

Abstract
Aims
Remodelling of gap junctions, involving reduction of total gap junction quantity and down-regulation of connexin43 (Cx43), contributes to the arrhythmic substrate in congestive heart failure. However, little is known of the underlying mechanisms. Recent studies from in vitro systems suggest that the connexin-interacting protein zonula occludens-1 (ZO-1) is a potential mediator of gap junction remodelling. We therefore examined the hypothesis that ZO-1 contributes to reduced expression of Cx43 gap junctions in congestive heart failure.
Methods and results
Left ventricular myocardium from healthy control human hearts (n = 5) was compared with that of explanted hearts from transplant patients with end-stage congestive heart failure due to idiopathic dilated cardiomyopathy (DCM; n = 5) or ischaemic cardiomyopathy (ICM; n = 5). Immunoconfocal and immunoelectron microscopy showed that ZO-1 is specifically localized to the intercalated disc of cardiomyocytes in control and failing ventricles. ZO-1 protein levels were significantly increased in both DCM and ICM (P = 0.0025), showing a significant, negative correlation to Cx43 levels (P = 0.0029). There was, however, no significant alteration of ZO-1 mRNA (P = 0.537). Double immunolabelling demonstrated that a proportion of ZO-1 label is co-localized with Cx43, and that co-localization of Cx43 with ZO-1 is significantly increased in the failing ventricle (P = 0.003). Interaction between the two proteins was confirmed by co-immunoprecipitation. The proportion of Cx43 that co-immunoprecipitates with ZO-1 was significantly increased in the failing heart.
Conclusion
Our findings suggest that ZO-1, by interacting with Cx43, plays a role in the down-regulation and decreased size of Cx43 gap junctions in congestive heart failure.
Keywords: Gap junction remodelling, Connexin43, ZO-1, Conjestive heart failure
1. Introduction
Gap junctions, clusters of cell-to-cell channels composed of connexins, mediate the orderly spread of electrical excitation throughout the heart.1 Experimental evidence from transgenic murine models,2–5 combined with findings from the diseased human heart,6–12 indicate that altered ventricular gap junction organization and connexin expression are key contributors to rhythm disturbances and contractile dysfunction. These alterations, referred to as gap junction remodelling, may take the form of structural modifications, involving disturbances in the distribution of gap junctions and/or changes in the amount or type of connexin(s) expressed.1,13 In patients with end-stage congestive heart failure [due to ischaemic cardiomyopathy (ICM) or idiopathic dilated cardiomyopathy (DCM)], the most prominent features of remodelling involve down-regulation of connexin43 (Cx43), reduction in gap junction plaque size and increased heterogeneity of gap junction distribution.6,8,10–12 While some mediators of Cx43 gap junction remodelling have been explored,14,15 our knowledge of the underlying mechanisms in human heart failure remains limited.
Recent evidence from various cell types has highlighted a role for connexin-interacting proteins,16 and in particular, the membrane-associated guanylate kinase, zonula occludens-1 (ZO-1), in regulating the formation, extent and size of Cx43 gap junctions.17–19 The interaction between ZO-1 and Cx43 is mediated through the second PDZ domain of ZO-1 and a PDZ-binding motif at the extreme carboxy terminus of Cx43.20,21 ZO-1 was originally identified as a tight junction protein, where it functions to tether transmembrane proteins to the actin cytoskeleton.22,23 Initially, it was assumed that ZO-1 forms a link between Cx43 and the cytoskeleton, thereby stabilizing gap junctions at the plasma membrane,21 but recent data suggest an additional, more dynamic role. In particular, inhibition of Cx43/ZO-1 interaction in a HeLa cell model results in the formation of abnormally large gap junctions, an effect rescued by restoration of the interaction.18 These findings raise the possibility that the reduction in ventricular Cx43 gap junction content in human congestive heart failure could be mediated, at least in part, via altered Cx43/ZO-1 interaction.
Accordingly, in this study, we set out to explore the hypothesis that ZO-1 plays a role in disease-related gap junction remodelling in the human heart. We show that ZO-1 is localized at the intercalated disc of human ventricular myocytes where it interacts with Cx43, and that the proportion of Cx43 that interacts with ZO-1 is significantly enhanced in the left ventricle of patients with congestive heart failure. A novel feature of our study is separate analysis of this interaction in the fraction of Cx43 within gap-junctional plaques (junctional fraction) and that elsewhere in the cell (non-junctional fraction).
2. Methods
2.1. Human heart samples
The study was conducted on explanted hearts of patients with ICM (n = 5) or DCM (n = 5) undergoing orthotopic cardiac transplantation at the Royal Brompton and Harefield NHS Trust Hospitals (Table 1). Samples of human left ventricles (encompassing the full thickness of the free wall) were snap-frozen in liquid nitrogen. As controls, tissues from five normal hearts initially harvested for use as transplantation donor organs, but rejected for technical reasons, were used. The study was approved by the Brompton, Harefield and NHLI Research Ethics Committee (#04–101 and #01–194). The investigation conforms with the principles outlined in the Declaration of Helsinki.24
Table 1.
