

Abstract
INTRODUCTION
Whether co-occurring neuropathologies interact or independently affect clinical disease progression is uncertain. We estimated rates of clinical progression and tested whether associations between clinical progression and Alzheimer’s disease neuropathology (ADNP) were modified by co-occurring Lewy body disease (LBD) or vascular brain injury (VBI).
METHODS
Linear mixed effects models evaluated longitudinal trends in the Clinical Dementia Rating Sum of Boxes on 2,046 autopsied participants seen at a U.S. Alzheimer’s Disease Center.
RESULTS
Annual clinical progression was slightly faster for ADNP+LBD compared to ADNP only (p=0.06) and slightly slower for ADNP+VBI (p=0.003). Differences in progression were less than expected if each neuropathology independently contributed to progression; ADNP interacted with LBD (p=0.002) and VBI (p=0.003). In secondary models, the effect of additional pathologies on clinical progression was greater in those with intermediate compared to high levels of ADNP.
DISCUSSION
The impact of co-occurring pathologies on progression may depend on severity of ADNP.
Keywords: Alzheimer’s disease neuropathology, Lewy body disease, cerebrovascular disease, mixed neuropathology, clinical progression
1. INTRODUCTION
Up to 75% of autopsied older adults have multiple brain pathologies, known as mixed neuropathologies [1–3]. Research focusing on Alzheimer’s disease, whether based on clinical criteria, biomarkers, or neuropathologic diagnosis, may ignore other relevant brain comorbidities [4]. Evidence as to whether neuropathologies interact synergistically or act independently to influence the dementia syndrome, is inconsistent.
Focus has traditionally centered on concomitant vascular brain injury (VBI), such as infarcts, and Alzheimer’s disease neuropathology (ADNP) [5–7]. Although one study reported a synergistic interaction between ADNP and VBI for memory scores [8], other studies suggest that ADNP and VBI do not interact [9,10]. ADNP in more severe stages may overwhelm the effects of VBI [11,12]. Lewy body disease (LBD) and ADNP also commonly coexist [13–16] and are associated with cognitive decline [17,18]. Lewy body development may be enhanced by ADNP [13,19]. Only one study reported testing whether concomitant LBD modified the association between ADNP and cognition, but they found no significant interactions [16].
To our knowledge, no prior studies have used statistical modelling to extensively examine whether LBD or VBI interact with ADNP in association with clinical progression over time. Such research could help clarify the role of mixed neuropathologies in clinical progression, with implications for prevention and treatment strategies. Testing interactions requires a large sample size. Thus, we used National Alzheimer’s Coordinating Center (NACC) data on autopsied participants who were clinically evaluated at a U.S. National Institute on Aging-funded Alzheimer’s Disease Center (ADC). We evaluated whether autopsied older adults with ADNP with co-occurring LBD or VBI had faster overall clinical progression compared to those with single and low neuropathologies. We test whether LBD or VBI modified the association between ADNP and progression.
2. METHODS
2.1 Study sample
NACC maintains the Uniform Data Set (UDS) on participants who had been prospectively evaluated and autopsied by an ADC since September 2005. Participants enrolled with any level of cognition and were examined annually in-person using a standard protocol, described in detail elsewhere [20,21]. Neuropathologic data was collected on participants who had died and consented to autopsy. All participants provided written informed consent and institutional review board approval was obtained from all individual ADCs.
This analysis focused on autopsied UDS participants with at least one clinical visit between September 2005 and September 2015. Participants were excluded based on the following criteria: 1.) rare cause of dementia that may conflict with neuropathologic assessment of ADNP or confound clinical conditions, such as Down’s syndrome, autosomal dominant genetic diseases, or frontotemporal lobar degeneration; 2.) missing information on covariates and/or neuropathologic information on ADNP, LBD, or VBI; and 4.) no ADNP, LBD, or VBI but presence of other pathologic burden such as hippocampal sclerosis, Braak stage V–VI with sparse or no neuritic plaques, frequent neuritic plaques but Braak stage 0–II, other major pathologies, or white matter disease. Additionally, in the main analyses we excluded participants without a clinical visit proximal death (e.g. last visit > 2 years prior to death) due to concern that the rate of progression and level of impairment may change closer to death but would have been unobserved. Given these exclusions, 2,046 participants with at least one clinical visit remained for analyses (see Supplementary Figure 1 for sample flow chart). Individuals with only one visit (n=475) were included in analytic models at baseline but did not contribute to longitudinal estimates.
In order to utilize additional information on VBI that was not available for all NACC participants, we conducted a sub-analysis in ADC participants seen at the Oregon Health & Science University (OHSU) (n=211) and University of Washington (UW) (n=82). These two ADCs have a joint agreement as part of the Pacific Northwest Dementia and Aging Neuropathology Group (PANDA) to follow the same neuropathologic assessment protocol, an additional benefit of this sub-analysis. Both ADCs recruit patients seen in clinic for enrollment into the UDS; however, OHSU also recruited participants from a number of cohort studies focusing on healthy aging and described elsewhere [22–25]. Subsequently, we will use the term PANDA ADCs to refer to OHSU and UW ADCs combined.
2.2 Neuropathological features
ADCs follow consensus guidelines but conduct neuropathologic assessments according to their own protocols, which vary between sites. ADNP was defined by Consortium to Establish a Registry for Alzheimer’s Disease scores of neuritic plaque density (none, sparse, moderate, and frequent) [26] and Braak stage for tau neurofibrillary pathology (none, I–II, III–IV, V–VI) [27]. ADNP was defined regardless of a participant’s cognitive status and was categorized semi-quantitatively as: low (no/sparse neuritic plaques & any Braak stage OR any neuritic plaques & Braak stage 0–II), intermediate (moderate/frequent CERAD plaques & Braak stage III–IV), and high (moderate/frequent plaques & Braak stage V–VI). This classification overlaps with the 2012 NIA-Alzheimer’s Association criteria [28]; however, Thal phasing [29] for amyloid plaques was not available for most participants. Assessment for Lewy bodies followed recognized guidelines [30]. LBD was defined as presence of Lewy bodies in any brain region examined. LBD subtype was classified as none, brainstem predominant, limbic (transitional), neocortical (diffuse), or other or unknown region.
