108–115. [DOI] [PubMed] [Google Scholar]
- Striessnig J., Koschak A., Sinnegger‐Brauns M. J., Hetzenauer A., Nguyen N. K., Busquet P., Pelster G. and Singewald N. (2006) Role of voltage‐gated L‐type Ca2 + channel isoforms for brain function. Biochem. Soc. Trans. 34, 903–909. [DOI] [PubMed] [Google Scholar]
- Subramaniam M., Althof D., Gispert S., Schwenk J., Auburger G., Kulik A., Fakler B. and Roeper J. (2014) Mutant alpha‐synuclein enhances firing frequencies in dopamine Substantia nigra neurons by oxidative impairment of A‐type potassium channels. J. Neurosci. 34, 13586–13599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D. (2007) Multiple hit hypotheses for dopamine neuron loss in Parkinson's disease. Trends Neurosci. 30, 244–250. [DOI] [PubMed] [Google Scholar]
- Sulzer D. and Surmeier D. J. (2013) Neuronal vulnerability, pathogenesis, and Parkinson's disease. Mov. Disord. 28, 41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier D. J. (2007) Calcium, ageing, and neuronal vulnerability in Parkinson's disease. Lancet Neurol 6, 933–938. [DOI] [PubMed] [Google Scholar]
- Surmeier D. J. and Schumacker P. T. (2013) Calcium, bioenergetics, and neuronal vulnerability in Parkinson's disease. J. Biol. Chem. 288, 10736–10741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier D. J., Guzman J. N., Sanchez‐Padilla J. and Goldberg J. A. (2010) What causes the death of dopaminergic neurons in Parkinson's disease? Prog. Brain Res. 183, 59–77. [DOI] [PubMed] [Google Scholar]
- Surmeier D. J., Guzman J. N., Sanchez‐Padilla J. and Goldberg J. A. (2011) The origins of oxidant stress in Parkinson's disease and therapeutic strategies. Antioxid. Redox Signal. 14, 1289–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier D. J., Guzman J. N., Sanchez J. and Schumacker P. T. (2012) Physiological phenotype and vulnerability in Parkinson's disease. Cold Spring Harb. Perspect Med. 2, a009290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaab D. F., Dubelaar E. J., Hofman M. A., Scherder E. J., van Someren E. J. and Verwer R. W. (2002) Brain aging and Alzheimer's disease; use it or lose it. Prog. Brain Res. 138, 343–373. [DOI] [PubMed] [Google Scholar]
- Tan Y. L., Genereux J. C., Pankow S., Aerts J. M., Yates J. R., 3rd and Kelly J. W. (2014) ERdj3 is an endoplasmic reticulum degradation factor for mutant glucocerebrosidase variants linked to Gaucher's disease. Chem. Biol. 21, 967–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y. and Shen J. (2012) Genetic analysis of Parkinson's disease‐linked leucine‐rich repeat kinase 2. Biochem. Soc. Trans. 40, 1042–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y., Pisani A., Martella G., Karouani M., Yamaguchi H., Pothos E. N. and Shen J. (2009) R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc. Natl Acad. Sci. USA 106, 14622–14627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toulorge D., Guerreiro S., Hirsch E. C. and Michel P. P. (2010) KATP channel blockade protects midbrain dopamine neurons by repressing a glia‐to‐neuron signaling cascade that ultimately disrupts mitochondrial calcium homeostasis. J. Neurochem. 114, 553–564. [DOI] [PubMed] [Google Scholar]
- Toulorge D., Guerreiro S., Hild A., Maskos U., Hirsch E. C. and Michel P. P. (2011) Neuroprotection of midbrain dopamine neurons by nicotine is gated by cytoplasmic Ca2 + . FASEB J. 25, 2563–2573. [DOI] [PubMed] [Google Scholar]
- Tovar S., Paeger L., Hess S. et al (2013) K(ATP)‐channel‐dependent regulation of catecholaminergic neurons controls BAT sympathetic nerve activity and energy homeostasis. Cell Metab. 18, 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traaseth N., Elfering S., Solien J., Haynes V. and Giulivi C. (2004) Role of calcium signaling in the activation of mitochondrial nitric oxide synthase and citric acid cycle. Biochim. Biophys. Acta 1658, 64–71. [DOI] [PubMed] [Google Scholar]