Patient details
| Heart no. | Age (years) | Sex (M/F) | Cardiac disease | Other diagnoses | Echocardiography | NYHA class | |
|---|---|---|---|---|---|---|---|
| LVEDD (mm) | FS (%) | ||||||
| Controls | |||||||
| 1 | 22 | F | None | ICH | |||
| 2 | 25 | F | None | ICH | |||
| 3 | 48 | F | None | ||||
| 4 | 19 | F | None | CF (dominoa) | |||
| 5 | 52 | M | None | Head trauma | |||
| Idiopathic dilated cardiomyopathy | |||||||
| 6 | 41 | M | DCM | 74 | 5.4 | 4 | |
| 7 | 52 | M | DCM | 50 | 16 | 4 | |
| 8 | 38 | M | DCM | 100 | 4 | 4 | |
| 9 | 46 | F | DCM | NIDDM | 76.8 | 19.6 | 4 |
| 10 | 65 | F | DCM | 4 | |||
| Ischemic cardiomyopathy | |||||||
| 11 | 50 | M | ICM, 3VD | 80 | 14.4 | 4 | |
| 12 | 61 | M | ICM, 3VD | NIDDM | 74.2 | 15.4 | 4 |
| 13 | 54 | M | ICM | 65 | 13.8 | 4 | |
| 14 | 52 | M | ICM | Asthma | 47 | 19 | 4 |
| 15 | 46 | M | ICM | Hodgkin's | 43 | <30b | 4 |
aDomino = patient receiving dual heart/lung transplant (for lung disease), the explanted healthy heart being intended for transplantation in another patient; bPrecise figure not available. NIDDM, non-insulin dependent diabetes mellitus; LVEDD, left ventricle end-diastolic diameter (normal range: 37–56); FS, fraction shortening (normal range: 34–44); ICH, intra-cranial haemorrhage; CF, cystic fibrosis; 3VD; triple valve disease. Some patients were used in a previous study.8
2.2. Immunoconfocal and electron microscopy
Immunoconfocal microscopy was conducted on cryosections of tissue that was directly frozen, without prior aldehyde fixation.6 For immunoelectron microscopy, tissue was processed by low denaturation procedures and thin sections were immunogold labelled.25,26 Primary antibodies used were anti-Cx43 (Chemicon International Ltd), anti-ZO-1 (Zymed, 40–2200), and anti-CD31 (Sigma). For details of antibody characterization and specificity, see Supplementary material.
2.3. Quantification of protein levels by western blotting
Western blotting and quantification were carried out as described previously8 using anti-ZO-1 (Zymed, 61–7300) or anti-Cx43 (Sigma) antibodies.
2.4. Quantification of mRNA levels by northern blotting
RNA extraction and northern blotting were carried out as previously described.27 A cDNA encompassing nucleotides 3394–5548 of the human ZO-1 coding sequence was generated by PCR from human DNA, and cloned into pGEM®-T Easy vector (Promega). Following release from the vector, the probe was used at a concentration of 2.5 ng ml−1 after random primer labelling with 32P dCTP. Northern blots were quantified following normalization against methylene blue stained 18S RNA.8
2.5. Quantitative analysis of immunolabelled Cx43 and ZO-1 by confocal microscopy
Quantification of ZO-1 and Cx43 immunolabelling was performed using a Leica TCS 4D confocal microscope and PC Image™ software (Foster Findlay Associates).6 Blind measurements were taken of the fluorescent label in 10 individual en face intercalated discs per section, selected by random scanning across the tissue. Serial optical sections (0.5 µm intervals) were acquired at ×63 magnification, with a setting of zoom4 and a 512 × 512-pixel image size, yielding a pixel size of 0.08 µm. Using PC Image™ software, individual complete discs were outlined by hand for separate quantification and thresholds for detection on a 255-point grey scale adjusted to reduce any background cell outlines. A pixel-by-pixel analysis was used to assess label for Cx43, ZO-1, and both Cx43 and ZO-1 (co-localized) label.
2.6. Extraction of non-junctional and junctional proteins
To enable separate analysis of Cx43 within gap junctions (the ‘junctional’ fraction) and that elsewhere in the cell (the ‘non-junctional’ fraction), Triton X-100 (TX-100) extraction was performed. The non-junctional fraction, comprising cytoplasmic Cx43 en route to the plasma membrane, Cx43 in the plasma membrane as undocked connexons, and Cx43 en route to degradation, is soluble in TX-100, whereas the Cx43 in the docked connexons of gap-junctional plaques is not. Samples of frozen left ventricle were pulverized,8 then homogenized by sonication in immunoprecipitation (IP) buffer (10 µl mg−1 frozen tissue: 25 mM Tris, pH7.4; 150 mM NaCl; 1/250 mammalian tissue extract protease inhibitors [Sigma]; 1% TX-100). The samples were incubated on ice for 30 min, with vortexing every 5 min, and then centrifuged (15 000 rpm, 30 min, 4oC). Non-junctional proteins in the supernatant were collected. Junctional proteins in the pellet were re-suspended in an equal volume (i.e. 10 µl mg−1 tissue) of IP buffer containing 4 M urea. Samples were sonicated, incubated at room temperature for 30 min with vortexing, centrifuged (15 000 rpm, 30 min, 4°C), and junctional proteins collected in the supernatant. Urea (8 M) separates the component membranes of gap junctions,28 thereby undocking connexons and making them soluble in TX-100 for processing. We have checked the validity of using 4 M urea to extract junctional connexins from non-purified gap-junctional membranes by solubilizing the final pellet with 20% SDS. Negligible connexin levels were detectable in this fraction by western blot.
Prior to IP, junctional proteins were desalted using Bio-Gel® P-6 DG desalting gel (Bio-Rad) and IP buffer in Zeba spin columns (Pierce).
2.7. Cross-linking
Proteins were extracted in IP buffer containing PBS instead of Tris. Prior to the first centrifugation, an equal volume of 0.2 M ethanolamine, pH 8, containing 0.5 mM dimethyl-3-3′-dithiobispropionimidate (DTBP; Sigma) was added. Following incubation for 30 min at room temperature, an equal volume of 50 mM Tris, pH 7.4, was added to quench the reaction. Non-junctional and junctional protein extraction was then carried out as described above.
