

Pulmonary artery smooth muscle cell (PASMC) hyperproliferation is typically associated with a shift to Warburg metabolism in end-stage pulmonary arterial hypertension (PAH). In pulmonary overcirculation-induced PAH, however, we find that PASMC hyperproliferation is associated with a unique metabolism characterized by mitochondrial dysfunction without enhanced glycolysis. Instead, Increased NADPH oxidase (Nox) activity and enhanced pentose phosphate pathway (PPP) flux help drive PASMC proliferation.
Keywords: glycolysis, mitochondria, oxygen consumption, pulmonary overcirculation, ROS
Abstract
Vascular cell hyperproliferation and metabolic reprogramming contribute to the pathophysiology of pulmonary arterial hypertension (PAH). An important cause of PAH in children with congenital heart disease (CHD) is increased pulmonary blood flow (PBF). To better characterize this disease course we studied early changes in pulmonary artery smooth muscle cell (PASMC) proliferation and metabolism using a unique ovine model of pulmonary overcirculation. Consistent with PAH in adults, PASMCs derived from 4-wk-old lambs exposed to increased PBF (shunt) exhibited increased rates of proliferation. While shunt PASMCs also exhibited significant decreases in mitochondrial oxygen consumption, membrane potential, and tricarboxylic acid (TCA) cycle function, suggesting a switch to Warburg metabolism as observed in advanced PAH in adults, they unexpectedly demonstrated decreased glycolytic lactate production, likely due to enhanced flux through the pentose phosphate pathway (PPP). This may be a response to the marked increase in NADPH oxidase (Nox) activity and decreased NADPH/NADP+ ratios observed in shunt PASMCs. Consistent with these findings, pharmacological inhibition of Nox activity preferentially slowed the growth of shunt PASMCs in vitro. Our results therefore indicate that PASMC hyperproliferation is observed early in the setting of pulmonary overcirculation and is accompanied by a unique metabolic profile that is independent of HIF-1α, PDHK1, or increased glycolytic flux. Our results also suggest that Nox inhibition may help prevent pulmonary overcirculation-induced PAH in children born with CHD.
NEW & NOTEWORTHY
Pulmonary artery smooth muscle cell (PASMC) hyperproliferation is typically associated with a shift to Warburg metabolism in end-stage pulmonary arterial hypertension (PAH). In pulmonary overcirculation-induced PAH, however, we find that PASMC hyperproliferation is associated with a unique metabolism characterized by mitochondrial dysfunction without enhanced glycolysis. Instead, Increased NADPH oxidase (Nox) activity and enhanced pentose phosphate pathway (PPP) flux help drive PASMC proliferation.
while pulmonary hypertension is simply defined by an elevated pulmonary artery pressure at rest, the biological mechanisms that underlie this hemodynamic derangement are multifactorial and biologically complex. Genetic, physiological, and environmental factors interact to produce a spectrum of vascular disease, ranging from reversible endothelial dysfunction to irrevocable remodeling of the vasculature. While relatively rare in the pediatric population, these patients suffer from high morbidity and mortality. Currently available therapies target the imbalance of vasoconstrictive and vasodilatory signals, but there are few modalities to treat underlying causes and no treatment options are available that reverse the late structural vascular changes (31). The majority of the pediatric burden of disease falls within the subgroup of pulmonary arterial hypertension (PAH) (31). The characteristic histological findings of late-stage PAH are proliferative abnormalities of the vasculature, including “medial hypertrophy” of the smooth muscle, and endothelial “plexiform lesions” (17). Investigation of the molecular and cellular characteristics of these lesions has revealed evidence of proliferation of apoptosis-resistant cell populations (42), monoclonal proliferation of endothelial cells (22), overexpression of vascular growth factors (45), and expression of anti-apoptotic markers (27). This has drawn analogies to malignant growth (16) and even spurred a biological model of these proliferative vascular lesions as “quasi-neoplastic” (46). This conceptual framework has drawn particular attention to the altered metabolism observed in the cells from this remodeled vasculature that parallels the changes observed in transformed cancer cells (16).
Pulmonary artery smooth muscle and endothelial cells (PASMCs and PAECs) from animal models of advanced PAH and explanted human tissue have been found to exhibit a consistent pattern of altered cellular metabolism consisting of decreased mitochondrial glucose oxidation paired with increased rates of glycolysis (6, 46, 50). This phenotype closely aligns with the Warburg effect, initially described in cancer tissue as early as 1924 and now widely recognized in many tumors (18). The molecular and biochemical changes driving this metabolic shift in PAH are incompletely understood but several important regulatory pathways that contribute to it have been identified. Perhaps the best-studied of these relates to increased stabilization of hypoxia-inducible factor (HIF) 1α under hypoxic and normoxic conditions in late-stage PAH (13). HIF-1α is a prolific transcriptional regulator and affects the expression of many genes related to carbohydrate metabolism including glucose transporters, glycolytic enzymes, and the regulatory kinase of pyruvate dehydrogenase (PDHK) (46). Through increased transcription of this inhibitory kinase, pyruvate dehydrogenase activity is suppressed, preventing conversion of pyruvate into acetyl-CoA and blocking the flow of glucose-derived carbons into the mitochondrial tricarboxylic acid cycle (TCA) (20). Notably, this regulatory pathway has been selectively targeted for therapy using the drug dichloroacetate (DCA). DCA inhibits PDHK, functionally derepressing mitochondrial glucose oxidation. In several rodent models of PAH, treatment with DCA has been shown to improve pulmonary hemodynamics and survival (6, 28). At the cellular level, these animals showed decreased proliferation and increased apoptosis of the pulmonary arterial smooth muscle cells (28). These experiments strongly suggest a functional link between observed alterations in cellular metabolism and clinically relevant hemodynamic dysfunction. Moreover, they suggest that therapies specifically aimed at reversing or redirecting these metabolic alterations may represent a viable treatment strategy for the disease.
Congenital heart defects resulting in pulmonary overcirculation are an important contributor to the pathophysiology of PAH in children (30). Almost 20 years ago, Reddy et al. (38) developed an ovine model of human congenital heart disease that utilizes fetal surgical techniques to establish postnatal left-to-right shunting and pulmonary overcirculation. Via a surgical conduit placed between the aorta and the main pulmonary artery (shunt), postnatal pulmonary blood flow is increased dramatically above normal levels. These lambs develop pulmonary vascular changes that recapitulate the morphological and physiological features of human disease (1). This includes smooth muscle cell hyperplasia of the proximal and peripheral pulmonary arteries, correlating with the medial thickening and histological abnormalities observed in adult patients with CHD and PAH (34). This large animal model therefore represents an invaluable resource for investigating the biochemical, cellular, and physiological derangements stemming from increased pulmonary blood flow, a specific and clinically relevant factor in pediatric pulmonary vascular disease. In addition, it provides unique insight into the biological changes occurring at early stages of pulmonary vascular dysfunction as opposed to the advanced disease states common to other animal models and explanted human tissues. This is due to the fact that animals can be studied at early ages before increased PBF triggers large-scale morphological and physiological derangements. We sought to take advantage of this system to identify and carefully define any metabolic and biochemical alteration in PASMCs exposed to chronically increased blood flow. We hypothesized that pulmonary overcirculation would induce cellular metabolic alterations of the pulmonary arterial smooth muscle similar to that observed in advanced disease in humans. We further postulated that these metabolic changes would be detectable at an early stage of flow-induced pulmonary vascular disease and that the phenotype would resemble the Warburg metabolism described in advanced PAH.
MATERIALS AND METHODS
Lamb model.
As described in detail previously (38), an 8.0-mm Gore-tex vascular graft, ∼2 mm in length, was anastomosed between the ascending aorta and main pulmonary artery in anesthetized late-gestation fetal lambs (137–141 days gestation, term = 145 days) from three mixed-breed Western ewes. Four weeks after spontaneous delivery, normal, age-matched controls (n = 3) or shunt lambs (n = 3) were anesthetized, mechanically ventilated, and instrumented to continuously measure hemodynamics and harvest tissue. Animals' vital signs, including core temperature, were monitored throughout the study, and they were given intravenous fluids and prophylactic antibiotics per protocol. At the end of each protocol, all lambs were euthanized with a lethal injection of pentobarbital sodium followed by bilateral thoracotomy as described in the NIH Guidelines for the Care and Use of Laboratory Animals. All protocols and procedures were approved by the Committees on Animal Research of the University of California, San Francisco and University of California, Davis.
Cell isolation and culture.