Cognitive impairment due to vascular disease is usually considered a result of VBI caused by vessel disorders and other vascular mechanisms [31]. We focused on VBI, as defined by gross and microscopic infarcts, because these are associated with cognitive impairment in other studies [32,33]. VBI was defined as any gross infarcts (small or large artery) or any cortical microinfarcts (infarcts in the cortex only seen microscopically) regardless of age. Some studies suggest multiple VBI, in particular microinfarcts, may be needed to affect cognition [32,34]. Information on number of microinfarcts were not available for most NACC participants. To address this limitation, we abstracted additional data on the number of microinfarcts from neuropathology reports of autopsy PANDA ADC participants. The number (0,1,2,3, or 4 or more) of cortical and subcortical microinfarcts was assessed separately following methods developed in the Honolulu Asia Aging Study [35]. Cerebral amyloid angiopathy, atherosclerosis, and arteriolosclerosis were recorded as none, mild, moderate, or severe. Participants with subcortical leukoencephalopathy or white matter rarefaction were considered to have white matter disease. Hippocampal sclerosis was categorized as present based on neuropathologic diagnosis (forms prior to 2014) or if recorded as unilateral, bilateral, or laterality unknown (forms after 2014). Participants were defined as having a low level of neuropathology (low NP) if they were without ADNP, LBD, VBI, or other major pathologic burden.
2.3 Clinical impairment
Clinical impairment was quantified at each study visit with the Clinical Dementia Rating Scale Sum of Boxes (CDR-SB) [36], a composite measure of the overall level of cognitive impairment and functional disability that is based on clinical judgment and study co-participant report. The CDR-SB ranges from 0–18, with increasing score for increasing impairment. It is a sensitive measure for staging dementia on a continuum from normal to severe dementia in heterogeneous samples [35]. The Mini-Mental State Examination (MMSE) [37], a neuropsychological test of overall cognitive function, was also administered to able participants.
2.4 Other clinical characteristics
Demographic characteristics included in analyses were age, sex, education, race/ethnicity, and ADC. History of comorbidities and clinical judgement of motor and behavioral problems was recorded at each visit. APOE genotyping was performed on consenting participants. APOE ε4 allele status was classified as at least one or none. At all ADCs, either a single clinician or consensus group of clinicians made at each visit a diagnosis of normal cognition, impaired but not mild cognitive impairment (MCI), MCI, or dementia.
2.5 Statistical analyses
2.5.1 Modelling clinical progression
To model longitudinal trends in clinical impairment we used multivariable regression modelling via linear mixed effects models. The primary outcome was the CDR-SB score (0 to 18) at each visit, modelled as a continuous measure. Following the approach of other studies [17,18] we modelled longitudinal trends such that visits work backwards from last visit to the initial clinical visit. Models included random intercepts to account for correlation of CDR-SB scores within the same participant as well as within the same ADC. In addition, random slopes were included for participants to allow for heterogeneity in rate of change in CDR-SB over time between participants. Primary predictors were dichotomous variables for ADNP, LBD, VBI, and time. We added interaction terms between time and each primary pathology term and 3-way interactions of ADNP×LBD×time and ADNP×VBI×time to describe modifications to the relationship between time and CDR-SB that were attributable to individual pathologies and their combinations. Primary models also included adjustment for potential confounders: age at death, sex, race/ethnicity, education, and interval between last visit and death. To assess model fit we examined plots of residuals by fitted values, which did not show any systematic lack of fit. Additionally, empirical plots suggested change in CDR-SB over time was approximately linear.
We conducted several sensitivity analyses to assess potential biases and subgroup differences. We reran the primary model including additional adjustment for comorbidities and APOE ε4 allele. We also reran the model including participants whose last visit was greater than 2 years prior to death to examine the effects of this restriction criteria. Due to concern that trends in those with severe dementia drove trends, we examined progression in a subset of participants without dementia at baseline. We also examined rates of progression stratified by high and low education (high school or less vs some college or more) as higher educational attainment may indicate cognitive or brain reserve [38]. Finally, we examined results of the primary model using the MMSE as an alternative outcome. In secondary models, we examined associations with semi-quantitative measures instead of dichotomous measures for pathologies. In PANDA ADC participants, we examined associations with number of cortical and subcortical microinfarcts, modelled as continuous, separately.
2.5.2 Inverse probability weighting
An important consideration in this study is that participants who have died and consented to autopsy may differ in characteristics from the overall study populations. In certain cases autopsy findings may be biased, specifically when determinants of autopsy are also potential confounders of the exposure-outcome association [39]. We investigated predictors of autopsy and used an analytic tool, inverse-probability weighting (IPW) [40], that can account for potential selection bias [41,42]. Among 6,507 NACC participants 41% (n=2,672) were not autopsied. Participants who had died were relatively similar regardless of autopsy status, however autopsied participants were more likely to be white, demented, male, older, and had undergone genetic testing than living/withdrawn participants (Supplementary Table 1).
IPW uses weights to adjust for observable differences between the overall study sample and the autopsy sample. Weights were created by first modelling autopsy selection using logistic regression. Initially, all factors potentially associated with autopsy as well as potential confounders of the relationship between neuropathology and clinical progression were included as predictors. Backward selection was used to select variables for final models. In NACC, predictors included in the model were: baseline age, sex, race, education, early birth year (pre 1928), presence of 1 or more comorbidities, CDR-SB at last visit, atypical dementia diagnosis (not clinical AD or vascular dementia), presence of motor symptoms, and volunteering for genotype assessment. In the PANDA ADC subset, predictors were: baseline age, sex, race, education, presence of 1 or more comorbidities, CDR-SB at last visit, and volunteering for genotype assessment. Predicted probability of selection into the autopsy sample was calculated for each participant from the selection model and inverse probabilities were incorporated into the main analytic models described above as weights [39]. To stabilize the weights for individuals with low autopsy selection probability, we truncated weights at the 95th percentile [42] (20.7 for NACC overall and 11.7 for PANDA ADCs). Predictive model fit was assessed via ROC curves; the area under the curve was 0.85 for both NACC and the PANDA ADCs. We used bias-corrected and accelerated (BCa) bootstrap CIs in our analyses in order to ensure that uncertainty attributable to the estimated weights was reflected in our parameter estimates [43]. Analyses were conducted using R (version 3.2.1). We used eulerAPE[44] to create Venn/Euler diagrams that accurately illustrate the overlap of ADNP, LBD, and VBI. All tests were two-sided with α = 0.05.