2.8. Co-immunoprecipitation
Protein samples were incubated with 2.5 µg of anti-ZO-1 (Zymed, 61–7300) or anti-Cx43 (Sigma) antibody overnight at 4°C with rotation, followed by 1 h at 4°C with 25 µl protein-A coated agarose beads (Pierce). Beads were washed with ice-cold IP buffer by centrifugation (2 min, 2500 rpm)/re-suspension. Bound proteins were desorbed by re-suspension in 20% SDS, containing 2.5% 2-mercaptoethanol. ZO-1/Cx43 interactions were quantified by western blotting using a polyclonal anti-Cx43 antibody (Sigma; favoured over the monoclonal anti-Cx43 (Chemicon) because of its higher sensitivity and ability to recognize all phosphorylated forms of Cx43), following normalization against Coomassie blue-stained actin. Control experiments, in which primary antibodies were omitted, excluded non-specific interactions.
2.9. Statistical analysis
All analysis was done using GraphPad Prism 4 (GraphPad Software Inc.). Data are expressed as mean ± SEM. Statistical significance was evaluated with ANOVA. Statistical differences were judged significant at P ≤ 0.05.
3. Results
3.1. Localization of ZO-1 in human ventricular cardiomyocytes
Immunoconfocal localization of ZO-1 in longitudinal sections of left ventricular tissue shows prominent signal which, at first sight, appears to outline individual myocytes, thereby suggesting the presence of ZO-1 throughout the entire myocyte plasma membrane (Figure 1A). However, careful comparison with the labelling patterns obtained for the endothelial marker, CD31, in both longitudinal and transverse planes, demonstrates that the apparent lateral labelling of the myocytes is, in fact, attributable to capillaries (Figures 1A–F). In longitudinal sections, CD31 highlights capillaries running alongside the myocytes (Figure 1B); transverse sections correspondingly show capillaries as small, prominently labelled circular profiles or streaks, with no signal of the myocytes that occupy the spaces between (Figure 1E). ZO-1 labelling of transverse sections showed similar small capillary profiles (Figure 1D), accounting for the apparent lateral label in longitudinal sections. There was no label circumscribing the myocyte bodies as would be expected if ZO-1 were present throughout the lateral plasma membrane. Periodic large discs were observed filled with positive ZO-1 signal occupying the entire myocyte cross-sectional area; these represented intercalated discs (Figure 1D). As confirmed by the merged images (Figures 1C and F), these observations indicate that, in human ventricular myocytes, ZO-1 is confined to the intercalated disc plasma membranes. This pattern of ZO-1 localization was not altered in the failing heart.
Figure 1.
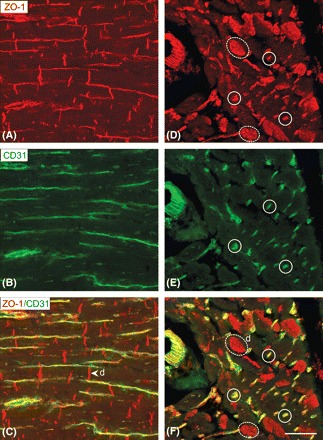
Localization of ZO-1 in human left ventricular myocardium. Immunoconfocal microscopy comparing labelling patterns for ZO-1 (red) and CD31 (green) as seen in double-labelled longitudinal (A–C) and transverse (D–F) sections. CD31 labelling identifies capillaries that run alongside the myocytes. When the images are merged, the ZO-1 labelling pattern can be seen to arise in part from capillaries (examples ringed by continuous lines) and from specific staining of myocyte intercalated discs (examples ringed by dashed lines). Merging the images (C and F) confirms that there is no detectable ZO-1 label in the lateral plasma membranes of the myocytes. In the myocytes, only the intercalated discs (d) contain red ZO-1 label. Sale bar = 50 µm.
Immunogold labelling at the higher resolution afforded by electron microscopy confirmed and extended these conclusions. ZO-1 gold label was localized specifically to intercalated disc membranes; no label was apparent at the lateral plasma membrane or membranes of intracellular organelles (Figure 2). Within the intercalated disc, ZO-1 label was observed both in association with gap junctions, particularly at the junctional edge, and in intervening segments of disc membrane.
Figure 2.

Thin-section electron micrograph of Lowicryl-embedded tissue immunogold labelled for ZO-1. Gold label occurs specifically at the intercalated discs, notably at the edges of gap junctions (GJ; arrowheads), as well as in non-gap-junctional membranes (arrows). Sale bar = 100 nm.
3.2. ZO-1 expression levels
To determine whether the expression levels of ZO-1 differed between control and failing ventricles, western and northern blot analyses were conducted. ZO-1 was detected by western blot as a prominent band migrating at ∼225 kDa (Figure 3A). Visual examination indicated increased levels of ZO-1 in both DCM and ICM vs. controls, which was confirmed by densitometric analysis following normalization against Coomassie blue-stained actin (P = 0.0025).
Figure 3.

Quantification of ZO-1 in human left ventricular tissue. (A) Typical western blots for ZO-1 and Cx43 and parallel Coomassie-blue staining for actin. Patient numbers (Table 1) and sample categories are indicated. (B) ZO-1 and Cx43 were quantified following normalization with the densitometric values of actin. ZO-1 is significantly elevated compared with controls (100 ± 11.40%) in DCM (175.3 ± 11.58%) and ICM (148.7 ± 12.62%; P = 0.0025). Quantification of Cx43 immunoblots confirmed significant down-regulation in failing samples (DCM = 65.85 ± 4.64; ICM = 53.40 ± 1.69%) compared with controls (100 ± 5.17%; P < 0.0001). (C) There is a significant, negative correlation between Cx43 and ZO-1 protein levels (P = 0.0029; r2 = 0.51). ***P < 0.0001 **P < 0.01; *P < 0.05 vs. controls.