Primary smooth muscle cells from shunt (n = 3) and control (n = 3) lambs were isolated from sections of the proximal (main, left, and right) pulmonary arteries by explant method as previously published (19). Identity was confirmed as PASMC by immunostaining (>99% positive) with established smooth muscle markers (40) SMC actin, smooth muscle myosin heavy chain, and calponin as described previously (26). The cells were maintained in culture with a medium of low-glucose DMEM (1 g/l) with sodium pyruvate (110 mg/l) and glutamine supplemented with 10% fetal bovine serum (Lonza), penicillin/streptomycin, and fungizone. They were grown in an incubator at 5% CO2 and 37°C and passaged between 70–90% confluence. All experiments were performed on passage-matched (within ± 1 passage) primary cells between passages 4–8.
Immunofluorescence.
PASMCs from shunt and control animals were grown on cover glass to 80% confluence, fixed with 4% paraformaldehyde (Fisher) for 30 min, and permeabilized with 0.1% Triton X-100 (Merck KGaA) for 5 min. After that, cells were incubated with anti-calponin IgG (1:400 rabbit, Abcam) primary antibody for 30 min, conjugated with a goat anti-rabbit IgG TRITC (1:200, ProteinTech Group) secondary antibody, and stained with 4′,6-diamidino-2-phenylindole (DAPI, 1:10.000, Invitrogen). As negative controls, the primary antibody was replaced by bovine serum albumin (BSA, Serva Electrophoresis GmbH). Finally, cells were mounted on histological slices using mounting medium (Cell Signaling Technologies) and imaged by a Leica SP5 inverted confocal microscope (Zeiss).
Baseline proliferation and drug treatment assays.
SMCs from shunt and control animals were trypsinized with 0.25% trypsin solution, washed with PBS and counted using a desktop coulter based cell counter (Moxi Z, Orflo). Uniform cell counts were then seeded into 24-well cell culture plates with standard cell culture medium (as above). Three distinct clonal cell lines from shunts and three from controls were plated in replicates of 8 per line for each experimental growth time point. At sequential 24-h time points after seeding, cells from each line were trypsinized and counted (as per above) out to 72 h. This procedure was repeated in triplicate. Total cell counts were averaged for all shunt cell lines and all control cell lines and plotted along with error bars corresponding to 1 SD in both the positive and negative direction. Total cell counts for all control and all shunt cell lines at each experimental time point were compared with two-tailed Student's t-test. To further control for differences in starting cell number due to natural variability and any differences in efficiency or time of adherence after trypsinization, all cell counts were log transformed and linear regression analysis was performed during the time period of 24–72 h representing the log growth phase. The slopes of the lines, corresponding to the actual rate of growth, were then compared using multiple regression/correlation analysis as outlined previously (10). Cell doubling times were calculated by use of least-squares fitting exponential.
For treatment with dichloroacetate (DCA) and dimethyl malate (DMM), and VAS 3947, cells were counted as above and seeded with uniform density into 24 well culture plates using standard growth media. At 24 h after cells had been allowed adequate time to adhere, media was changed to standard growth media with the drug and dose of interest (experimental) or standard media with an equivalent volume of drug solvent (control). For dichloroacetate experiments, 0.5, 1, or 5 mM DCA (Sigma Aldrich 634522) were used for experimental groups. For dimethyl malate experiments, 5 mM DMM (Sigma Aldrich 374318) was used for experimental groups. For VAS 3947, 10 μM (CalBiochem 532336) was used for experimental groups. At 72 h cells from all groups were counted in replicates of 8. All counts in treatment groups were normalized to control 72 h growth averages for comparison. Comparison between shunt and control growth was performed using unpaired, two-tailed Student's t-test.
Extracellular flux analysis.
For our experiments we used the Seahorse XF-24 Extracellular Flux Analyzer (Seahorse Bioscience, Copenhagen, Denmark) to compare shunt and control PASMCs. Per manufacturer instructions, initial optimization assays were conducted using both shunt and control cell lines to establish optimum cellular density for the assay. Experimental assays were then performed at optimal cell density (∼10,000–30,000 cells/well). Basal metabolism comparisons were performed as per manufacturer recommendations. Media for all extracellular flux assays was DMEM with sodium pyruvate (110 mg/l), glutamine, and glucose (1 g/l) without NaHCO3 and without phenol red (Sigma). After reconstitution from powder, media was pH adjusted to 7.40 and sterile filtered. Cells from each line were plated in replicates of 5. Immediately following basal metabolism assays, cells from all wells were counted and averaged per experimental group. OCR and ECAR data for each experimental group were normalized to average cell number for analysis. Given that there is variation between assay plates, to perform comparison across multiple experiments, all values were normalized to a single cell line included in all assays.
OCR measurements with DMM addition were performed using a modification of the basal metabolism measurements. Cells and media were prepared as above. Cells from each line were plated in replicates of 5 for 2 shunt cell lines and 2 control cell lines. Following measurement of basal OCR, DMM was injected into the assay medium to reach a total final concentration of 5 mM, and measurements of OCR were repeated in triplicate following a 15 min mixing cycle. OCR with DMM was subsequently normalized to OCR under basal conditions within each group for comparison.
Protein isolation.
For whole cell protein extraction, PASMCs were seeded and grown on 10-cm culture plates to 70–80% confluence. Whole cell lysates were prepared on ice using urea lysis buffer [8 M urea, 10% glycerol, 5 mM DTT, 10 mM Tris-HCl pH 6.8, 1% SDS, and 1 × Proteinase Inhibitor Cocktail (Roche Diagnostics)]. Cells were scraped and sonicated and protein lysates were subsequently stored at −80°C. Protein was quantified using the Nanodrop microvolume spectrophotometer (Thermo Scientific).
Western blots.
For Western blot analysis of HIF-1α and PDHK, 30 μg of total PASMC protein lysate was loaded for each sample onto an 8–16% SDS-PAGE Gel (Lonza) and transferred to a PVDF membrane using the semi-dry transfer method. The membranes were then blocked using Li-COR blocking buffer (LI-COR Biosciences) with 0.1% Tween and probed with primary antibodies directed against HIF-1α (Cayman Chemical, 10006421), PDHK 1 (Cell Signaling 3820), and α-Tubulin (Sigma Aldrich T9026), which served as a loading control. This was followed by incubation with IRDye 800CW goat anti-rabbit and IRDye 680 goat anti-mouse (LI-COR Biosciences) at a dilution of 1:5,000. Blots were visualized using the Odyssey Imaging System (LI-COR) and quantitated with Image Studio software in accordance with manufacturer's protocol.
For Western Blot analysis of Nox 2, Nox 4, p47 phox, and Rac-1, 20 μg of total PASMC protein lysate was loaded for each sample onto a 10% SDS-PAGE and transferred to a PVDF membrane using the wet transfer method. The membranes were then blocked with 5% nonfat dried milk in 130 mM NaCl and 25 mM Tris (TBS, pH 7.5) and probed with primary antibodies against Gp91-phox (H-60) (NOX2) (Santa Cruz Biotechnology, Sc-20782), NOX4 (Abcam, ab109225), p47-phox (Cell Signaling, 4301), RAC-1 (Sigma, GW22212F), and β-actin (Abcam), which served as a loading control. This was followed by incubation with the appropriate horseradish peroxidase-conjugated secondary antibodies. Chemiluminescence was then used to detect bands (SuperSignal West Pico Chemiluminescent Substrate kit, Pierce Biotechnology, Rockford, IL). Densitometry was performed using a public domain Java image processing program, ImageJ (NIH Image).
Metabolomics.
For metabolite quantification, 1 line each of shunt and control cells were selected and grown in 10-cm culture plates in replicates of five each (n = 5). Once cells reached 75–80% confluence, media was aspirated and cells were briefly rinsed with ∼10 ml deionized water before metabolism was quenched by direct addition of ∼10 ml of liquid N2 to the plates. The plates were then stored at −80°C. Quantification was performed by the Michigan Regional Comprehensive Metabolomics Resource Core, supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK-097153 to the University of Michigan. For extraction, plates were transferred to a 4°C cold room, and 1.5 ml of ice-cold 90% 9:1 MeOH:CHCl3 was immediately added to each plate and cells scraped/suspended with a cell lifter. The extraction solvent was spiked with 13C-labeled internal standards. Extracts were transferred to 1.5 ml microcentrifuge tubes and pelleted at 4°C. Supernatants were then assayed by HPLC/mass spectrometry. Samples were separated on an Agilent 1200 HPLC on a Phenomenex Luna NH2 column, then analyzed either in positive or negative ESI by a coupled Agilent 6530 QTOF mass spectrometer (24). Compounds were identified based on retention time and mass/charge ratio match to injections of authentic standards. Quantitation was performed using Agilent Technologies MassHunter Quantitative software. Peak areas were measured from extracted ion chromatograms of metabolite ions with detection windows centered on the theoretical mass. Peak areas from internal standards were measured using an identical procedure. Data processing was performed using Agilent Technologies MassHunter Qualitative software for peak picking and MassProfiler Professional for data alignment, statistical analysis, and visualization as described (24). Total protein content of each sample was measured concurrently and all metabolite measurements for each sample were normalized to protein. Normalized levels were compared between shunt and control lines by unpaired, two-tailed Student's t-test.