3. RESULTS
3.1 Participant characteristics
Among 2,046 autopsied NACC participants, the mean age at death was 82.1 years (standard deviation [SD]: 10.3), the average interval between last clinical visit and death was 9.1 months (SD: 6.1). There were 1,571 participants with multiple visits (ranging from 2 to 10 visits), including 1,196 participants with 3 or more visits. The median follow-up of those with 2 or more visits was 3.2 years (IQR: 2.0–5.0); 872 participants were followed for 3 or more years. Co-occurrence of ADNP, LBD, and VBI at autopsy was common (Figure 1). Characteristics of participants grouped by ADNP, LBD, and VBI neuropathologies are described in Table 1. There were relatively few participants with LBD+VBI and ADNP+LBD+VBI, so these groupings were not examined separately in analytic models.
Figure 1. Co-occurrence of Alzheimer’s disease neuropathology (ADNP), Lewy body disease (LBD), and vascular brain injury (VBI).
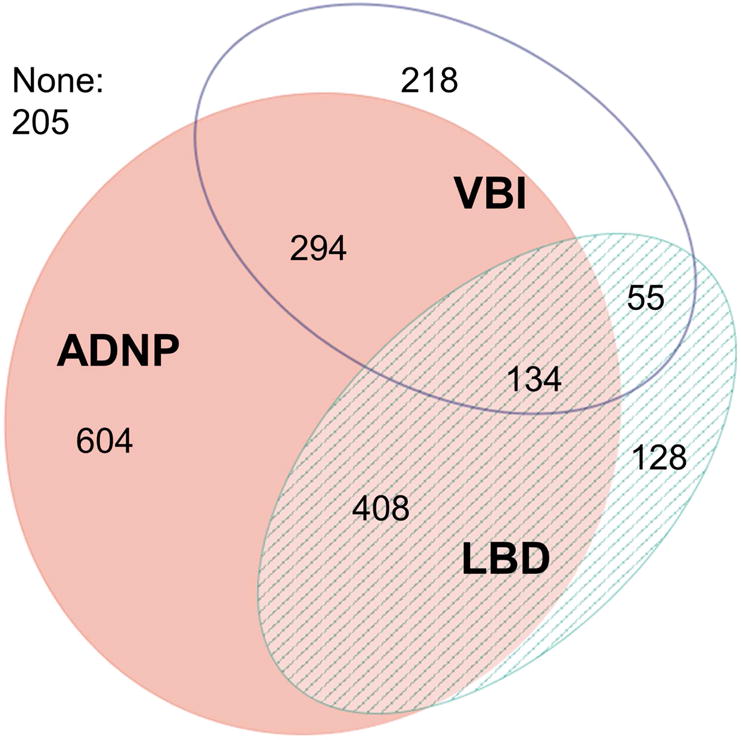
ADNP = moderate/frequent neuritic plaques & Braak stage III–VI; LBD = Lewy bodies in any brain region examined; VBI = gross infarcts and cortical microinfarcts.
Table 1.
Study participant characteristics stratified by ADNP, LBD, and VBI neuropathologic groupings*
| Characteristics† | Low NP | VBI only | LBD only | VBI+LBD | ADNP only | ADNP+ LBD | ADNP+ VBI | ADNP+ LBD+VBI |
|---|---|---|---|---|---|---|---|---|
| Total autopsies, N | 205 | 218 | 128 | 55 | 604 | 408 | 294 | 134 |
| Clinical | ||||||||
| Age at death, mean (SD) | 86.4 (10.6) | 88.2 (9.2) | 78.6 (9.8) | 84.8 (8.9) | 80.3 (10.6) | 77.8 (9.4) | 84.8 (8.6) | 82.9 (8.6) |
| Female | 112 (54.6) | 130 (59.6) | 32 (25.0) | 20 (36.4) | 281 (46.5) | 157 (38.5) | 137 (46.6) | 54 (40.3) |
| Non – white | 13 (6.3) | 12 (5.5) | 4 (3.1) | 4 (7.3) | 27 (4.5) | 20 (4.9) | 26 (8.8) | 17 (12.7) |
| College graduate | 114 (55.6) | 109 (50) | 81 (63.3) | 30 (54.5) | 349 (57.8) | 233 (57.1) | 153 (52.0) | 71 (53.0) |
| History of stroke | 17 (8.3) | 75 (34.4) | 3 (2.3) | 9 (16.7) | 41 (6.9) | 19 (4.7) | 81 (27.9) | 19 (14.3) |
| APOE ε4 allele | 34 (18.1) | 40 (20.4) | 32 (33) | 16 (34) | 284 (54.2) | 216 (62.6) | 136 (54.8) | 73 (62.4) |
| Cognitively impaired | 93 (45.4) | 147 (67.4) | 108 (84.4) | 46 (83.6) | 583 (96.5) | 403 (98.8) | 279 (94.9) | 132 (98.5) |
| Motor problems | 52 (25.4) | 91 (41.7) | 91 (71.1) | 35 (63.6) | 364 (60.3) | 305 (74.8) | 180 (61.2) | 100 (74.6) |
| Psychosis | 11 (5.4) | 20 (9.2) | 50 (39.1) | 14 (25.9) | 143 (23.9) | 165 (40.6) | 71 (24.2) | 50 (37.6) |
| Last visit – death (months) | 8.2 (5.5) | 9.1 (5.3) | 9.1 (6.4) | 10.3 (6.8) | 9.0 (6.2) | 9.3 (6.2) | 9.5 (6.2) | 9.2 (6.5) |
| Longitudinal data | 166 (81.0) | 168 (77.1) | 102 (79.7) | 43(78.2) | 458 (75.8) | 310 (76.0) | 220 (74.8) | 104 (78.2) |
| Initial visit – death (years)‡ | 4.5 (2.1) | 4.5 (2.1) | 3.8 (1.7) | 4.3 (2.1) | 4.4 (2.0) | 3.9 (1.9) | 4.3 (2.0) | 4.5 (2.0) |
| Pathological | ||||||||
| Braak stage V – VI | NA | 13 (5.9) | 7 (5.5) | 1 (1.8) | 493 (81.6) | 308 (75.5) | 208 (70.7) | 98 (73.1) |
| Cortical LBD | NA | NA | 59 (46.1) | 23 (41.8) | NA | 184 (45.1) | NA | 45 (33.6) |
| Severe atherosclerosis | 17 (8.4) | 63 (29) | 11 (8.6) | 8 (14.5) | 68 (11.4) | 36 (8.9) | 46 (15.8) | 22 (16.4) |
| Severe arteriolosclerosis | 3 (1.7) | 54 (25.8) | 5 (4.7) | 13 (24.5) | 43 (8.8) | 31 (9.2) | 54 (20.8) | 37 (30.6) |
| Severe CAA | 8 (4.0) | 7 (3.3) | 4 (3.2) | 5 (9.1) | 101 (17.1) | 52 (13.0) | 54 (19.1) | 25 (20) |
| White matter disease | NA | 37 (17.1) | 10 (7.9) | 10 (18.2) | 72 (12.0) | 48 (12.0) | 48 (16.6) | 19 (14.6) |
| Hippocampal sclerosis | NA | 17 (7.9) | 8 (6.3) | 3 (5.5) | 47 (7.9) | 29 (7.1) | 26 (8.9) | 21 (16.2) |
ADNP, Alzheimer’s disease neuropathology; CAA, cerebral amyloid angiopathy; LBD, Lewy body disease; NA, not applicable; NP, neuropathology; VBI, vascular brain injury
ADNP = moderate/frequent neuritic plaques & Braak stage III–VI; LBD = Lewy bodies in any brain region examined; low NP = no ADNP, no VBI, no LBD, and no other major pathologies; VBI = gross infarcts and cortical microinfarcts.