Western blot analyses of Cx43 protein levels accorded with previous studies showing down-regulation in the failing heart (P < 0.0001; Figure 3A and B). Plotting these data against ZO-1 protein levels revealed a significant negative correlation between Cx43 and ZO-1 (Figure 3C), i.e. low Cx43 levels correspond to high levels of ZO-1 (P = 0.0029; r2 = 0.51).
ZO-1 mRNA was detected by northern blot as a single ∼8 kb band after 3–5 h exposure (Figure 4). Densitometric analysis following normalization against methylene blue stained 18S RNA showed no significant difference between transcript levels in control and failing hearts (P = 0.537).
Figure 4.
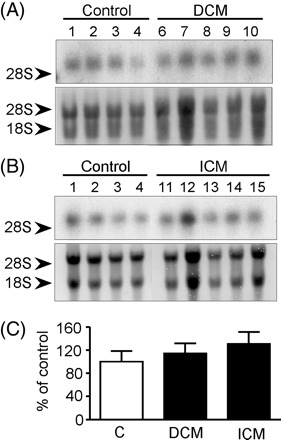
Northern analysis of ZO-1 mRNA expression. (A and B) Typical hybridizations with a probe for ZO-1 (upper panels) and corresponding methylene blue staining (lower panels). Patient numbers (Table 1), sample categories, and the migration of the 28S and/or 18S rRNA are indicated. (C) ZO-1 mRNA levels in failing hearts expressed relative to control samples following normalization with 18S mRNA. No significant differences are apparent between failing and control hearts (P = 0.537).
3.4. Co-localization of ZO-1 and Cx43 at the intercalated disc
To assess the possibility of interaction between ZO-1 and Cx43, quantitative double-label immunoconfocal microscopy was carried out in serial confocal reconstructions of en face intercalated discs, and the extent of co-localization determined.
Cx43 labelling revealed the typical pattern comprising large gap junctions at the periphery of the disc enclosing smaller junctions in the interior zone (Figure 5A).6,29,30 ZO-1 label was apparent across the entire face of the disc in both control and failing hearts. Double-labelled sections revealed substantial areas with Cx43 or ZO-1 signal only. However, a small proportion of co-localized signal was consistently apparent, indicating the existence of a pool of Cx43 situated where it could potentially interact with ZO-1.
Figure 5.
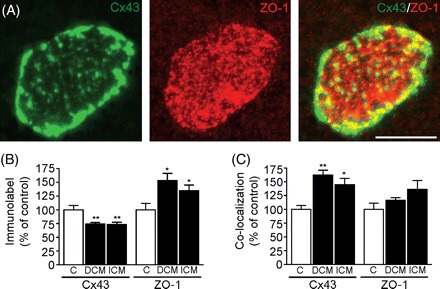
Co-localization of Cx43 and ZO-1 at the intercalated disc. (A) Example of an en-face viewed intercalated disc immunolabelled for Cx43 and ZO-1. Although Cx43 is seen predominantly as large gap junctions at the disc periphery whereas ZO-1 is spread evenly across the disc, a small fraction of each signal overlaps (yellow), indicating co-localization. Sale bar =10 µm. (B and C) Quantification of Cx43 and/or ZO-1 immunolabel at the intercalated disc. PC Image™ software uses the relative grayscale intensity to select pixels specific for immunolabelling to determine a total pixel count. (B) The proportion of the disc labelled for Cx43 is reduced compared with controls (C; 100 ± 7.52%) in DCM (74.17 ± 2.58%) and ICM (73.44 ± 3.91%; P = 0.004). ZO-1 label is increased compared with controls (C; 100 ± 11.61%) in DCM (153.1 ± 13.21%) and ICM (134.6 ± 10.23%; P = 0.031). (C) Cx43 co-localization with ZO-1 is increased compared with controls (C; 100 ± 6.91%) in DCM (161.8 ± 9.06%) and ICM (144.6 ± 11.30%; P = 0.003). **P < 0.01; *P < 0.05 vs. controls. Only P-values <0.05 are shown.
Quantitative analysis demonstrated that Cx43 label per unit area of disc was reduced in failing ventricles compared with controls (P = 0.004; Figure 5B). In contrast, ZO-1 label was increased in failing ventricles compared with controls (P = 0.031; Figure 5B). The proportion of Cx43 signal co-localizing with ZO-1 was increased in failing ventricles compared with controls (P = 0.003; Figure 5C). The proportion of ZO-1 signal co-localizing with Cx43 did not differ between failing and control hearts owing to the overall increase in ZO-1 observed in the former (P = 0.155).
3.5. Co-immunoprecipitation of Cx43 and ZO-1
Increased co-localization of Cx43 with ZO-1 raises the possibility of, but does not strictly demonstrate, an increased Cx43/ZO-1 interaction in the failing heart. To determine the extent of any interaction, co-immunoprecipitation experiments were carried out.
Using anti-ZO-1 or anti-Cx43 antibodies, ZO-1 and Cx43 were co-immunoprecipitated from human ventricular samples (Figure 6). Owing to the limited availability of human samples, low levels of ZO-1/Cx43 interaction and high sensitivity of the anti-Cx43 antibody (Sigma) in western blot analysis, co-immunoprecipitation was quantified by western blotting for Cx43 following IP with anti-ZO-1 antibody (Figure 7A). Remnants of the IgG heavy chain from the antibody used for co-immunoprecipitation are visible, but are readily distinguishable from Cx43 signal (Figure 7A). Densitometric analyses of Cx43 brought down with ZO-1 following normalization against total actin (Figure 7B) indicated that in both non-junctional and junctional fractions, slightly more Cx43 interacts with ZO-1 in the failing ventricles compared with controls, although this difference between absolute levels did not attain statistical significance (P = 0.397 and P = 0.209, respectively; Figure 7B). However, as Cx43 is down-regulated in the failing left ventricle (Figure 3) and this occurs in both non-junctional and junctional fractions (data not shown; P = 0.053 and P = 0.023, respectively), Cx43 co-immunoprecipitation with ZO-1 is more meaningfully quantified relative to Cx43 levels in each sample (Figure 7C). This demonstrated that the proportion of Cx43 that interacts with ZO-1 is significantly increased in the failing ventricle compared with controls in both non-junctional and junctional fractions (P = 0.01 and P = 0.01, respectively).