Cellular ROS measurement by flow cytometry.
To compare total cellular ROS, three shunt and three control cell lines were grown to 70–90% confluency in 6-cm plates. Prior to analysis by flow cytometry, cells were incubated per manufacturer's recommendations in media with CellROX Deep Red at a concentration of 5 μM at 37°C for 30 min. Media with Deep Red was washed off with PBS × 3 and the cells were then trypsinized with 0.25% Trypsin EDTA solution and resuspended in PBS for analysis. After preparation, cells were immediately analyzed on a BD LSR II flow cytometer using FACSDiva software. The dye was excited using a 640-nm red laser, and emission from CellROX was measured in the 650–670 nm spectra. Data were analyzed using FlowJo V10 software. Cells were gated according to forward and sidescatter parameters to exclude acellular debris and isolate singlet events. Mean and median cellular fluorescence intensity were calculated along with parameters of variance for each sample. Median fluorescence intensity between groups was compared using two-tailed unpaired Student's t-test. To generate overlay graphic, all gated cells from shunt and control samples, respectively, were concatenated into histograms based on fluorescence in the 650–670 nm spectra and plotted using FlowJo software.
Determination of mitochondrial superoxide levels and mitochondrial membrane potential.
MitoSOX Red mitochondrial superoxide indicator (Molecular Probes, Grand Island, NY), a fluorogenic dye for selective detection of superoxide in the mitochondria of live cells, was used. Briefly, cells (1 × 105) of the same passage (p8) from three control and three shunt PASMC lines were plated on into six-well plates. Twenty-four hours later, cells were washed with fresh media, incubated in media containing MitoSOX Red (5 μM) or TMRM (50 nM), for 30 min at 37°C in dark conditions, then subjected to fluorescence microscopy using an excitation of 510 nm and an emission at 580 nm (for MitoSOX) or an excitation of 548 nm and an emission at 575 nm (for TMRM). An Olympus IX51 microscope equipped with a CCD camera (Hamamatsu Photonics) was used for acquisition of fluorescent images. The average fluorescent intensities (to correct for differences in cell number) were quantified using ImagePro Plus version 5.0 imaging software (Media Cybernetics).
Measurement of NADPH oxidase activity.
Measurement of NADPH oxidase activity was performed as described previously (25). Briefly, cells were trypsinized, pelleted, then homogenized with Tris-sucrose buffer [10 mM Tris base (Fisher, Pittsburgh, PA), 340 mM sucrose (Mallinkrodt Baker, Philipsburg, NJ), 1 mM EDTA (Mallinkrodt Baker), 10 μg/ml protease inhibitor mixture (Sigma)]. The homogenate protein concentration was measured for each sample and 100 μg of homogenate protein + 100 μM NADPH substrate (Sigma) was added to an 8-mm test tube with 500 μl reaction buffer (5 μM lucigenin, 1 mM EGTA, and 50 mM phosphate buffer, pH 7.0) and then incubated at 37°C for 5 min. NADPH oxidase activity was measured by luminescence with emission measured at 15 s in a luminometer (Model TD-20/20; Turner Designs, Sunnyvale, CA).
Measurement of superoxide levels using electron paramagnetic resonance (EPR) spectroscopy.
To detect superoxide generation in intact cells, we performed electron paramagnetic resonance (EPR) measurements as described previously (49). Briefly, we used the spin probe 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrrolidine·HCl (CMH; Alexis Biochemicals, San Diego, CA). Twenty microliters of a spin-trap stock solution consisting of CMH [20 μM in Dulbecco's PBS (DPBS) plus 25 μM desferrioxamine (Calbiochem) and 5 μM diethyldithiocarbamate (Alexis Biochemicals, Lausen, Switzerland)] plus 2 μl of DMSO were added to each well before a 2 h apocynin (100 μm) treatment. Adherent cells were trypsinized and pelleted at 500 g after a 45-min incubation at 37°C following the treatment to allow entrapment of superoxide by the spin trap. Cell pellet was washed and suspended in a final volume of 35 μl of DPBS (plus desferrioxamine and diethyldithiocarbamate), loaded into a 50-μl capillary tube, and analyzed with a MiniScope MS200 EPR (Magnettech, Berlin, Germany) at a microwave power of 40 mW, modulation amplitude of 3,000 mG, and modulation frequency of 100 kHz EPR spectra. EPR signal amplitudes were analyzed using ANALYSIS 2.0 software (Magnettech). To quantitate the amount of superoxide per milligram of protein, we performed a standard reaction of the superoxide-generating enzyme xanthine oxidase as described previously (37). After the experiment, protein concentrations in mitochondria samples were measured. Final superoxide generation rate was expressed as picomoles per 40 min per milligram of protein.
RESULTS
PASMC lines from shunt lambs proliferate faster than those from controls.
Using previously described explant methods (41) primary PASMCs were derived from the proximal pulmonary arteries of shunt lambs (exposed to pulmonary overcirculation) and healthy control lambs with normal cardiac anatomy. Smooth muscle lineage was confirmed by immunohistochemical analysis of each cell line with antibodies directed against established smooth muscle lineage markers (40) including isoforms SM1 and SM2 (Sigma Aldrich M7786), and calponin (Fig. 1). Quantification of cell proliferation indicated that when plated at identical density and counted at sequential 24-h time points, PASMCs derived from shunt animals consistently exhibited significantly greater proliferation rates at all time points (Fig. 2).
Fig. 1.

Analysis of primary cell lines for smooth muscle cell markers. A: immunoblot analysis performed on whole cell protein lysates from shunt and control primary PASMC lines as well as control, non-smooth muscle cell lines with primary antibodies specifically targeting α smooth muscle actin (from mouse) and NH2-terminal actin (from rabbit), followed by incubation with anti-mouse and anti-rabbit secondary antibodies conjugated to red and green fluorophores, respectively. B: the smooth muscle lineage of each cell line was confirmed by immunofluorescence staining with anti-Calponin. All isolated cell lines from control and shunt animals express the cell lineage typical marker.
Fig. 2.
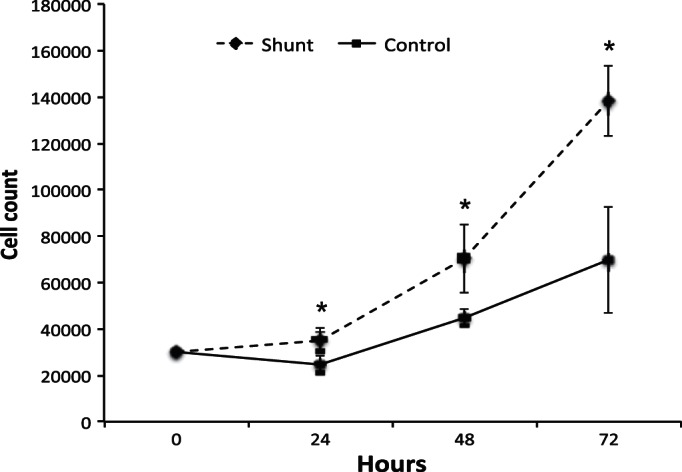
Proliferation of primary PASMCs derived from shunt animals compared with PASMCs from control animals under standard growth conditions. Cell proliferation assay comparing cell counts for primary PASMCs from shunt vs. control animals (n = 3 per group) at sequential 24-h time points. Error bars correspond to 1 SD. *Significance with P value < 0.05 by Student's t-test.
PASMCs from shunt lambs exhibit metabolic alterations distinct from advanced PAH.