N,% unless otherwise specified. Participants missing data: stroke=15 (<1%), APOE genotype=284 (13.9%), psychosis=12 (<1%), atherosclerosis=15 (<1%), arteriolosclerosis=296 (14.5%), CAA=53 (2.6%), white matter disease=31 (1.5%), hippocampal sclerosis=309 (16.4%).
Among those with longitudinal data (2+ visits) only.
3.2 Clinical progression in primary models
On average, participants with ADNP+LBD had the fastest rate of progression followed by ADNP only and then ADNP +VBI (Table 2). However, average trajectories of those with ADNP, regardless of co-occurring pathology were relatively similar upon visualization (Figure 2). Significant negative interactions were present between ADNP and LBD (β: −0.47 points; 95% confidence interval [CI]: −0.76, −0.22; p=0.002) and between ADNP and VBI (β: −0.34; 95% CI: −0.53, −0.10; p=0.003), such that participants with ADNP and co-occurring LBD or VBI had a lower rate of progression than would be expected if each pathology independently (additively) contributed to progression.
Table 2.
Estimated annual change in CDR-SB for single and mixed neuropathologies
| Neuropathologies* | Annual change in CDR-SB (95%CI)† |
|---|---|
| Low neuropathology | 0.45 (0.35, 0.57) |
| VBI only | 0.54 (0.42, 0.65) |
| LBD only | 1.07 (0.90, 1.30) |
| ADNP only | 1.70 (1.60, 1.81) |
| ADNP+VBI | 1.45 (1.32, 1.60) |
| ADNP+LBD | 1.85 (1.71, 2.00) |
ADNP, Alzheimer’s disease neuropathology; CDR-SB, Clinical Dementia Rating Sum of Boxes; LBD, Lewy body disease; VBI, vascular brain injury
Based on models with adjustment for age at baseline, sex, non-white race, years of education, and last visit-death interval and weighted by inverse probability of autopsy. Note: A positive value corresponds to increasing impairment over time.
ADNP = moderate/frequent neuritic plaques & Braak stage III–VI; LBD = Lewy bodies in any brain region examined; low NP = no ADNP, no VBI, no LBD, and no other major pathologies; VBI = gross infarcts and cortical microinfarcts.
Figure 2. Model based population mean trajectories of clinical impairment (CDR-SB) prior to death.

ADNP, Alzheimer’s disease neuropathology = moderate/frequent neuritic plaques & Braak III–VI; CDR-SB, Clinical Dementia Rating Sum of Boxes; LBD, Lewy body disease = Lewy bodies in any brain region examined; low NP, low neuropathology = no ADNP, no VBI, no LBD, and no other major pathologies; VBI, vascular brain injury = any gross infarcts or cortical microscopic infarcts. Progression was faster for those with LDB only (p=0.002) and ADNP only (p=0.002) but not VBI only (p=0.2) compared to those with low NP. Compared to ADNP only progression for ADNP+LBD was borderline faster (p=0.06) while progression was slower for ADNP+VBI (p=0.003). Significant negative interactions were present between ADNP and LBD (p=0.002) and between ADNP and VBI (p=0.003), such that participants with ADNP and co-occurring LBD or VBI had a lower rate of progression than would be expected if each pathology independently contributed to progression.
In sensitivity analyses, results did not differ substantially with further adjustment for comorbidities and APOE ε4 allele and in unweighted models that did not use IPW to adjust for potential selection bias (Supplementary Table 2). Among those without dementia at baseline, rates of progression were slower overall but the rate of progression diverged further between those with ADNP only and those with ADNP+LBD (Supplementary Table 2), additional adjustment for comorbidities and APOE ε4 allele did not substantially alter results (data not shown). Inclusion of participants without a clinical visit proximal to death did not substantially change rates of progression (Supplementary Table 3). Compared to those with high education; participants with low education had faster progression of low neuropathology, VBI only, and LBD only but slower progression of ADNP with or without VBI or LBD (Supplementary Table 4); trends otherwise remained similar to primary models. Trends were also similar when using the MMSE as the outcome measure (Supplementary Table 5).
3.3 Microinfarcts and clinical progression in PANDA ADCs
Among 293 PANDA ADC participants 10.9% (n=32) had 2 or more cortical microinfarcts and 34.1% (n=100) had 2 or more subcortical microinfarcts. Among participants without ADNP the number of subcortical microinfarcts was associated with a significantly faster rate of progression compared to those with low neuropathology (β: 0.10; 95% CI: 0.02, 0.18; p=0.002) but the number of cortical microinfarcts was not associated with faster progression (β: 0.05; 95%CI: −0.09, 0.26; p=0.6). The rate of progression of those with ADNP and subcortical microinfarcts was lower than would be expected if effects were additive (β for interaction: −0.22; 95% CI: −0.34, −0.08; p=0.003).
