


Abstract
The T cell compartment must contain diversity in both T cell receptor (TCR) repertoire and cell state to provide effective immunity against pathogens. However, it remains unclear how differences in the TCR contribute to heterogeneity in T cell state. Single cell RNA-sequencing (scRNA-seq) can allow simultaneous measurement of TCR sequence and global transcriptional profile from single cells. However, current methods for TCR inference from scRNA-seq are limited in their sensitivity and require long sequencing reads, thus increasing the cost and decreasing the number of cells that can be feasibly analyzed. Here we present TRAPeS, a publicly available tool that can efficiently extract TCR sequence information from short-read scRNA-seq libraries. We apply it to investigate heterogeneity in the CD8+ T cell response in humans and mice, and show that it is accurate and more sensitive than existing approaches. Coupling TRAPeS with transcriptome analysis of CD8+ T cells specific for a single epitope from Yellow Fever Virus (YFV), we show that the recently described ‘naive-like’ memory population have significantly longer CDR3 regions and greater divergence from germline sequence than do effector-memory phenotype cells. This suggests that TCR usage is associated with the differentiation state of the CD8+ T cell response to YFV.
INTRODUCTION
The population of antigen-specific CD8+ T cells formed in response to infection or vaccination is highly heterogeneous in terms of function and phenotype (1,2). Efforts to deconvolve this cellular heterogeneity have used flow cytometry, mass spectrometry, and more recently, single-cell RNA-sequencing (3). These approaches have identified a reliable set of phenotypic markers that can classify antigen-specific T cells into a large number of subsets, and distinguish them from antigen-naive T cells. However, recent work also suggests that some antigen-experienced CD8+ T cells can have a naive-like phenotype, meaning that despite their potential to effectively respond to an antigen, they show transcriptomic and surface marker similarities to antigen-naïve T cells (4–6). The cellular heterogeneity in the T cell compartment is thought to arise from different exposure to differentiation cues such as antigen dose, duration of contact, and cytokines. How the T cell receptor (TCR) sequence expressed by each T cell contributes to that cellular heterogeneity is not fully understood.
The T cell receptor is a heterodimer of two chains—alpha and beta, each consisting of three types of genomic segments—variable (V), joining (J) and constant (C) (the beta chain includes an additional short diversity (D) segment; Methods) (7). The V and J segments are selected out of a pool of several dozen loci encoded in the germline genome, through a recombination process. The diversity of the TCR repertoire (estimated at ∼107 in humans (7)) is further enhanced by random insertions and deletions into the complementarity determining region 3 (CDR3)—the junction between the V and J segments, which largely determines the ability of the cell to recognize specific antigens. However despite this diversity, some T cell responses can include TCRs that are identical between individuals - known as ‘public’ clonotypes, while other T cell responses use TCRs that are unique to each individual (‘private’ clonotypes). Previous studies have shown that these public clonotypes tend to appear at a higher frequency and have a shorter CDR3 region, possibly as a result of a more efficient recombination process (7–10).
Unlike analysis of the cell state, the clonal diversity of the TCR repertoire has to date been studied mostly in aggregated samples from pools of T cells rather than individual cells (7,11,12). This approach has two significant limitations: (i) since each chain of the TCR (alpha, beta) is a separate transcript, it cannot determine which chains are co-expressed in the same cell, leading to a partial view of the TCR identity; (ii) the sequence of the TCR and the global transcriptional state of the cell that expresses it cannot be simultaneously determined. Previous studies have profiled TCR use in single cells, but these studies were limited in the number of transcripts that were quantified (11,13).
Single cell RNA-seq can generate full-length sequence information for many transcripts in individual cells including the alpha and beta chains of the TCR. However, standard methods to map sequence fragments to the genome (14) cannot be directly used for reconstructing and estimating the abundance of TCRs because of the highly variable nature of the CDR3 regions. One approach to address this challenge is to rely on scRNA-seq with long sequencing reads (>100 bp), which can cover the entire CDR3 region along with the flanking V and J sub-segments (15). The underlying TCR (along with the junctional diversification events) can then be reconstructed using methods similar to TCR-seq population repertoire analysis (7,16). However, sequencing with long reads is costly and time consuming, thus a method to successfully reconstruct TCRs from shorter, paired-end reads is desirable. Another approach (15,17,18) relies on previous methods for de-novo transcriptome or genome assembly to reconstruct the CDR3 region (19,20). In general, de-novo assemblers were designed with a very large input data set and long reads in mind, and use the concept of de-bruijn graphs to achieve high efficiency. Indeed the TCR reconstruction methods that use this approach have mainly been tested on long RNA-seq libraries (except scTCRseq which was also tested on simulated short reads (17)). However, more accurate yet possibly more computationally intensive algorithms are feasible and may be more appropriate for the smaller target of reconstructing only the TCR.