Given that shunt PASMCs proliferate much more rapidly than their control counterparts, we asked whether they would also display metabolic alterations similar to what has been described in transformed cancer cells or in the PASMCs of humans with advanced PAH. To this end, we performed extracellular flux analyses using the Seahorse XF-24 Extracellular Flux Analyzer (Seahorse Bioscience, Copenhagen, Denmark). This assay provides a multiplexed platform to obtain measurements of the cellular microenvironment in culture and provides simultaneous readings of cellular oxygen consumption and extracellular acidification. The oxygen consumption rate (OCR) represents a direct measurement of mitochondrial respiration while the extracellular acidification rate (ECAR) is an indirect measure of glycolytic lactate production (12). However, other acidification mechanisms may also contribute to ECAR, including carbonic acid produced by conversion of TCA cycle-derived CO2 by carbonic anhydrases (29). Figure 3A shows the basal OCR readings at sequential time points within a single representative assay for one shunt and one control cell line. Figure 3B shows the composite average of 3 independently derived shunt and control cell lines under basal conditions normalized across multiple experiments. Fitting with our initial hypothesis, shunt PASMCs showed a 66% decrease in the rate of oxygen consumption compared with control cells (P < 0.001). Since the standard oxygen consumption measurements were performed in specially prepared media lacking the supplemental BSA in which the cells were routinely cultured, we were concerned that the assay may be missing a significant fraction of oxygen consumption related to the oxidation of exogenous fatty acids. We therefore repeated basal measurements with the addition of conjugated palmitate:albumin and found that although OCR did indeed increase by approximately 25% and 35% in controls and shunts, respectively (data not shown), the shunt PASMCs continued to have significantly lower O2 consumption rates than control PASMCs, and the relative rate of decrease was maintained. Surprisingly, however, concurrent measurements of ECAR indicated that PASMCs from the shunted animals also exhibited substantially lower rates of basal extracellular acidification. As seen in Fig. 3, C and D, PASMCs derived from shunt animals consistently exhibited a 76% decrease in ECAR (P < 0.001) compared with controls. While some of the decrease in extracellular acidification may be related to decreased CO2 production from the suppression of mitochondrial oxygen consumption, there is clearly no countervailing increase from glycolytic lactate production. Notably, the relative decrease in ECAR in these cells is even greater than the decrease in oxygen consumption. This finding represents a major deviation from the expected Warburg phenotype, which would couple a decrease in aerobic metabolism with a substantial increase in glycolytic activity, presumably to help supply biosynthetic precursor production required for enhanced cell division.
Fig. 3.

Primary PASMC metabolism in shunt vs. control cells. A: oxygen consumption rate (OCR) measurement under basal conditions with standard media comparing one shunt (dashed) and one control (solid) cell line from a single experiment. Error bars correspond to 1 SD. B: normalized oxygen consumption rate measurements comparing shunt and control (n = 3 per group) cell lines under basal conditions in standard media. Error bars correspond to 1 SD. *Significance with P value <0.001 by Welch's t-test. C: extracellular acidification rate (ECAR) measurement under basal conditions with standard media comparing one shunt (dashed) and one control (solid) cell line from a single experiment. Error bars correspond to 1 SD. D: normalized extracellular acidification rate measurements comparing shunt and control (n = 3 per group) cell lines under basal conditions in standard media. Error bars correspond to 1 SD. *Significance with P value <0.001 by Welch's t-test.
Despite the evidence that both mitochondrial oxidation and glycolytic flux are decreased in shunt compared with control cells, these cells do not exhibit any evidence of bioenergetic compromise. In addition to generating sufficient energy to carry out the biosynthetic processes required to facilitate their increased rate of proliferation, the shunt cells showed no deficit in the levels of the energetic intermediates ATP or NADH and no increase in the respective monophosphate or oxidized forms of these cofactors (Fig. 4). In fact, the level of AMP is slightly decreased in the shunt cells, resulting in an increased ATP-to-AMP ratio in these cells, typically an indicator of abundant energy supplies.
Fig. 4.

Targeted analysis of energetic intermediate metabolites in shunt and control primary PASMCs. Selected cofactors were measured in shunt and control PASMCs using a targeted approach with tandem HPLC/QTOF mass spectrometry. Metabolites were quantified by comparison with 13C labeled internal standards included at known concentration during the experimental extraction and analysis procedures. Results are normalized to total protein content of the sample with concentrations of metabolites expressed in picomoles/microgram of protein. Error bars indicate 1 SD. *Significance by Student's t-test with P value < 0.05.
Glycolytic intermediates are decreased while pentose phosphate pathway intermediates are increased in shunt PASMCs.
Given that extracellular flux analyses can be limited, particularly when attempting to investigate glycolytic flux, we performed HPLC-mass spectrometry-based quantitative metabolite analyses in shunt and control PASMCs. We screened targeted metabolites from three of the central carbohydrate catabolic pathways: glycolysis, the pentose phosphate shunt, and the TCA cycle. Figure 5A shows results for specific intermediates of the glycolytic pathway in shunt and control cells. Fructose-1,6-bisphosphate is the first energetically committed metabolite of glycolysis, with preceding intermediates able to flow freely into other catabolic and anabolic pathways. Phosphoenolpyruvate (PEP) is the final pathway intermediate before the glycolytic production of pyruvate. Both were found at significantly lower levels in the shunt cells compared with controls (Fig. 5A). When interpreted in the context of the overall decrease in extracellular acidification rates, these results are consistent with an overall reduction in glycolytic flux in shunt PASMCs. While the ratio of fructose-6P to glucose-6-P does not reach statistical significance due to large variance in the control cells, the increased ratio in control cells is similarly consistent with this model. In contrast, Fig. 5B shows that specific intermediates of the pentose phosphate pathway (PPP) are increased in shunt PASMCs. d-Sedoheptulose-7-P is a unique seven-carbon sugar of the nonoxidative branch of the PPP, and erythritol is a metabolic product/by-product of this pathway. There is also an increased ratio of ribose-5-P relative to xylulose-5-P, suggesting a directionality of flow within the pathway towards production of ribose, a necessary precursor for DNA synthesis. Taken together, our results indicate an overall shift in glucose catabolism away from glycolysis and towards the PPP. We additionally measured pertinent energetic cofactors including NAD+ and NADH, ATP/AMP, and GTP/GMP (Fig. 4). Importantly, these results indicate that overall cellular bioenergetic status is not compromised in shunt PASMCs.
Fig. 5.

Targeted metabolite analysis in shunt vs. control primary PASMCs. Selected metabolites were measured in shunt and control PASMCs using a targeted approach with tandem HPLC/QTOF mass spectrometry. Metabolites were quantified by comparison with 13C labeled internal standards included at known concentration during the experimental extraction and analysis procedures. Results are normalized to total protein content of the sample with concentrations of metabolites expressed in picomoles/microgram of protein. Error bars indicate 1 SD. *Significance by Student's t-test with P value < 0.05. A: targeted analysis of intermediates from the glycolytic pathway including fructose-1,6-bisphosphate, phosphoenolpyruvate and the ratio of fructose-6-phosphate to glucose-6-phosphate. B: targeted analysis of intermediates from the pentose phosphate pathway including d-sedoheptulose-7-phosphate, erythritol and the ratio of ribose-5-phosphate to xylulose-5-phosphate. C: comparison of acetyl-CoA levels in shunt and control PASMCs. D: targeted analysis of intermediate metabolites from the TCA cycle including succinate, malate, and the ratio of citrate to isocitrate.
Shunt PASMCs exhibit accumulation of acetyl CoA and alterations in the TCA metabolite malate.
From the same targeted metabolic analysis, we found that shunt PASMCs have increased levels of acetyl-CoA (Fig. 5C). While levels of acetyl-CoA are below the detection threshold of the assay (<0.1 nanomoles per plate) in all of the control cell lines analyzed, the shunt cells exhibit a significant accumulation. This finding suggests that control PASMCs, with their relatively high oxygen consumption, are more rapidly utilizing acetyl-CoA as a fuel source for mitochondrial respiration while the shunt cells, and their corresponding lower oxygen consumption rates, are accumulating acetyl-CoA due to downstream suppression of the TCA cycle and/or mitochondrial electron transport chain inhibition. Of particular note, this elevation in acetyl-CoA levels is inconsistent with the observed decrease in this metabolite in PASMCs from advanced disease due to HIF-1α mediated suppression of mitochondrial respiration through regulation of pyruvate dehydrogenase (20, 32), which occurs upstream of acetyl-CoA production. It is thus quite interesting that there are distinct differences found within the TCA cycle itself. While the ratio of citrate to isocitrate, and succinate levels are not significantly different between control and shunt PASMC lines, the late cycle intermediate malate is significantly decreased in shunt cells (Fig. 5D).