3.4 Semi-quantitative measures and clinical progression
Finally, we examined progression by severity of ADNP (none/low, intermediate, and high) and interactions with VBI and cortical LBD (Table 3). In this analysis we focused on cortical LBD, rather than LBD in general since cortical LBD was most strongly associated with progression of all LBD subtypes (Supplementary Table 6). Both Braak Stage and neuritic plaque density were associated with progression (Supplementary Table 7), thus we used a combined measure of ADNP when fitting interactions. Rates of progression were fastest for those with intermediate ADNP+cortical LBD followed by those with high ADNP with or without cortical LBD or VBI. Rates of progression were similar between those with and without VBI when stratified by level of ADNP (Table 3). Trajectories of those with intermediate ADNP+cortical LBD resulted in a similar level of impairment as those with high ADNP at the last visit (Figure 3). We found a suggestive synergistic or positive interaction between intermediate ADNP and cortical LBD, which was not significant (p=0.37; estimates for interactions shown in Supplementary Table 8). The interaction between intermediate ADNP and VBI was not significant (p=0.4). Trends in those with high ADNP were similar to primary models: negative interactions were significant (p=0.003 for high ADNP×cortical LBD and p=0.03 for high ADNP×VBI). In comparison, in a model without interactions between pathologies the difference in progression for high ADNP+cortical LBD compared to high ADNP only was overestimated while difference in rates for intermediate ADNP×cortical LBD compared to intermediate ADNP only were underestimated (Table 3).
Table 3.
Estimated annual change in CDR-SB according to level of ADNP and co-occurring cortical LBD or VBI
| Neuropathologies* | Modelling interactions | Additive model† |
|---|---|---|
| Annual change in CDR-SB (95%CI)‡ | ||
| Low ADNP | 0.52 (0.42, 0.65) | 0.63 (0.54, 0.75) |
| Low ADNP + cLBD | 1.44 (1.16, 1.75) | 1.25 (1.05, 1.46) |
| Low ADNP + VBI | 0.56 (0.48, 0.70) | 0.53 (0.43, 0.63) |
| Intermediate ADNP | 1.10 (0.91, 1.31) | 1.21 (1.06, 1.39) |
| Intermediate ADNP + cLBD | 2.27 (1.83, 2.64) | 1.83 (1.60, 2.07) |
| Intermediate ADNP + VBI | 1.01 (0.77, 1.25) | 1.11 (0.96, 1.29) |
| High ADNP | 1.82 (1.72, 1.93) | 1.75 (1.65, 1.84) |
| High ADNP + cLBD | 2.02 (1.75, 2.29) | 2.37 (2.18, 2.58) |
| High ADNP + VBI | 1.62 (1.48, 1.76) | 1.65 (1.53, 1.76) |
ADNP, Alzheimer’s disease neuropathology; CDR-SB, Clinical Dementia Rating Sum of Boxes; cLBD, cortical Lewy body disease; VBI, vascular brain injury
Intermediate ADNP = moderate/frequent neuritic plaques & Braak III–IV; high ADNP= moderate or frequent plaques & Braak V–VI; VBI = any gross infarcts or cortical microscopic infarcts.
Interactions between pathologies not included in model
Models included adjustment for age at baseline, sex, non-white race, years of education, and interval between last visit and death as well as inverse probability of autopsy selection weights. Note: A positive value corresponds to increasing impairment over time.
Figure 3. Model based population mean trajectories of clinical impairment (CDR-SB) prior to death associated with intermediate and high ADNP with and without co-occurring cortical LBD.

ADNP, Alzheimer’s disease neuropathology; CDR-SB, Clinical Dementia Rating Sum of Boxes; cLBD, cortical Lewy body disease. Intermediate ADNP = moderate/frequent neuritic plaques & Braak III–IV; high ADNP= moderate or frequent plaques & Braak V–VI. Trajectories for ADNP+VBI were similar to ADNP only and are not shown. Compared to high ADNP, progression in intermediate ADNP+cLBD was significantly faster (p=0.04), but progression in high ADNP+cLBD was not significantly different (p=0.2). Significant negative interactions were present between high ADNP and cLBD (p=0.003) such that participants with ADNP and co-occurring LBD or VBI had a lower rate of progression than would be expected if each pathology independently contributed to progression.
4. DISCUSSION
This study reports on novel findings regarding interactions of ADNP and LBD or VBI in the association with clinical progression in a large autopsy sample. In primary models, compared to ADNP only, progression in those with ADNP+LBD was slightly faster while progression in those with ADNP+VBI was slightly slower. However, trajectories of those with ADNP were relatively similar regardless of co-occurring pathology, and there were significant negative interactions between both ADNP and LBD and ADNP and VBI. Findings were similar across sensitivity analyses. In a subset of those without dementia, however, trajectories diverged more from those with ADNP only. In secondary analyses, we found clinical progression to be fastest among participants with co-occurring intermediate ADNP+cortical LBD. Meanwhile, trajectories of progression were similar among those with high ADNP regardless of co-occurring LBD or VBI. Together these findings suggest that concomitant pathologies, in particular LBD, may have a larger effect on progression in those with intermediate compared to high ADNP.
Our primary findings of negative interactions between ADNP and LBD or VBI were unexpected, but were limited to those with high ADNP (e.g. those with higher Braak Stages), in secondary analyses. A prior study using NACC data also found a significant negative interaction between cerebrovascular disease and Braak stage in association with dementia [28]. Severe ADNP, in particular tau neurofibrillary pathology in cortical regions (Braak Stage V/VI), may clinically overwhelm the effect of co-occurring neuropathologies, especially closer to death. Co-occurring vascular lesions were not associated with cognitive decline beyond that of ADNP in individuals with high ADNP in several other studies [11,12,45]. Considering that NACC participants tended to have severe ADNP there may also have been a ceiling effect in the rates of progression observable.
On the other hand, our secondary findings also suggest there may be synergistic interactions between intermediate ADNP and cortical LBD on the rate of clinical progression. ADNP and LBD interact to produce more rapid cognitive decline in mice [46]. In prior autopsy studies rates of decline were faster in those with ADNP+LBD than ADNP only [47,48]; but another study did not find evidence for interactions [16]. LBD in ADNP may be the result of a distinct pathogenesis and clinical course compared to ADNP only [47,49]. Individuals with intermediate ADNP+cortical LBD may have a worse prognosis as they had more rapid progression and ended with similar levels of impairment as those with high ADNP by the last study visit. Future research with prospective biomarkers is needed to elucidate further the temporal effects of these pathologies in cases with mixed pathologies.