- Tucker S. J., Gribble F. M., Zhao C., Trapp S. and Ashcroft F. M. (1997) Truncation of Kir6.2 produces ATP‐sensitive K+ channels in the absence of the sulphonylurea receptor. Nature 387, 179–183. [DOI] [PubMed] [Google Scholar]
- Turner R. W. and Zamponi G. W. (2014) T‐type channels buddy up. Pflugers Arch. 466, 661–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungless M. A. and Grace A. A. (2012) Are you or aren't you? Challenges associated with physiologically identifying dopamine neurons. Trends Neurosci. 35, 422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente E. M., Abou‐Sleiman P. M., Caputo V. et al (2004) Hereditary early‐onset Parkinson's disease caused by mutations in PINK1. Science 304, 1158–1160. [DOI] [PubMed] [Google Scholar]
- Valenzuela M. J., Matthews F. E., Brayne C. et al (2012) Multiple biological pathways link cognitive lifestyle to protection from dementia. Biol. Psychiatry 71, 783–791. [DOI] [PubMed] [Google Scholar]
- Van Laar V. S., Roy N., Liu A., Rajprohat S., Arnold B., Dukes A. A., Holbein C. D. and Berman S. B. (2015) Glutamate excitotoxicity in neurons triggers mitochondrial and endoplasmic reticulum accumulation of Parkin, and in the presence of N‐acetyl cysteine, mitophagy. Neurobiol. Dis. 74, 180–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhratsky A. (2005) Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol. Rev. 85, 201–279. [DOI] [PubMed] [Google Scholar]
- Vila M., Ramonet D. and Perier C. (2008) Mitochondrial alterations in Parkinson's disease: new clues. J. Neurochem. 107, 317–328. [DOI] [PubMed] [Google Scholar]
- Virgili N., Mancera P., Wappenhans B., Sorrosal G., Biber K., Pugliese M. and Espinosa‐Parrilla J. F. (2013) K(ATP) channel opener diazoxide prevents neurodegeneration: a new mechanism of action via antioxidative pathway activation. PLoS ONE 8, e75189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volman S. F., Lammel S., Margolis E. B., Kim Y., Richard J. M., Roitman M. F. and Lobo M. K. (2013) New insights into the specificity and plasticity of reward and aversion encoding in the mesolimbic system. J. Neurosci. 33, 17569–17576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlqvist M. L., Lee M. S., Hsu C. C., Chuang S. Y., Lee J. T. and Tsai H. N. (2012) Metformin‐inclusive sulfonylurea therapy reduces the risk of Parkinson's disease occurring with Type 2 diabetes in a Taiwanese population cohort. Parkinsonism Relat. Disord. 18, 753–758. [DOI] [PubMed] [Google Scholar]
- Waldeck‐Weiermair M., Jean‐Quartier C., Rost R., Khan M. J., Vishnu N., Bondarenko A. I., Imamura H., Malli R. and Graier W. F. (2011) Leucine zipper EF hand‐containing transmembrane protein 1 (Letm1) and uncoupling proteins 2 and 3 (UCP2/3) contribute to two distinct mitochondrial Ca2 + uptake pathways. J. Biol. Chem. 286, 28444–28455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. and Schwarz T. L. (2009) The mechanism of Ca2 + ‐dependent regulation of kinesin‐mediated mitochondrial motility. Cell 136, 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Qu L., Wang X. L., Gao L., Li Z. Z., Gao G. D. and Yang Q. (2015) Firing pattern modulation through SK channel current increase underlies neuronal survival in an organotypic slice model of Parkinson's disease. Mol. Neurobiol. 51, 424–436. [DOI] [PubMed] [Google Scholar]
- Watabe‐Uchida M., Zhu L., Ogawa S. K., Vamanrao A. and Uchida N. (2012) Whole‐brain mapping of direct inputs to midbrain dopamine neurons. Neuron 74, 858–873. [DOI] [PubMed] [Google Scholar]