Shunt PASMCs do not exhibit increased basal levels of HIF-1α or PDHK and are insensitive to treatment with dichloroacetate.
While shunt PASMCs show evidence of suppressed mitochondrial respiration consistent with the Warburg phenotype described in advanced pulmonary hypertension, the finding of elevated acetyl-CoA led us to question a similar role of HIF-1α mediated regulation in our model. Toward that end, we evaluated levels of HIF-1α and PDHK proteins in shunt and control cells in culture at atmospheric oxygen tension. The results, shown in Fig. 6, A and B, reveal that HIF-1α levels are nearly undetectable under these conditions in both shunt and control cells. Consistent with this, PDHK levels are also not increased in shunt PASMCs. To test whether pharmacological PDHK inhibition would affect PASMC proliferation, we cultured shunt and control PASMCs with the PDHK inhibitor DCA. As shown in Fig. 6C, at standard dosing concentrations described for cell culture (0.5 mM to 1 mM), there is no growth inhibitory effect of DCA on either shunt or control PASMCs. At higher doses, however (5 mM), cell line-independent repression of cell proliferation was observed, likely due to toxic effects.
Fig. 6.

Evaluation of HIF-1α/PDHK activity in shunt and control primary PASMCs. A: immunoblot analysis of whole cell lysates from shunt and control PASMCs with antibodies specifically targeting HIF-1α and tubulin. B: immunoblot analysis of whole cell lysates from shunt and control PASMCs with antibodies specifically targeting PDHK and tubulin. Quantification was performed using LiCor ImageStudio software and intensities of PDHK were normalized to intensity of tubulin per lane. Averages were compared by Student's t-test and not significantly different. Error bars correspond to 1 SD. C: cell proliferation assay comparing growth of shunt vs. control primary PASMCs when cultured in standard assay medium with addition of dichloroacetate over a dose range from 0.5 to 5 mM. All growth results are normalized to growth in standard medium with drug vehicle only. Error bars correspond to 1 SD. P value > 0.05 for all comparisons of shunt vs. control.
Supplementation with cell-permeable dimethyl malate (DMM) does not alter the oxygen consumption or growth phenotype of shunt PASMCs.
These findings support the implication that the decrease in mitochondrial respiration we observe in the shunt PASMCs is not driven by the HIF-1α/PDHK upregulation found in advanced disease, but rather due to some limitation of the TCA cycle or intrinsic to shunt mitochondria themselves. In this context, the finding of decreased malate in these cells becomes an intriguing finding that may point to TCA cycle insufficiency as an underlying cause of the observed mitochondrial suppression. Malate and its metabolism have also been implicated in abnormal cell growth. Hereditary deficiency of fumarase, the enzyme that converts fumarate to malate, results in a cancer syndrome characterized by renal cell carcinoma and benign smooth muscle tumors (23). Conversely, in non-small cell lung cancers, overexpression of an abnormal mitochondrial malic enzyme diverts malate away from the TCA cycle and facilitates rapid cell growth. Malic enzyme knockdown or supplementation with cell permeable dimethyl malate (DMM) enhances O2 consumption and slows proliferation (39). To test the hypothesis that malate deficiency may underlie mitochondrial repression in our model, we supplemented shunt and control cells with 5 mM DMM and evaluated proliferation and O2 consumption. Figure 7A shows that the addition of DMM to standard assay media did not significantly alter O2 consumption, by either shunt or control cells. Similarly, supplementation of growth media with DMM resulted in no significant change in proliferation of either shunt or control cells (Fig. 7B).
Fig. 7.

Analysis of growth and oxygen consumption with addition of cell-permeable malate analog DMM. A: oxygen consumption rate of shunt and control cell lines under basal conditions and following addition of dimethyl malate to standard media at a final concentration of 5 mM. Error bars correspond to 1 SD. No significant difference between groups by Student's t-test. B: cell proliferation assay comparing growth of shunt vs. control primary PASMCs when cultured in standard assay medium with addition of dimethyl malate at a concentration of 5 mM. All growth results are normalized to growth in standard medium with drug vehicle only. Error bars correspond to 1 SD. No significant difference between groups by Student's t-test.
Shunt PASMCs exhibit increased cellular ROS and oxidative stress responses.
The oxidative arm of the PPP, controlled primarily by activity of the rate-limiting enzyme glucose-6-phosphate dehydrogenase (G6PDH), serves as a major source of reduced NADPH within the cell (Fig. 8A). Despite evidence that metabolic intermediates of the pentose phosphate pathway are increased, the level of the oxidized cofactor NADP+ is markedly elevated and the ratio of NADPH/NADP+ is significantly decreased in the shunt PASMCs (Fig. 8B). It initially seems paradoxical that the cofactor is skewed so heavily towards the oxidized form if overall PPP activity is increased; however, the shift towards NADP+ at the expense of NADPH has been shown to be a potent activator of G6PDH and PPP activity (7). Furthermore, other authors have speculated that this ratio is an important determinant of G6PDH activity in the pulmonary arteries (15), and in rodent models of PAH, G6PDH has been shown to play a role in the regulation of PASMC growth and contractile phenotypes (8). In its reduced form, NADPH is an important enzymatic cofactor in a number of essential cellular processes including the reductive biosynthesis of fatty acids, nucleotides, amino acids and cholesterols, generation of superoxides via NADPH oxidases, and protection of cells from free radical damage through the regeneration of reduced glutathione (7). It has been shown previously in this animal model that lung tissue shows an increase in NADPH dependent superoxide generation (44), and other investigators have established that cyclic stretch increases levels of cytosolic ROS in PASMCs (48). We therefore hypothesized that the alteration in the NADPH/NADP+ in our cells may represent a state of increased NADPH consumption secondary to cellular oxidative stress. To evaluate the oxidative state of the cells we treated them with CellROX Deep Red, an agent that fluoresces in proportion to the amount of total cellular ROS, and analyzed the cellular fluorescence intensity by flow cytometry. As shown in Fig. 8C, the mean fluorescence intensity of the shunt PASMCs was indeed elevated by 24.7% compared with that of controls (P < 0.05). This is consistent with other models of pulmonary hypertension which have described increased cellular ROS originating from a variety of sources (33, 48).
Fig. 8.

Cellular oxidative stress and response in shunt and control primary PASMCs. A: initial reactions of the oxidative arm of the pentose phosphate pathway showing the conversion of glucose to ribulose-5-phosphate with the generation of 2 equivalents of the reduced cofactor NADPH per oxidized glucose. B: targeted comparison of NADP and NADPH in shunt and control primary PASMCs by HPLC/QTOF mass spectrometry. Metabolites were quantified by comparison with 13C labeled internal standards with results normalized to total protein content of the sample with concentrations of metabolites expressed in picomoles/microgram of protein. Error bars indicate 1 SD. *Significance by Student's t-test with P value < 0.05. C: comparison of total cellular ROS of shunt and control cells following treatment with CellROX. Fluorescence intensity in the 650-607 nm spectra of gated singlet events is shown on a logarithmic scale along the x-axis. The shunt primary cell population is shown in the histogram with darker outline, and control cells in histogram with the lighter outline.
Cellular ROS may originate from various sources, but in the lungs and pulmonary vasculature they primarily arise from the NADPH oxidase (Nox) complexes, uncoupled nitric oxide synthase (NOS) and the mitochondrial electron transport chain (2). Both mitochondria and Nox complexes have been implicated as sources of increased smooth muscle cell ROS in other models of PAH (2, 4). Given our findings of altered mitochondrial respiration, we first sought to determine whether the mitochondria accounted for the increase in cellular ROS levels. We specifically measured mitochondrial ROS with the selective dye MitoSox Red, and found that the shunt PASMCs exhibited reduced mitochondrial ROS levels (Fig. 9A). Consistent with their diminished activity, mitochondria in shunt PASMCs also had lower membrane potentials (Fig. 9B). Thus the increase in cellular ROS observed in shunt PASMCs does not appear to originate from their mitochondria.
Fig. 9.

Analysis of mitochondrial ROS and membrane potential in shunt and control primary PASMCs. A: immunofluorescence imaging and quantification of MitoSox mean fluorescence intensity in shunt and control PASMCs under basal conditions. B: immunofluorescence imaging and quantification of mitochondrial membrane potential using TMRM in shunt and control primary PASMCs under basal conditions. (n = 3, *P < 0.05).
Shunt PASMCs exhibit increased NADPH oxidase expression and activity which contributes to their hyperproliferative phenotype.