In primary models, participants with ADNP+VBI had slightly slower rates of progression than those with ADNP only. This small but unusual result likely reflects differences in level of ADNP between those with and without VBI. Braak Stage was lower in those with ADNP+VBI compared to ADNP only and rates were more similar between those with and without VBI when split by level of ADNP. Some prior studies found no interactions between ADNP and VBI using pathologic [9] or imaging data [10], but that VBI was associated with more rapid cognitive decline. Information on number of infarcts was not available for most participants in NACC, and may have reduced our ability to detect associations between VBI and clinical progression. Our findings in PANDA ADC participants highlight that clinical impairment due to VBI may be detectable only in those with multiple VBI, similar to other studies [9,32]. Interestingly, only subcortical microinfarcts were associated with progression, in contrast to other studies finding associations between cortical microinfarcts and cognitive impairment [34,35]. Future studies may be useful to assess the impact of other types of vascular pathologies, such as specific vessel disorders, on progression in mixed ADNP.
This study has several important limitations. Neuropathologic assessments may not reflect burden of pathology when clinical progression was measured. Excluding participants whose last visit was more than 2 years prior to death may help minimized this issue. We focused on mixes of three common pathologies; however other overlapping pathologic features may impact cognition. Information was not available on more recently identified pathologies such as TDP-43 [50]. Biological interactions between pathologies may not have been captured in this analysis. The CDR-SB is based on overall clinical assessment and measures of specific symptoms may be more sensitive to detecting differences between each type of pathology. NACC participants were predominantly Caucasian, well-educated older adults with a relatively high prevalence of the APOE ε4 allele and dementia; future studies maybe needed in diverse populations. As participants would not have known their APOE ε4 allele status prior to enrollment the increased prevalence may be reflective of the high prevalence of dementia and ADNP. Follow-up was over later disease stages prior to death; in studies with lower prevalence of dementia, longer followup, or earlier disease stages, trajectories may be more similar to those found in participants without dementia at baseline or those with intermediate ADNP, where ADNP+LBD trajectories further diverged from ADNP only.
This study also has appreciable strengths. We used a large data set of standardized clinical evaluations that could provide power to detect statistical interactions. Few samples can rival the size (and thus statistical power) of NACC data. We modelled progression allowing for differences between participants with mixed pathologies and those with individual pathologies. We conducted a variety of sensitive analyses using alternative measures, restriction criteria, and covariate adjustment to explore potential biases and extend generalizability of our findings. We investigated associations in a subset of participants without dementia, and with the same neuropathologic assessment protocols (PANDA ADCs) and we also explored relationships between clinical progression and semi-quantitative measures of pathologic burden. Finally, we incorporated methods that can account for potential bias due to autopsy sampling.
Overall we found evidence that ADNP+LBD is associated with faster rate of progression than ADNP only, but that this effect is most evident in those with intermediate levels of ADNP. Modelling individual pathologies as independent factors may overestimate differences in the rate of progression between those with single and mixed ADNP, particularly in those with high ADNP. Given the high prevalence of mixed pathologies among participants with dementia, effective treatments may need to target multiple etiologies. However, as highlighted by our complex findings, differentiating patients with mixed vs. single ADNP may be challenging in practice without accurate biomarkers for multiple pathologies.
Supplementary Material
Research in Context.
Systemic review
While many studies document that multiple brain pathologies (aka mixed neuropathologies) are common in autopsied older adults; it is unclear whether coexisting pathologies interact or independently affect clinical disease progression. Large studies comparing premortem trends in clinical progression between individuals with single and mixed neuropathologies are lacking.
Interpretation
Among older adults with Alzheimer’s disease neuropathology (ADNP), the effect of additional pathologies on clinical progression may be greater in those with intermediate levels of ADNP than in those with severe ADNP. Modelling individual pathologies as independent factors may overestimate differences in the rate of progression between those with single and mixed ADNP, particularly in those with high ADNP.
Future directions
Considering interactions may be important for understanding the impact of multiple pathologies on clinical progression. Future research with prospective biomarkers will be needed to elucidate further the temporal effects of pathologies in cases with mixed pathologies.
Acknowledgments
Conflicts/Funding Sources
We are deeply grateful to all of the study participants, clinicians, and other workers at the ADCs, NACC, and ACT that made this research possible. We also thank NACC, ACT, UW, and OHSU staff including Allison Beller, Robin Guariglia, Lilah Besser, and Mark Bollenbeck for help obtaining data. Preliminary versions of this work were included in a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy from the Department of Epidemiology, University of Washington (WDB). Analyses were run using the UW Center for Studies in Demography and Ecology’s computer simulation cluster.