- Weiser T. (2008) Ambroxol: a CNS drug? CNS Neurosci. Ther. 14, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westbroek W., Gustafson A. M. and Sidransky E. (2011) Exploring the link between glucocerebrosidase mutations and parkinsonism. Trends Mol. Med. 17, 485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wet H. and Proks P. (2015) Molecular action of sulphonylureas on KATP channels: a real partnership between drugs and nucleotides. Biochem. Soc. Trans. 43, 901–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C. J. and Callaway J. C. (2000) Coupled oscillator model of the dopaminergic neuron of the Substantia nigra . J. Neurophysiol. 83, 3084–3100. [DOI] [PubMed] [Google Scholar]
- Winklhofer K. F. and Haass C. (2010) Mitochondrial dysfunction in Parkinson's disease. Biochim. Biophys. Acta 1802, 29–44. [DOI] [PubMed] [Google Scholar]
- Wise R. A. (2009) Roles for nigrostriatal–not just mesocorticolimbic–dopamine in reward and addiction. Trends Neurosci. 32, 517–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfart J. and Roeper J. (2002) Selective coupling of T‐type calcium channels to SK potassium channels prevents intrinsic bursting in dopaminergic midbrain neurons. J. Neurosci. 22, 3404–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfart J., Neuhoff H., Franz O. and Roeper J. (2001) Differential expression of the small‐conductance, calcium‐activated potassium channel SK3 is critical for pacemaker control in dopaminergic midbrain neurons. J. Neurosci. 21, 3443–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H. and Ren D. (2015) Lysosomal physiology. Annu. Rev. Physiol. 77, 57–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K. and Inagaki N. (2005) Neuroprotection by KATP channels. J. Mol. Cell. Cardiol. 38, 945–949. [DOI] [PubMed] [Google Scholar]
- Yamada T., McGeer P. L., Baimbridge K. G. and McGeer E. G. (1990) Relative sparing in Parkinson's disease of Substantia nigra dopamine neurons containing calbindin‐D28K. Brain Res. 526, 303–307. [DOI] [PubMed] [Google Scholar]
- Yang F., Feng L., Zheng F., Johnson S. W., Du J., Shen L., Wu C. P. and Lu B. (2001) GDNF acutely modulates excitability and A‐type K(+) channels in midbrain dopaminergic neurons. Nat. Neurosci. 4, 1071–1078. [DOI] [PubMed] [Google Scholar]
- Youle R. J. and van der Bliek A. M. (2012) Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaghloul K. A., Blanco J. A., Weidemann C. T., McGill K., Jaggi J. L., Baltuch G. H. and Kahana M. J. (2009) Human Substantia nigra neurons encode unexpected financial rewards. Science 323, 1496–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi G. W., Striessnig J., Koschak A. and Dolphin A. C. (2015) The Physiology, Pathology, and Pharmacology of Voltage‐Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol. Rev. 67, 821–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarow C., Lyness S. A., Mortimer J. A. and Chui H. C. (2003) Neuronal loss is greater in the locus coeruleus than nucleus basalis and Substantia nigra in Alzheimer and Parkinson diseases. Arch. Neurol. 60, 337–341. [DOI] [PubMed] [Google Scholar]
- Zhang P. and Tian B. (2014) Metabolic Syndrome: an Important Risk Factor for Parkinson's Disease. Oxid. Med. Cell Longev. 2014, 729194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucca F. A., Segura‐Aguilar J., Ferrari E., Munoz P., Paris I., Sulzer D., Sarna T., Casella L. and Zecca L. (2015) Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson's disease. Prog. Neurobiol. pii: S0301‐0082(15)00101‐X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuhlke R. D., Pitt G. S., Deisseroth K., Tsien R. W. and Reuter H. (1999) Calmodulin supports both inactivation and facilitation of L‐type calcium channels. Nature 399, 159–162. [DOI] [PubMed] [Google Scholar]