Figure 6.
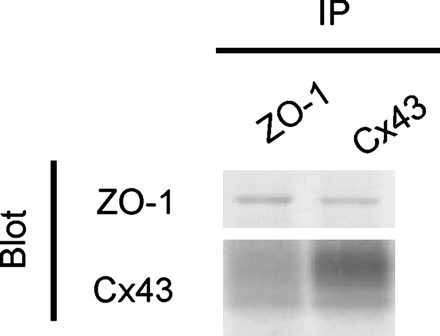
Co-immunoprecipitation of Cx43 and ZO-1. Using anti-ZO-1 or anti-Cx43 antibodies, ZO-1 and Cx43 were co-immunoprecipitated from human heart samples.
Figure 7.
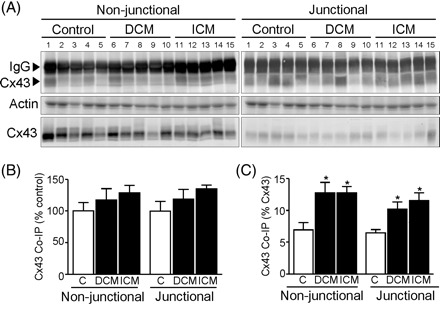
(A) Typical western blots for Cx43 co-immunoprecipitated with ZO-1 (upper panel), parallel Coomassie-blue stained actin (middle panel) and Cx43 in the starting fraction (lower panel). Patient heart numbers (Table 1) and sample categories are indicated and the migration of the IgG heavy chain and Cx43 are shown. (B) Cx43 co-immunoprecipitation with ZO-1, following normalization to total actin, was quantified relative to the controls. In both non-junctional and junctional fractions slightly more Cx43 was brought down by ZO-1 in DCM (117.5 ± 17.73 and 118.7 ± 15.37%, respectively) and ICM (128.8 ± 11.72 and 134.8 ± 6.30%, respectively) relative to controls (100 ± 13.44 and 100 ± 15.74%) but this did not attain statistical significance (P = 0.397 and P = 0.209, respectively). (C) Since Cx43 is down-regulated in congestive heart failure, Cx43 co-immunoprecipitated with ZO-1 was additionally quantified relative to Cx43 levels in each sample. In both non-junctional and junctional fractions a greater proportion of Cx43 was brought down by ZO-1 in DCM (12.76 ± 1.65 and 10.22 ± 1.11%, respectively) and ICM (12.76 ± 1.02 and 11.54 ± 1.25%, respectively) relative to controls (6.93 ± 1.14 and 6.47 ± 0.52%; P = 0.011 and P = 0.011, respectively). *P < 0.05 vs. controls.
Tissue was treated with the reversible cross-linking reagent DTBP to examine the Cx43/ZO-1 association in the in vivo setting, and to establish whether the interaction was altered during protein extraction. Densitometric analysis of western blots indicated that cross-linking did not alter the amount of Cx43 co-immunoprecipitated with ZO-1 in the non-junctional or junctional fractions from control (P = 0.759 and P = 0.246, respectively), DCM (P = 0.363 and P = 0.629, respectively), or ICM (P = 0.304 and P = 0.111, respectively) samples (data not shown).
4. Discussion
Evidence that gap junction remodelling and altered connexin expression are contributory factors in the development of arrhythmia in human heart disease is now compelling. It is well established that heterogeneous reduction in Cx43 expression and disordering in gap junction distribution occur in defined forms of ventricular disease and correlate with electrophysiologically identified arrhythmic changes and contractile dysfunction in animal models.1–13,31,32 Although disease-related down-regulation of Cx43 involves reduction in gap junction size,6,12 the cellular mechanisms responsible for this remodelling remain largely unexplored. This study sheds new light on this aspect of human congestive heart failure by demonstrating that (i) ZO-1 is specifically localized at the intercalated disc of human cardiac myocytes, (ii) a pool of this ZO-1 is closely associated with gap junctions and interacts with Cx43, (iii) ZO-1 is up-regulated in parallel with the reduced expression of Cx43 in heart failure, and (iv) this change is accompanied by an increase in the proportion of Cx43 interacting with ZO-1.
Several previous studies have claimed that ZO-1 is present at both the intercalated discs and lateral plasma membrane of mammalian cardiomyocytes.33–35 By comparing the distribution pattern of ZO-1 with the endothelial marker, CD31, in transverse and longitudinal sections, we demonstrate unequivocally here that ZO-1 is confined to the intercalated disc plasma membranes of human ventricular cardiomyocytes, as also concluded in studies on rat myocytes.21,36–39 The apparent lateral labelling previously described arises from labelling of endothelial cell plasma membranes of the numerous capillaries running adjacent to and parallel with myocytes in longitudinal section view. We found that the restriction of ZO-1 to the intercalated disc did not alter in the failing left ventricle, but in contrast to a recent report,40 the overall level of ZO-1 protein expression was increased. ZO-1 transcript levels, however, remained unchanged, indicating post-transcriptional control of ZO-1 levels, for example through increased half-life or increased efficiency of translation.