The NACC database is funded by NIA/NIH Grant U01 AG016976. NACC data are contributed by the NIA-funded ADCs: P30 AG019610 (PI Eric Reiman, MD), P30 AG013846 (PI Neil Kowall, MD), P50 AG008702 (PI Scott Small, MD), P50 AG025688 (PI Allan Levey, MD, PhD), P50 AG047266 (PI Todd Golde, MD, PhD), P30 AG010133 (PI Andrew Saykin, PsyD), P50 AG005146 (PI Marilyn Albert, PhD), P50 AG005134 (PI Bradley Hyman, MD, PhD), P50 AG016574 (PI Ronald Petersen, MD, PhD), P50 AG005138 (PI Mary Sano, PhD), P30 AG008051 (PI Steven Ferris, PhD), P30 AG013854 (PI M. Marsel Mesulam, MD), P30 AG008017 (PI Jeffrey Kaye, MD), P30 AG010161 (PI David Bennett, MD), P50 AG047366 (PI Victor Henderson, MD, MS), P30 AG010129 (PI Charles DeCarli, MD), P50 AG016573 (PI Frank LaFerla, PhD), P50 AG016570 (PI Marie-Francoise Chesselet, MD, PhD), P50 AG005131 (PI Douglas Galasko, MD), P50 AG023501 (PI Bruce Miller, MD), P30 AG035982 (PI Russell Swerdlow, MD), P30 AG028383 (PI Linda Van Eldik, PhD), P30 AG010124 (PI John Trojanowski, MD, PhD), P50 AG005133 (PI Oscar Lopez, MD), P50 AG005142 (PI Helena Chui, MD), P30 AG012300 (PI Roger Rosenberg, MD), P50 AG005136 (PI Thomas Montine, MD, PhD), P50 AG033514 (PI Sanjay Asthana, MD, FRCP), P50 AG005681 (PI John Morris, MD), and P50 AG047270 (PI Stephen Strittmatter, MD, PhD). PANDA ADCs are supported by NIH P50 AG005136 (UW ADC) and NIH grants P30 AG008017, R01 AG024059, M01 RR000334, UL1 RR024140 as well as Intel Corporation, and Department of Veterans Affairs (OHSU ADC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69:2197–204. doi: 10.1212/01.wnl.0000271090.28148.24. [DOI] [PubMed] [Google Scholar]
- 2.Montine TJ, Sonnen JA, Montine KS, Crane PK, Larson EB. Adult Changes in Thought Study: Dementia is an Individually Varying Convergent Syndrome with Prevalent Clinically Silent Diseases that may be Modified by Some Commonly Used Therapeutics. Current Alzheimer Research. 2012;9:718. doi: 10.2174/156720512801322555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawas CH, Kim RC, Sonnen JA, Bullain SS, Trieu T, Corrada MM. Multiple pathologies are common and related to dementia in the oldest-old: The 90+ Study. Neurology. 2015;85:535–42. doi: 10.1212/WNL.0000000000001831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Attems J, Jellinger K. Neuropathological correlates of cerebral multimorbidity. Curr Alzheimer Res. 2013;10:569–77. doi: 10.2174/15672050113109990002. [DOI] [PubMed] [Google Scholar]
- 5.Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA. 1997;277:813–7. [PubMed] [Google Scholar]
- 6.Zekry D, Duyckaerts C, Moulias R, Belmin J, Geoffre C, Herrmann F, et al. Degenerative and vascular lesions of the brain have synergistic effects in dementia of the elderly. Acta Neuropathol. 2002;103:481–7. doi: 10.1007/s00401-001-0493-5. [DOI] [PubMed] [Google Scholar]
- 7.Riekse RG, Leverenz JB, McCormick W, Bowen JD, Teri L, Nochlin D, et al. Effect of vascular lesions on cognition in Alzheimer’s disease: a community-based study. J Am Geriatr Soc. 2004;52:1442–8. doi: 10.1111/j.1532-5415.2004.52405.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schneider JA, Boyle PA, Arvanitakis Z, Bienias JL, Bennett DA. Subcortical infarcts, Alzheimer’s disease pathology, and memory function in older persons. Ann Neurol. 2007;62:59–66. doi: 10.1002/ana.21142. [DOI] [PubMed] [Google Scholar]
- 9.Schneider JA, Wilson RS, Bienias JL, Evans DA, Bennett DA. Cerebral infarctions and the likelihood of dementia from Alzheimer disease pathology. Neurology. 2004;62:1148–55. doi: 10.1212/01.WNL.0000118211.78503.F5. [DOI] [PubMed] [Google Scholar]
- 10.Vemuri P, Lesnick TG, Przybelski SA, Knopman DS, Preboske GM, Kantarci K, et al. Vascular and amyloid pathologies are independent predictors of cognitive decline in normal elderly. Brain. 2015;138:761–71. doi: 10.1093/brain/awu393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chui HC, Zarow C, Mack WJ, Ellis WG, Zheng L, Jagust WJ, et al. Cognitive impact of subcortical vascular and Alzheimer’s disease pathology. Annals of Neurology. 2006;60:677–687. doi: 10.1002/ana.21009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jellinger KA, Attems J. Prevalence and impact of cerebrovascular pathology in Alzheimer’s disease and parkinsonism. Acta Neurol Scand. 2006;114:38–46. doi: 10.1111/j.1600-0404.2006.00665.x. [DOI] [PubMed] [Google Scholar]
- 13.Wirths O, Weickert S, Majtenyi K, Havas L, Kahle PJ, Okochi M, et al. Lewy body variant of Alzheimer’s disease: alpha-synuclein in dystrophic neurites of A beta plaques. Neuroreport. 2000;11:3737–41. doi: 10.1097/00001756-200011270-00029. [DOI] [PubMed] [Google Scholar]
- 14.Hamilton RL. Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 2000;10:378–84. doi: 10.1111/j.1750-3639.2000.tb00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jellinger KA, Attems J. Prevalence and impact of vascular and Alzheimer pathologies in Lewy body disease. Acta Neuropathol. 2008;115:427–36. doi: 10.1007/s00401-008-0347-5. [DOI] [PubMed] [Google Scholar]
- 16.Schneider JA, Arvanitakis Z, Yu L, Boyle PA, Leurgans SE, Bennett DA. Cognitive impairment, decline and fluctuations in older community-dwelling subjects with Lewy bodies. Brain. 2012;135:3005–14. doi: 10.1093/brain/aws234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boyle PA, Wilson RS, Yu L, Barr AM, Honer WG, Schneider JA, et al. Much of late life cognitive decline is not due to common neurodegenerative pathologies. Annals of Neurology. 2013;74:478–489. doi: 10.1002/ana.23964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boyle PA, Yu L, Wilson RS, Schneider JA, Bennett DA. Relation of neuropathology with cognitive decline among older persons without dementia. Front Aging Neurosci. 2013;5:50. doi: 10.3389/fnagi.2013.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jellinger KA. Interaction between α-synuclein and other proteins in neurodegenerative disorders. ScientificWorldJournal. 2011;11:1893–907. doi: 10.1100/2011/371893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beekly DL, Ramos EM, van Belle G, Deitrich W, Clark AD, Jacka ME, et al. The National Alzheimer’s Coordinating Center (NACC) Database: an Alzheimer disease database. Alzheimer Dis Assoc Disord. 2004;18:270–7. [PubMed] [Google Scholar]