We then turned our attention to the Nox system as a potential source of increased ROS in shunt PASMCs. The Nox complexes are transmembrane multimers that catalyze the formation of superoxide (O2−) by electron transfer from NADPH to molecular O2. There are several catalytic isoforms, but Nox 1, 2, 4 and 5 have been described in the pulmonary vasculature. Some isoforms, such as 4, are constitutively active, while others require stabilization and activation by other subunits to function (21). We first looked at expression of the Nox complexes and found increased protein levels of both the Nox 4 and Nox 2 catalytic domains, along with the Rac1 and p47phox subunits of the Nox 2 complex (Fig. 10A). We then evaluated total NADPH oxidase activity in cellular homogenates using an established chemiluminescence assay (23) and demonstrated a fivefold increase in total Nox activity in shunt compared with control PASMCs (Fig. 10B). To validate this result in intact cells, we performed electron paramagnetic resonance (EPR) measurement as previously described (49) to measure Nox-dependent superoxide generation. As seen in Fig. 10C, shunt PASMCs exhibited a 115% increase in Nox-dependent superoxide production compared with control cells. We then sought to determine what effect, if any, this increase in cellular Nox expression and activity was exerting on the cellular proliferation phenotype. To this effect we utilized the small molecule VAS 3947 to pharmacologically inhibit Nox activity and evaluate the impact on PASMC proliferation. VAS 3947 is a commercially available triazolo-pyrimidine compound that inhibits Nox activity and is notable for its lack of off-target effects (notably including NOS and xanthine oxidase) and the absence of intrinsic antioxidant activity (3). VAS 3947 (10 μM) preferentially slowed the growth of shunt PASMCs in vitro (17.3% slower than standard growth conditions) with no significant effect on the growth of control PASMCs (Fig. 10D). We therefore conclude that increased Nox activity may be a driver of PASMC hyperproliferation and metabolic shift in the setting of pulmonary overcirculation.
Fig. 10.

NADPH oxidase expression, activity, and effect on proliferation in shunt and control primary PASMCs. A: immunoblot analysis of whole cell lysates from shunt and control PASMCs with antibodies specifically targeting Nox 2, p47phox, and Rac-1 with β-actin control. Densitometry was performed using a public domain Java image processing program, ImageJ (NIH Image), and intensities of were normalized to intensity β-actin per lane. Error bars indicate 1 SD. *Significance by Student's t-test with P value < 0.05. B: NADPH oxidase activity of shunt and control cell homogenate by luminescence assay. Error bars indicate 1 SD. *Significance by Student's t-test with P value < 0.05. C: comparison of NADPH-dependent superoxide production in intact shunt and control PASMCs using electron paramagnetic resonance. Error bars indicate 1 SD. *Significance by Student's t-test with P value < 0.05. D: cell proliferation assay comparing growth of shunt vs. control primary PASMCs when cultured in standard assay medium and assay medium with addition of VAS 3947 at 10 μM. Growth is normalized to controls in standard media at 72 h. *Significant differences by Student's t-test with P value < 0.05 between shunt cells in standard medium compared with VAS medium, and between shunt and control cells in VAS medium. There was no significant difference between growth of control cells in standard compared with VAS medium.
DISCUSSION
In this set of studies, we sought to characterize whether metabolic alterations similar to those observed in advanced PAH would also be observed in a unique model of early pulmonary vascular disease induced by pulmonary overcirculation. It is well established that in advanced pulmonary hypertension of various etiologies, PASMC and PAEC hyperproliferation is frequently associated with aberrations in the central metabolic pathways of glycolysis and mitochondrial respiration. Furthermore, recent studies by Bonnett et al. (6) and McMurtry et al. (28), wherein pharmacologic de-repression of mitochondrial glucose oxidation improves pulmonary hemodynamics and mortality, strongly suggest that cellular metabolic changes may be an important driver of pathological vascular remodeling in the setting of PAH. In addition, these studies provide proof of concept that targeted intervention to reverse these metabolic alterations may be able to slow or even reverse disease progression. Importantly, a similar alteration in central metabolic pathways in transformed cancer cells has suggested parallels between the hyperproliferative nature of malignant cells and vascular cells in PAH (46) and these pathways are currently under intense investigation in the search for novel chemotherapeutic agents (9, 16, 18). Our model of pulmonary overcirculation-induced pulmonary vascular remodeling enables us to study varied time points in disease evolution. By investigating and characterizing metabolic alterations at an early stage of disease we hope to gain insights into the proximal regulatory stimuli and pathways to potentially identify novel therapeutic targets that can be utilized prior to evolution of irreversible pathology. Importantly, this is a highly feasible, and currently unavailable treatment strategy for children born with congenital heart lesions that will ultimately lead to PAH as a result of increased pulmonary blood flow.
Consistent with vascular changes observed in vivo (17), we observed that PASMCs derived from shunt animals exposed to increased pulmonary blood flow proliferated at an increased rate compared with those derived from control animals. This phenotype was retained following multiple passages in vitro, indicating that the observed alterations represent an intrinsically altered state that is programmed in vivo and maintained ex vivo. Our large animal model of congenital heart disease therefore provides a robust platform for investigation of the mechanisms driving the hyperplastic behavior of vascular smooth muscle cells in response to increased PBF.
Based on our hypothesis that PASMC hyperproliferation would be accompanied by metabolic changes similar to those observed in advanced PAH in rat and human models (13, 50), we undertook a targeted metabolomic analysis of shunt vs. control PASMCs. We found an abnormal but unique metabolic phenotype in this model of early pulmonary vascular dysfunction induced by pulmonary overcirculation. While shunt PASMCs clearly show evidence of repressed mitochondrial glucose oxidation, we did not observe the typical commensurate increase in glycolytic lactate production. On the contrary, the data from our extracellular flux analyses suggested that glycolytic lactate production in these cells was also suppressed. Our targeted metabolic analyses confirmed alterations in glucose catabolism and, in conjunction with the changes in extracellular flux, pointed to a shift in glucose flux away from glycolysis and towards the PPP. Our data also suggested an altered directionality within the PPP, favoring production of ribose-5-phosphate. This shift favoring production of metabolites required for the synthesis of ribonucleic acids and other macromolecules is a long hypothesized function of Warburg metabolism (18) and may be an important factor supporting the hyperproliferative nature of PASMCs in the setting of pulmonary overcirculation. Furthermore, the activity of G6PDH, the first and rate-limiting step of the oxidative PPP, has been directly implicated in promoting PASMC growth in models of hypoxic PAH (8). The observed shift in the NADP+/NADPH ratio towards the oxidized state in the shunt PASMCs is expected to act as a potent activator of G6PDH, driving glucose-6-P towards the PPP at the expense of glycolysis, consistent with our observations. This raises the interesting possibility that the observed shifts in glucose metabolism in these cells may be modulated, at least in part, by the relative oxidation state of this cofactor.
The presence of a decreased NADPH/NADP+ ratio despite evidence of increased glucose flux through the oxidative PPP led us to further investigate this seemingly incongruous finding. We hypothesized that this pattern could be consistent with rapid ongoing consumption of NADPH and turned our attention to the role of the reduced cofactor. NADPH serves important functions in the reductive biosynthesis of macromolecules, particularly complex lipids and fatty acids, as well as in the cellular antioxidant response. As the cofactor responsible for regenerating both reduced glutathione and thioredoxin, the level of NADPH is tightly linked to the intracellular oxidative environment (7). Abnormalities of redox status and ROS are ubiquitous in the PH literature (2, 4) and have been previously established in this model of pulmonary vascular dysfunction (1), suggesting that this might be a good initial focus for our investigation. We demonstrated an increase in total cellular ROS in the shunt PASMCs but when we looked at mitochondria as a potential source we found that mitochondrial ROS were actually decreased. We next turned our attention to the NADPH oxidase system, which had been previously shown to exhibit increased activity in PASMCs exposed to mechanical stressors (48). We found increased expression of multiple Nox isoforms, including the constitutively active Nox 4, along with increased overall Nox activity and superoxide production. When we subjected the PASMCs to targeted pharmacological inhibition of the Nox system, we demonstrated a selective slowing effect on the hyperproliferative shunt PASMCs. From these findings, we believe that the cofactor NADPH, and specifically the NADPH/NADP+ ratio, may represent a central convergence point linking cellular signaling induced by mechanical forces with the intracellular redox status and central carbohydrate metabolism. Through both active consumption of NADPH as a cofactor, and indirect requirement via the increase in cellular superoxide production, the Nox system is heavily favoring NADP+ at the expense of NADPH and creating a potent stimulus driving the oxidative arm of the PPP. Furthermore, this constellation of changes is actively promoting the abnormal, hyperproliferative nature of these PASMCs that have been exposed to increased pulmonary blood flow. Further study is required to determine if this effect on cellular proliferation is mediated through a direct impact of increased superoxide on redox sensitive pathways (5), or via induced activity of G6PDH and the PPP (8).