Immunoconfocal analysis shows that although the majority of Cx43 in the human ventricle is not associated with ZO-1, a small but significant pool of each protein co-localizes with the other. While previous studies have reported low levels of co-localization between Cx43 and ZO-1 in non-myocyte cell types41–43 and rat myocytes,36,44 only two of these studies quantified the extent of the overlap.36,44 Barker et al.36 reported that at intercalated discs of adult rat ventricle, ∼15% of ZO-1 signal co-localizes with Cx43. This is comparable to our finding of ∼21% in control human ventricle. Our immunoelectron microscopy data confirm that ZO-1 signal is associated preferentially with the edges of gap junctions, in keeping with earlier reports.44,45
More crucial to potential regulatory mechanisms, however, is the extent of Cx43 co-localization with ZO-1, and whether this represents interaction between the proteins. In human congestive heart failure, we show a significant increase in co-localization of Cx43 with ZO-1. Co-immunoprecipitation analyses confirmed an association and demonstrate that, while only a small proportion of Cx43 interacts with ZO-1, this proportion is significantly increased in the failing left ventricle. There was no significant difference in the amount of Cx43 co-immunoprecipitated with ZO-1 following cross-linking, demonstrating that the protein extraction procedure does not disrupt or initiate the interaction.
Our data show that the increased proportion of Cx43 interacting with ZO-1 accompanies the down-regulation of Cx43 that has been consistently reported in human congestive heart failure.1,8,10–13,31,32 Interaction of ZO-1 with Cx43 has been previously implicated in regulation of the size and distribution of gap junctions. In an experimental HeLa cell system, blocking the interaction leads to the formation of exceptionally large gap junctions, whose growth is apparently unconstrained.17,18 A current hypothesis is that ZO-1 regulates gap junction plaque size.18 The localization of ZO-1 at the periphery of gap junction plaques44 places it in an ideal position to inhibit further recruitment of connexons18 or to favour the removal of gap junctions on reaching a certain size. Thus, increased interaction of Cx43 with ZO-1—as demonstrated here—could decrease the maximum size to which the gap junctions grow, thereby contributing to reduction in the overall levels of Cx43 in the failing heart.
Our co-immunoprecipitation experiments demonstrated, however, that Cx43 interaction with ZO-1 was not only detected in the junctional fraction (i.e. within gap junctions), but also in the non-junctional fraction. As the non-junctional fraction includes undocked connexons that lie outside gap junction plaques, this might reflect a role for Cx43/ZO-1 interaction at the edge of the gap junction, i.e. at the transition between junctional and non-junctional membrane. However, an effect at the cytoplasmic level cannot be excluded. Binding of ZO-1 to Cx43 connexons en route to the plasma membrane (possibly in a proportion dictated by the metabolic state of the cells) could conceivably be a signal by which gap junction size and overall quantity is limited. Increased Cx43/ZO-1 association is a prominent feature of some situations in which gap junctions are internalized by endocytosis, both in rat cardiac myocytes36 and non-myocyte cell types.42 Internalization of gap junctions, mediated via interactions with the spectrin/actin cytoskeleton,36,46 is a prelude to degradation, at which point any Cx43/ZO-1 interaction may be detected in the non-junctional as well as junctional fractions. Thus, our findings highlight the possibility of increased ZO-1-mediated degradation of Cx43 as an alternative/additional mechanism of reduced Cx43 expression in heart failure.
In summary, Cx43 and ZO-1 protein levels are negatively correlated in the failing human ventricle and a pool of Cx43 interacts with ZO-1 at the intercalated discs. Our data are consistent with the hypothesis that ZO-1, through interacting with Cx43, is involved in decreasing gap junction size and quantity in congestive heart failure, possibly by regulating the size of the Cx43 gap junction and/or mediating gap junction endocytosis.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Funding
British Heart Foundation (grant PG/05/003).
Supplementary Material
Acknowledgements
The authors thank Professor Sian Harding (NHLI, Imperial College) for donating some of the control tissue used in this study.
Conflict of interest: none declared.
References
- 1.Severs NJ, Coppen SR, Dupont E, Yeh HI, Ko YS, Matsushita T. Gap junction alterations in human cardiac disease. Cardiovasc Res. 2004;62:368–377. doi: 10.1016/j.cardiores.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 2.Gutstein DE, Morley GE, Tamaddon H, Vaidya D, Schneider MD, Chen J, et al. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res. 2001;88:333–339. doi: 10.1161/01.res.88.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gutstein DE, Morley GE, Vaidya D, Liu F, Chen FL, Stuhlmann H, et al. Heterogeneous expression of gap junction channels in the heart leads to conduction defects and ventricular dysfunction. Circulation. 2001;104:1194–1199. doi: 10.1161/hc3601.093990. [DOI] [PubMed] [Google Scholar]