- 21.Beekly DL, Ramos EM, Lee WW, Deitrich WD, Jacka ME, Wu J, et al. The National Alzheimer’s Coordinating Center (NACC) database: the Uniform Data Set. Alzheimer Dis Assoc Disord. 2007;21:249–58. doi: 10.1097/WAD.0b013e318142774e. [DOI] [PubMed] [Google Scholar]
- 22.Howieson DB, Holm LA, Kaye JA, Oken BS, Howieson J. Neurologic function in the optimally healthy oldest old Neuropsychological evaluation. Neurology. 1993;43:1882–1882. doi: 10.1212/WNL.43.10.1882. [DOI] [PubMed] [Google Scholar]
- 23.Kaye J, Michael Y, Calvert J, Leahy M, Crawford D, Kramer P. Exceptional brain aging in a rural population-based cohort. J Rural Health. 2009;25:320–5. doi: 10.1111/j.1748-0361.2009.00237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaye JA, Maxwell SA, Mattek N, Hayes TL, Dodge H, Pavel M, et al. Intelligent Systems For Assessing Aging Changes: home-based, unobtrusive, and continuous assessment of aging. J Gerontol B Psychol Sci Soc Sci. 2011;66(Suppl 1):i180–190. doi: 10.1093/geronb/gbq095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petersen J, Austin D, Mattek N, Kaye J. Time Out-of-Home and Cognitive, Physical, and Emotional Wellbeing of Older Adults: A Longitudinal Mixed Effects Model. PLoS ONE. 2015;10 doi: 10.1371/journal.pone.0139643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–86. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 27.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC, et al. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement. 2012;8:1–13. doi: 10.1016/j.jalz.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thal DR, Rüb U, Orantes M, Braak H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 30.McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65:1863–72. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 31.Gorelick PB, Scuteri A, Black SE, DeCarli C, Greenberg SM, Iadecola C, et al. Vascular Contributions to Cognitive Impairment and Dementia A Statement for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke. 2011;42:2672–713. doi: 10.1161/STR.0b013e3182299496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sonnen JA, Larson EB, Crane PK, Haneuse S, Li G, Schellenberg GD, et al. Pathological correlates of dementia in a longitudinal, population-based sample of aging. Ann Neurol. 2007;62:406–13. doi: 10.1002/ana.21208. [DOI] [PubMed] [Google Scholar]
- 33.Arvanitakis Z, Leurgans SE, Barnes LL, Bennett DA, Schneider JA. Microinfarct pathology, dementia, and cognitive systems. Stroke. 2011;42:722–7. doi: 10.1161/STROKEAHA.110.595082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corrada MM, Sonnen JA, Kim RC, Kawas CH. Microinfarcts are common and strongly related to dementia in the oldest-old: The 90+ study. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association. 2016;12:900–8. doi: 10.1016/j.jalz.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.White L, Petrovitch H, Hardman J, Nelson J, Davis DG, Ross GW, et al. Cerebrovascular pathology and dementia in autopsied Honolulu-Asia Aging Study participants. Ann N Y Acad Sci. 2002;977:9–23. doi: 10.1111/j.1749-6632.2002.tb04794.x. [DOI] [PubMed] [Google Scholar]
- 36.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–4. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- 37.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–98. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 38.Bennett DA, Wilson RS, Schneider JA, Evans DA, Mendes de Leon CF, Arnold SE, et al. Education modifies the relation of AD pathology to level of cognitive function in older persons. Neurology. 2003;60:1909–15. doi: 10.1212/01.wnl.0000069923.64550.9f. [DOI] [PubMed] [Google Scholar]
- 39.Haneuse S, Schildcrout J, Crane P, Sonnen J, Breitner J, Larson E. Adjustment for selection bias in observational studies with application to the analysis of autopsy data. Neuroepidemiology. 2009;32:229–39. doi: 10.1159/000197389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Little RJA, Rubin DB. Statistical analysis with missing data. New York: Wiley; 1987. [Google Scholar]
- 41.Hernán MA, Hernández-Díaz S, Robins JM. A structural approach to selection bias. Epidemiology. 2004;15:615–25. doi: 10.1097/01.ede.0000135174.63482.43. [DOI] [PubMed] [Google Scholar]
- 42.Cole SR, Hernan MA. Constructing Inverse Probability Weights for Marginal Structural Models. American Journal of Epidemiology. 2008;168:656–64. doi: 10.1093/aje/kwn164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Efron B, Tibshirani R. An introduction to the bootstrap. New York: Chapman & Hall; 1994. [Google Scholar]
- 44.Micallef L, Rodgers P. eulerAPE: Drawing Area-Proportional 3-Venn Diagrams Using Ellipses. PLoS ONE. 2014;9:e101717. doi: 10.1371/journal.pone.0101717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Esiri MM, Nagy Z, Smith MZ, Barnetson L, Smith AD. Cerebrovascular disease and threshold for dementia in the early stages of Alzheimer’s disease. Lancet. 1999;354:919–20. doi: 10.1016/S0140-6736(99)02355-7. [DOI] [PubMed] [Google Scholar]
- 46.Clinton LK, Blurton-Jones M, Myczek K, Trojanowski JQ, LaFerla FM. Synergistic Interactions between Abeta, tau, and alpha-synuclein: acceleration of neuropathology and cognitive decline. J Neurosci. 2010;30:7281–9. doi: 10.1523/JNEUROSCI.0490-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Olichney JM, Galasko D, Salmon DP, Hofstetter CR, Hansen LA, Katzman R, et al. Cognitive decline is faster in Lewy body variant than in Alzheimer’s disease. Neurology. 1998;51:351–7. doi: 10.1212/wnl.51.2.351. [DOI] [PubMed] [Google Scholar]
- 48.Kraybill ML, Larson EB, Tsuang DW, Teri L, McCormick WC, Bowen JD, et al. Cognitive differences in dementia patients with autopsy-verified AD, Lewy body pathology, or both. Neurology. 2005;64:2069–73. doi: 10.1212/01.WNL.0000165987.89198.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hansen L, Salmon D, Galasko D, Masliah E, Katzman R, DeTeresa R, et al. The Lewy body variant of Alzheimer’s disease: a clinical and pathologic entity. Neurology. 1990;40:1–8. doi: 10.1212/wnl.40.1.1. [DOI] [PubMed] [Google Scholar]
- 50.Wilson RS, Yu L, Trojanowski JQ, Chen E-Y, Boyle PA, Bennett DA, et al. TDP-43 pathology, cognitive decline, and dementia in old age. JAMA Neurol. 2013;70:1418–24. doi: 10.1001/jamaneurol.2013.3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.