Prevailing models of the aerobic glycolysis described in end-stage pulmonary hypertension highlight the role of HIF-1α in both the repression of mitochondrial oxidation as well as the upregulation of glycolytic flux (6, 13, 46). In our model, we witness a phenotype of repressed aerobic respiration that is independent of HIF-mediated inhibition of mitochondrial substrate flux. In contrast, these cells exhibit abnormally increased intracellular concentrations of acetyl-CoA, the basic molecular fuel for the TCA cycle. The diminished oxygen consumption observed in these cells therefore suggests a primary repression of mitochondrial respiratory activity. This is corroborated by the relative decrease in the mitochondrial membrane potential of these cells, suggesting an overall reduction in electron transport. These findings may stem from multiple underlying causes, such as altered capacity of the TCA cycle, decreased function of the mitochondrial electron transport chain, or both. It thus seems highly significant that we find distinctions in these cells with respect to intermediate metabolites of the TCA cycle. Malate levels may be decreased in these cells due to decreased production, increased flow through the TCA cycle catalyzed by malate dehydrogenase, or loss to another enzymatic pathway. Given that oxidation of malate to oxaloacetate (OAA) by malate dehydrogenase is highly energetically unfavorable and depends largely on the subsequent condensation of OAA with acetyl-CoA to drive the reaction forward (47), it seems unlikely that this is the underlying mechanism. Attempts to replenish mitochondrial malate stores with the membrane permeable analog DMM proved ineffective at restoring cellular oxygen consumption and attenuating growth, indicating that low malate concentration is not the sole rate-limiting deficiency. One possible explanation would be an induced or acquired dysfunction of the electron transport chain itself, which could result in high acetyl-CoA, low cellular O2 consumption, low mitochondrial membrane potential, and depletion of TCA cycle intermediates. This type of primary ETC dysfunction has been described previously by Rafikov et al. (36) in a monocrotaline model of PAH, in which mitochondrial repression was caused by a loss of Complex I assembly and activity. An alternative possibility is suggested by metabolic abnormalities observed in glioblastoma and melanoma cell lines. Using different adaptive mechanisms, both cell types subvert the typical TCA cycle progression to promote citrate efflux out of the mitochondria into the cytoplasm, where it gives rise to a pool of cytoplasmic acetyl-CoA required for de novo fatty acid synthesis (13a). Glioblastoma lines use glutamine as an alternative carbon source to maintain flow through the back half of the TCA cycle and provide a steady source of OAA (11), while melanoma lines exhibit reverse flux through the cycle to convert glutamine to citrate (14). Both mechanisms result in low cellular oxygen consumption and could potentially account for low mitochondrial membrane potential, diminished malate, and an abundance of acetyl-CoA sequestered in the cytoplasm where it cannot be oxidized through the TCA cycle.
Although physiological evidence of mitochondrial dysfunction is well established (4), to our knowledge, this description of alterations in the TCA cycle itself is novel in the field of pulmonary hypertension. Coupled with the findings of HIF-independent mitochondrial repression and decreased mitochondrial membrane potential, the metabolic phenotype we observe diverges significantly from what is widely described in advanced PAH in adults. A possible explanation for this distinction is that the metabolic and redox alterations at this early stage of pulmonary vascular dysfunction ultimately facilitate HIF accumulation and secondary metabolic reprogramming leading to the more typical aerobic glycolysis phenotype with advancing disease. Alternatively, the metabolic and cellular alterations observed in PASMCs derived from animals exposed to chronically increased PBF may represent a phenotype unique to this model of PAH.
In this study we describe a unique metabolic phenotype in PASMCs at an early stage of pulmonary vascular dysfunction from a clinically relevant model of pulmonary overcirculation. We show evidence of altered glucose catabolism through the glycolytic and PPP, with preferential redirection of flow towards the oxidative branch of the PPP. This is accompanied by an increase in NADPH oxidase expression and superoxide production that we believe is driving increased PPP activity and is contributing to the hyperproliferative phenotype of these cells. We have also shown the early emergence of suppressed mitochondrial respiration that seems to be mediated through primary alterations of the TCA cycle and/or mitochondrial electron transport chain. These findings suggest that proposed interventions targeting HIF activity and the associated increase in aerobic glycolysis may prove ineffective at reversing the underlying metabolic drivers of PASMC hyperproliferation in certain settings. Concurrently, the convergence of increased Nox activity, cellular oxidative stress, and PPP activation emerges as a promising therapeutic target in our model of pulmonary vascular dysfunction. Further investigation is required to understand this phenotype with greater clarity and appropriately target metabolic therapies. Importantly, this model directly relates to children born with CHD who are typically diagnosed prenatally or shortly after birth, and who do not develop pulmonary vascular pathology due to overcirculation until months of age at the earliest (35). This is a group of patients that would readily lend itself to early and even prophylactic intervention if such treatments were available.
GRANTS
This study was supported by Ruth L. Kirschstein National Research Service Award 5-T32-HD-049303-09, and National Institutes of Health Grants HD-072455, HL-60190, HL-061284, HL-67841, and HL-0101902.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
J.B., S.D., R.J.K., J.X.-J.Y., J.R.F., S.M.B., and E.M. conception and design of research; J.B., X.S., K.T., W.G., M.K., R.J.K., and G.W.R. performed experiments; J.B., X.S., K.T., W.G., M.K., S.D., R.J.K., J.X.-J.Y., J.R.F., S.M.B., and E.M. analyzed data; J.B., X.S., K.T., W.G., M.K., S.D., R.J.K., J.X.-J.Y., J.R.F., S.M.B., and E.M. interpreted results of experiments; J.B., X.S., K.T., M.K., S.M.B., and E.M. prepared figures; J.B. and E.M. drafted manuscript; J.B., S.D., R.J.K., J.X.-J.Y., J.R.F., S.M.B., and E.M. edited and revised manuscript; J.B. and E.M. approved final version of manuscript.