- 4.Morley GE, Danik SB, Bernstein S, Sun Y, Rosner G, Gutstein DE, et al. Reduced intercellular coupling leads to paradoxical propagation across the Purkinje-ventricular junction and aberrant myocardial activation. Proc Natl Acad Sci USA. 2005;102:4126–4129. doi: 10.1073/pnas.0500881102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lerner DL, Yamada KA, Schuessler RB, Saffitz JE. Accelerated onset and increased incidence of ventricular arrhythmias induced by ischemia in Cx43-deficient mice. Circulation. 2000;101:547–552. doi: 10.1161/01.cir.101.5.547. [DOI] [PubMed] [Google Scholar]
- 6.Kaprielian RR, Gunning M, Dupont E, Sheppard MN, Rothery SM, Underwood R, et al. Down-regulation of immunodetectable connexin43 and decreased gap junction size in the pathogenesis of chronic hibernation in the human left ventricle. Circulation. 1998;97:651–660. doi: 10.1161/01.cir.97.7.651. [DOI] [PubMed] [Google Scholar]
- 7.Smith JH, Green CR, Peters NS, Rothery S, Severs NJ. Altered patterns of gap junction distribution in ischemic heart disease. An immunohistochemical study of human myocardium using laser scanning confocal microscopy. Am J Pathol. 1991;139:801–821. [PMC free article] [PubMed] [Google Scholar]
- 8.Dupont E, Matsushita T, Kaba R, Vozzi C, Coppen SR, Khan N, et al. Altered connexin expression in human congestive heart failure. J Mol Cell Cardiol. 2001;33:359–371. doi: 10.1006/jmcc.2000.1308. [DOI] [PubMed] [Google Scholar]
- 9.Kostin S, Dammer S, Hein S, Klovekorn WP, Bauer EP, Schaper J. Connexin 43 expression and distribution in compensated and decompensated cardiac hypertrophy in patients with aortic stenosis. Cardiovasc Res. 2004;62:426–436. doi: 10.1016/j.cardiores.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 10.Kostin S, Rieger M, Dammer S, Hein S, Richter M, Klovekorn WP, et al. Gap junction remodeling and altered connexin43 expression in the failing human heart. Mol Cell Biochem. 2003;242:135–144. [PubMed] [Google Scholar]
- 11.Kitamura H, Ohnishi Y, Yoshida A, Okajima K, Azumi H, Ishida A, et al. Heterogeneous loss of connexin43 protein in nonischemic dilated cardiomyopathy with ventricular tachycardia. J Cardiovasc Electrophysiol. 2002;13:865–870. doi: 10.1046/j.1540-8167.2002.00865.x. [DOI] [PubMed] [Google Scholar]
- 12.Yamada KA, Rogers JG, Sundset R, Steinberg TH, Saffitz JE. Up-regulation of connexin45 in heart failure. J Cardiovasc Electrophysiol. 2003;14:1205–1212. doi: 10.1046/j.1540-8167.2003.03276.x. [DOI] [PubMed] [Google Scholar]
- 13.Severs NJ, Dupont E, Kaba RA, Thomas N. Gap junction and connexin remodeling in human heart disease. In: Winterhager E, editor. Gap Junctions in Development and Disease. Berlin Heidelberg: Springer-Verlag; 2005. pp. 57–82. [Google Scholar]
- 14.Petrich BG, Eloff BC, Lerner DL, Kovacs A, Saffitz JE, Rosenbaum DS, et al. Targeted activation of c-Jun N-terminal kinase in vivo induces restrictive cardiomyopathy and conduction defects. J Biol Chem. 2004;279:15330–15338. doi: 10.1074/jbc.M314142200. [DOI] [PubMed] [Google Scholar]
- 15.Saffitz JE, Kleber AG. Effects of mechanical forces and mediators of hypertrophy on remodeling of gap junctions in the heart. Circ Res. 2004;94:585–591. doi: 10.1161/01.RES.0000121575.34653.50. [DOI] [PubMed] [Google Scholar]
- 16.Giepmans BN. Role of connexin43-interacting proteins at gap junctions. Adv Cardiol. 2006;42:41–56. doi: 10.1159/000092561. [DOI] [PubMed] [Google Scholar]
- 17.Hunter AW, Jourdan J, Gourdie RG. Fusion of GFP to the carboxyl terminus of connexin43 increases gap junction size in HeLa cells. Cell Commun Adhes. 2003;10:211–214. doi: 10.1080/cac.10.4-6.211.214. [DOI] [PubMed] [Google Scholar]
- 18.Hunter AW, Barker RJ, Zhu C, Gourdie RG. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol Biol Cell. 2005;16:5686–5698. doi: 10.1091/mbc.E05-08-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laing JG, Chou BC, Steinberg TH. ZO-1 alters the plasma membrane localization and function of Cx43 in osteoblastic cells. J Cell Sci. 2005;118:2167–2176. doi: 10.1242/jcs.02329. [DOI] [PubMed] [Google Scholar]
- 20.Giepmans BN, Moolenaar WH. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr Biol. 1998;8:931–934. doi: 10.1016/s0960-9822(07)00375-2. [DOI] [PubMed] [Google Scholar]
- 21.Toyofuku T, Yabuki M, Otsu K, Kuzuya T, Hori M, Tada M. Direct association of the gap junction protein connexin-43 with ZO-1 in cardiac myocytes. J Biol Chem. 1998;273:12725–12731. doi: 10.1074/jbc.273.21.12725. [DOI] [PubMed] [Google Scholar]
- 22.Stevenson BR, Siliciano JD, Mooseker MS, Goodenough DA. Identification of ZO-1: A high molecular weight polypeptide associated with the tight junction (zonula occludens) in a variety of epithelia. J Cell Biol. 1986;103:755–766. doi: 10.1083/jcb.103.3.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Denker BM, Nigam SK. Molecular structure and assembly of the tight junction. Am J Physiol. 1998;274:F1–F9. doi: 10.1152/ajprenal.1998.274.1.F1. [DOI] [PubMed] [Google Scholar]
- 24.[Anon] World Medical Association Declaration of Helsinki–recommendations guiding physicians in biomedical research involving human subjects–Adopted by the 18th World Medical Assembly Helsinki, Finland, June, 1964. Cardiovasc Res. 1997;35:2–3. [PubMed] [Google Scholar]
- 25.Yeh H-I, Dupont E, Rothery S, Coppen SR, Severs NJ. Individual gap junction plaques contain multiple connexins in arterial endothelium. Circ Res. 1998;83:1248–1263. doi: 10.1161/01.res.83.12.1248. [DOI] [PubMed] [Google Scholar]