REFERENCES
- 1.Aggarwal S, Gross C, Fineman JR, Black SM. Oxidative stress and the development of endothelial dysfunction in congenital heart disease with increased pulmonary blood flow: lessons from the neonatal lamb. Trends Cardiovasc Med 20: 238–246, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aggarwal S, Gross CM, Sharma S, Fineman JR, Black SM. Reactive oxygen species in pulmonary vascular remodeling. Compr Physiol 3: 1011–1134, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Altenhöfer S, Radermacher KA, Kleikers PWM, Wingler K, Schmidt HH. Evolution of NADPH oxidase inhibitors: selectivity and mechanisms for target engagement. Antioxid Redox Signal 23: 406–427, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JGN, Weir EK. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol 294: H570–H578, 2008. [DOI] [PubMed] [Google Scholar]
- 5.Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, Svensson EC, Dyck JRB, Gomberg-Maitland M, Thébaud B, Husain AN, Cipriani N, Rehman J. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation 121: 2661–2671, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thébaud B, Bonnet S, Haromy A, Harry G, Moudgil R, McMurtry MS, Weir EK, Archer SL. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 113: 2630–2641, 2006. [DOI] [PubMed] [Google Scholar]
- 7.Chandel N. Navigating Metabolism (1st ed.). Cold Spring Harbor Laboratory Press, 2015. [Google Scholar]
- 8.Chettimada S, Gupte R, Rawat D, Gebb SA, McMurtry IF, Gupte SA. Hypoxia-induced glucose-6-phosphate dehydrogenase overexpression and -activation in pulmonary artery smooth muscle cells: implication in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 308: L287–L300, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452: 230–233, 2008. [DOI] [PubMed] [Google Scholar]
- 10.Cohen J. Applied Multiple Regression/Correlation Analysis for the Behavioral Sciences (vol. 1) [Online]. New York: Taylor & Francis, 1983. [Google Scholar]
- 11.Deberardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci USA 104: 19345–19350, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferrick a D, Neilson A, Beeson C. Advances in measuring cellular bioenergetics using extracellular flux. Drug Discov Today 13: 268–274, 2008. [DOI] [PubMed] [Google Scholar]
- 13.Fijalkowska I, Xu W, Comhair SA, Janocha AJ, Mavrakis LA, Krishnamachary B, Zhen L, Mao T, Richter A, Erzurum SC, Tuder RM. Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Pathol 176: 1130–1138, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13a.Filipp FV, Ratnikov B, De Ingeniis J, Smith JW, Osterman AL, Scott DA. Glutamine-fueled mitochondrial metabolism is decoupled from glycolysis in melanoma. Pigment Cell Melanoma Res 25: 732–739, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Filipp FV, Scott DA, Ronai ZA, Osterman AL, Smith JW. Glutamine-fueled mitochondrial metabolism is decoupled from glycolysis in melanoma. Proc Natl Acad Sci USA 25: 375–383, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupte SA, Wolin MS. Oxidant and redox signaling in vascular oxygen sensing: implications for systemic and pulmonary hypertension. Antioxid Redox Signal 10: 1137–1152, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 144: 646–674, 2011. [DOI] [PubMed] [Google Scholar]
- 17.Heath D, Edwards JE. The pathology of hypertensive pulmonary vascular disease; a description of six grades of structural changes in the pulmonary arteries with special reference to congenital cardiac septal defects. Circulation 18: 533–547, 1958. [DOI] [PubMed] [Google Scholar]
- 18.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324: 1029–33, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hoage T, Ding Y, Xu X. Cardiovascular development. Cardiovasc Dev Methods Protoc 843: 11–21, 2012. [Google Scholar]
- 20.Kim J, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3: 177–85, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Lassegue B, San Martin A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res 110: 1364–1390, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee SD, Shroyer KR, Markham NE, Cool CD, Voelkel NFTR. Monoclonal endothelial cell proliferation is present in primary but not secondary pulmonary hypertension. J Clin Invest 101: 927–934, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Linehan WM, Rouault TA. Molecular pathways: fumarate hydratase-deficient kidney cancer—targeting the Warburg effect in cancer. Clin Cancer Res 19: 3345–3352, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lorenz MA, Burant CF, Kennedy RT. Reducing time and increasing sensitivity in sample preparation for adherent mammalian cell metabolomics. Anal Chem 29: 997–1003, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu Q, Wainwright MS, Harris VA, Aggarwal S, Hou Y, Rau T, Poulsen DJ, Black SM. Increased NADPH oxidase-derived superoxide is involved in the neuronal cell death induced by hypoxia-ischemia in neonatal hippocampal slice cultures. Free Radic Biol Med 53: 1139–1151, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mata-Greenwood E, Grobe A, Kumar S, Noskina Y, Black SM. Cyclic stretch increases VEGF expression in pulmonary arterial smooth muscle cells via TGF-beta1 and reactive oxygen species: a requirement for NAD(P)H oxidase. Am J Physiol Lung Cell Mol Physiol 289: L288–L298, 2005. [DOI] [PubMed] [Google Scholar]
- 27.Mcmurtry MS, Archer SL, Altieri DC, Bonnet S, Haromy A, Harry G, Bonnet S, Puttagunta L, Michelakis ED. Gene therapy targeting survivin selectively induces pulmonary vascular apoptosis and reverses pulmonary arterial hypertension. J Clin Invest 115: 1479–1491, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McMurtry MS, Bonnet S, Wu X, Dyck JRB, Haromy A, Hashimoto K, Michelakis ED. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ Res 95: 830–840, 2004. [DOI] [PubMed] [Google Scholar]
- 29.Mookerjee SA, Goncalves RLS, Gerencser AA, Nicholls DG, Brand MD. The contributions of respiration and glycolysis to extracellular acid production. Biochim Biophys Acta 1847: 171–181, 2015. [DOI] [PubMed] [Google Scholar]
- 30.Oishi P, Datar SA, Fineman JR. Advances in the management of pediatric pulmonary hypertension. Respir Care 56: 1314-39; discussion 1339-40, 2011. [DOI] [PubMed] [Google Scholar]
- 31.Oishi P, Datar SA, Fineman JR. Advances in the management of pediatric pulmonary hypertension. Respir Care 56: 1314–1340, 2011. [DOI] [PubMed] [Google Scholar]
- 32.Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 3: 187–197, 2006. [DOI] [PubMed] [Google Scholar]
- 33.Perez-Vizcaino F, Cogolludo A, Moreno L. Reactive oxygen species signaling in pulmonary vascular smooth muscle. Respir Physiol Neurobiol 174: 212–220, 2010. [DOI] [PubMed] [Google Scholar]
- 34.Prapa M, Mccarthy KP, Dimopoulos K, Sheppard MN, Krexi D, Swan L, Wort SJ, Gatzoulis MA, Yen S. Histopathology of the great vessels in patients with pulmonary arterial hypertension in association with congenital heart disease: large pulmonary arteries matter too. Int J Cardiol 168: 2248–2254, 2013. [DOI] [PubMed] [Google Scholar]
- 35.Rabinovitch M. Pathobiology of pulmonary hypertension: impact on clinical management. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu 3: 63–81, 2000. [DOI] [PubMed] [Google Scholar]
- 36.Rafikov R, Sun X, Ra O, Louise M, Desai AA, Khalpey Z, Yuan JX, Fineman JR, Black SM. Redox Biology Complex I dysfunction underlies the glycolytic switch in pulmonary hypertensive smooth muscle cells. Redox Biol 6: 278–286, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rafikova O, Rafikova R, Kumar S, Sharma S, Aggarwal S, Schneider F, Jonigk D, Black SM, Tofovic S. Bosentan inhibits oxidative and nitrosative stress and rescues occlusive pulmonary hypertension. Free Radic Biol Med 56: 28–43, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reddy VM, Meyrick B, Wong J, Khoor A, Liddicoat JR, Hanley FL, Fineman JR. In utero placement of aortopulmonary shunts: a model of postnatal pulmonary hypertension with increased pulmonary blood flow in lambs. Circulation 92: 606–613, 1995. [DOI] [PubMed] [Google Scholar]
- 39.Ren JG, Seth P, Clish CB, Lorkiewicz PK, Higashi RM, Lane AN, Fan TWM, Sukhatme VP. Knockdown of malic enzyme 2 suppresses lung tumor growth, induces differentiation and impacts PI3K/AKT signaling. Sci Rep 4: 5414, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rensen SS, Doevendans PA, van Eys GJ. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth Heart J 15: 100–108, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ross R. The smooth muscle cell. II. Growth of smooth muscle in culture and formation of elastic fibers. J Cell Biol 50: 172–186, 1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sakao S, Taraseviciene-Stewart L, Lee JD, Wood K, Cool CD, Voelkel NF. Initial apoptosis is followed by increased proliferation of apoptosis-resistant endothelial cells. FASEB J 19: 1178–1180, 2005. [DOI] [PubMed] [Google Scholar]
- 44.Sharma S, Kumar S, Wiseman DA, Kallarackal S, Ponnala S, Elgaish M, Fineman JR, Black SM. Perinatal changes in superoxide generation in the ovine lung: Alterations associated with increased pulmonary blood flow. Vascul Pharmacol 18: 1199–1216, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tuder RM, Chacon M, Alger L, Wang J, Taraseviciene-Stewart L, Kasahara Y, Cool CD, Bishop EA, Geraci M, Semenza GL, Yacoub M, Polak JM, Voelkel NF. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: evidence for a process of disordered angiogenesis. J Pathol 195: 367–374, 2001. [DOI] [PubMed] [Google Scholar]
- 46.Tuder RM, Davis LA, Graham BB. Targeting energetic metabolism: a new frontier in the pathogenesis and treatment of pulmonary hypertension. Am J Respir Crit Care Med 185: 260–266, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Voet D, Voet JG. Biochemistry 4e [Online]. New York: Wiley, 2010. [Google Scholar]
- 48.Wedgwood S, Lakshminrusimha S, Schumacker PT, Steinhorn RH. Cyclic stretch stimulates mitochondrial reactive oxygen species and Nox4 signaling in pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 309: L196–L203, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wiseman DA, Wells SM, Wilham J, Hubbard M, Welker JE, Black SM. Endothelial response to stress from exogenous Zn2+ resembles that of NO-mediated nitrosative stress, and is protected by MT-1 overexpression. Am J Physiol Cell Physiol 291: C555–C568, 2006. [DOI] [PubMed] [Google Scholar]
- 50.Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, Koo M, Janocha AJ, Masri FA, Arroliga AC, Jennings C, Dweik RA, Tuder RM, Stuehr DJ, Erzurum SC. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci USA 104: 1342–1347, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]