- 26.Dupont E, Ko YS, Rothery S, Coppen SR, Baghai M, Haw M, et al. The gap-junctional protein, connexin40, is elevated in patients susceptible to post-operative atrial fibrillation. Circulation. 2001;103:842–849. doi: 10.1161/01.cir.103.6.842. [DOI] [PubMed] [Google Scholar]
- 27.Chomczynski P, Sacchi N. The single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction: twenty-something years on. Nat Protoc. 2006;1:581–585. doi: 10.1038/nprot.2006.83. [DOI] [PubMed] [Google Scholar]
- 28.El Aoumari A, Dupont E, Fromaget C, Jarry T, Briand J-P, Kreitman B, et al. Immunolocalization of an extracellular domain of connexin43 in rat heart gap junctions. Eur J Cell Biol. 1991;56:391–400. [PubMed] [Google Scholar]
- 29.Gourdie RG. A map of the heart: gap junctions, connexin diversity and retroviral studies of conduction myocyte lineage. Clin Sci. 1995;88:257–262. doi: 10.1042/cs0880257. [DOI] [PubMed] [Google Scholar]
- 30.Severs NJ. Cardiac muscle cell interaction: from microanatomy to the molecular make-up of the gap junction. Histol Histopathol. 1995;10:481–501. [PubMed] [Google Scholar]
- 31.Severs NJ, Dupont E, Coppen SR, Halliday D, Inett E, Baylis D, et al. Remodelling of gap junctions and connexin expression in heart disease. Biochim Biophys Acta. 2004;1662:138–148. doi: 10.1016/j.bbamem.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 32.Severs NJ, Dupont E, Thomas N, Kaba R, Rothery S, Jain R, et al. Alterations in cardiac connexin expression in cardiomyopathies. Adv Cardiol. 2006;42:228–242. doi: 10.1159/000092572. [DOI] [PubMed] [Google Scholar]
- 33.Gutstein DE, Liu FY, Meyers MB, Choo A, Fishman GI. The organization of adherens junctions and desmosomes at the cardiac intercalated disc is independent of gap junctions. J Cell Sci. 2003;116:875–885. doi: 10.1242/jcs.00258. [DOI] [PubMed] [Google Scholar]
- 34.Sanford JL, Edwards JD, Mays TA, Gong B, Merriam AP, Rafael-Fortney JA. Claudin-5 localizes to the lateral membranes of cardiomyocytes and is altered in utrophin/dystrophin-deficient cardiomyopathic mice. J Mol Cell Cardiol. 2005;38:323–332. doi: 10.1016/j.yjmcc.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 35.Li J, Patel VV, Kostetskii I, Xiong Y, Chu AF, Jacobson JT, et al. Cardiac-specific loss of N-cadherin leads to alteration in connexins with conduction slowing and arrhythmogenesis. Circ Res. 2005;97:474–481. doi: 10.1161/01.RES.0000181132.11393.18. [DOI] [PubMed] [Google Scholar]
- 36.Barker RJ, Price RL, Gourdie RG. Increased association of ZO-1 with connexin43 during remodeling of cardiac gap junctions. Circ Res. 2002;90:317–324. doi: 10.1161/hh0302.104471. [DOI] [PubMed] [Google Scholar]
- 37.Toyofuku T, Akamatsu Y, Zhang H, Kuzuya T, Tada M, Hori M. c-Src regulates the interaction between connexin-43 and ZO-1 in cardiac myocytes. J Biol Chem. 2001;276:1780–1788. doi: 10.1074/jbc.M005826200. [DOI] [PubMed] [Google Scholar]
- 38.Matsushita S, Kurihara H, Watanabe M, Okada T, Sakai T, Amano A. Alterations of phosphorylation state of connexin 43 during hypoxia and reoxygenation are associated with cardiac function. J Histochem Cytochem. 2006;54:343–353. doi: 10.1369/jhc.4A6611.2005. [DOI] [PubMed] [Google Scholar]
- 39.Sasano C, Honjo H, Takagishi Y, Uzzaman M, Emdad L, Shimizu A, et al. Internalization and dephosphorylation of connexin43 in hypertrophied right ventricles of rats with pulmonary hypertension. Circ J. 2007;71:382–389. doi: 10.1253/circj.71.382. [DOI] [PubMed] [Google Scholar]
- 40.Laing JG, Saffitz JE, Steinberg TH, Yamada KA. Diminished zonula occludens-1 expression in the failing human heart. Cardiovasc Pathol. 2007;16:159–164. doi: 10.1016/j.carpath.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 41.Duffy HS, Ashton AW, O’Donnell P, Coombs W, Taffet SM, Delmar M, et al. Regulation of connexin43 protein complexes by intracellular acidification. Circ Res. 2004;94:215–222. doi: 10.1161/01.RES.0000113924.06926.11. [DOI] [PubMed] [Google Scholar]
- 42.Segretain D, Fiorini C, Decrouy X, Defamie N, Prat JR, Pointis G. A proposed role for ZO-1 in targeting connexin 43 gap junctions to the endocytic pathway. Biochimie. 2004;86:241–244. doi: 10.1016/j.biochi.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 43.Jin C, Martyn KD, Kurata WE, Warn-Cramer BJ, Lau AF. Connexin43 PDZ2 binding domain mutants create functional gap junctions and exhibit altered phosphorylation. Cell Commun Adhes. 2004;11:67–87. doi: 10.1080/15419060490951781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu C, Barker RJ, Hunter AW, Zhang Y, Jourdan J, Gourdie RG. Quantitative analysis of ZO-1 colocalization with Cx43 gap junction plaques in cultures of rat neonatal cardiomyocytes. Microsc Microanal. 2005;11:244–248. doi: 10.1017/S143192760505049X. [DOI] [PubMed] [Google Scholar]
- 45.Akoyev V, Takemoto DJ. ZO-1 is required for protein kinase C gamma-driven disassembly of connexin 43. Cell Signal. 2007;19:958–967. doi: 10.1016/j.cellsig.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Segretain D, Falk MM. Regulation of connexin biosynthesis, assembly, gap junction formation, and removal. Biochim Biophys Acta. 2004;1662:3–21. doi: 10.1016/j.bbamem.2004.01.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.