To address this, we have developed ‘TCR Reconstruction Algorithm for Paired-End Single cell’ (TRAPeS), a software capable of accurately reconstructing TCRs from paired-end sequencing libraries of single cells, even at short (25 bp) read length. Unlike the previous methods, TRAPeS does not reduce the input sequences into k-mers, but rather works on the original reads - leading to increased sensitivity. We benchmarked TRAPeS on a diverse set of viral stimulations, and then demonstrate how simultaneous analysis of TCR properties and global expression profiling in individual cells helps relate specific TCR properties such as CDR3 length to heterogeneity of T cell state among CD8+ T cells that respond to YFV. TRAPeS is publicly available, and can be readily used to investigate the relationship between the TCR repertoire and cellular phenotype.
MATERIALS AND METHODS
TRAPeS
The TRAPeS algorithm has four main steps, each applied separately to the alpha and beta chains:
Identifying putative pairs of V and J segments. In order to recognize the V and J segments of the TCR, TRAPeS takes as input the alignment of the RNA-seq reads to the genome. TRAPeS searches for a paired-end read where one mate maps to a V segment while the other mate is mapped to a J segment, and takes those V–J pairs as putative candidates for the CDR3 reconstruction. In a case where there are no such pairings, TRAPeS takes all possible V-J combinations of V and J segments that have V–C and J–C pairing (i.e. reads where one mate maps to V or J and the other mate maps to the C segments). We note that reads are not successfully aligned to D segments of the beta chain due to their short length. Thus, for the beta chain, reconstruction of the CDR3 includes reconstruction of the D segment sequence. In addition, TRAPeS allows the user to specify the maximum number of reconstructions per chain. If the number of possible V–J pairs exceeds this number, TRAPeS ranks the pairs based on the number of reads initially mapped to them, and only attempts to reconstruct the top pairs.
Collecting putative CDR3-originating reads. TRAPeS finds the putative CDR3- originating reads by taking all the unmapped reads whose mates map to the V/J/C segments. In addition, since the first step of the CDR3 reconstruction includes alignment to the genomic V/J sequences (see below), TRAPeS also collects the reads that map to the V and J segments.
-
Reconstructing the CDR3. Using an iterative dynamic programming algorithm, TRAPeS extends the V and J regions. TRAPeS takes only the bases at the ends of the V and J segments closest to the CDR3 (3′ of the V segment and 5′ of the J segment). The number of initial bases is a parameter that can be tuned, set by default to min(length(V), length(J)). If the specified length is longer than the J segment, TRAPeS concatenates the J sequence to the beginning of the C sequence and use this extended segment as the initial J segment. In each iteration, TRAPeS aligns all the reads to the V and J segments separately with the Needleman–Wunsch algorithm, using the following scoring scheme: +1 for a match, –1 for a mismatch, –20 for gap opening and –4 for gap extension. In addition, we don’t penalize for having the read ‘flank’ the V and J toward their 3′ and 5′, respectively.
Next, TRAPeS takes all the reads that aligned to the V and J segments above a certain score threshold, and build the ‘extended’ V and J sequences based on the reads. For each position, we take the base that appears in most reads as the chosen sequence for this position. This way, we extend the V and J regions in each iteration and also correct for mutations or SNPs in the known genomic V and J segments. TRAPeS repeats this step until the extended V and extended J overlap, or until TRAPeS reaches a number of predefined iterations. If no overlap is found, TRAPeS also offers an optional ‘one-sided’ mode, where it will attempt to determine the productivity (see below) of only the extended V segment. For this work, we used a threshold score of 21 for the alignment of the reads. However, in some cases a lower threshold was required, thus if no sequence was reconstructed we run TRAPeS with a scoring threshold of 15.
Separating similar TCRs and determining chain productivity. Since some V and J segments have similar sequence, reads can be mapped to several segments, creating few similar putative V–J pairs. In addition, two alpha or beta chains can be created within a single cell. TRAPeS takes all possible pairing and attempts to reconstruct the CDR3 region for all pairs. After reconstruction, full-length TCR sequences are created by extending the reconstructed region with the known reference sequences. Then, TRAPeS runs RSEM (14) on all reconstructed TCRs and the set of reads used as input (and their mates) in order to rank the TCRs based on the relative abundance. Next, TRAPeS determines if the TCR is productive: V and J segments are in the same reading frame and the CDR3 does not contain a stop codon. TRAPeS outputs a file with a summary of all possible reconstructions (see Supplementary Table S1 for example) for all cells, as well as separate files for each cell with the full-length TCR sequences. For this paper we used the productive chain with the highest expression as the TCR sequence for each cell.
TRAPeS is implemented in python. To increase performance, the CDR3 reconstruction using the dynamic programming algorithm is implemented in C++, and uses the seqan package (21). TRAPeS is freely available and can be downloaded in the following link: https://github.com/YosefLab/TRAPeS
TRAPeS can be easily extended to work with single-end data. The reconstruction algorithm only requires the paired-end information for the recognition of V/J segments and CDR3-originating reads, which can be easily done in single-end reads by searching for partial alignment of the read edges to the V/J segments. This feature will be available in the next TRAPeS version.
Single cell sorting
Mouse LCMV experiments
Female C57BL/6 mice (The Jackson Laboratory), aged 7 weeks, were infected with 2 × 105 plaque forming units (PFU) LCMV Armstrong intraperitoneally i.p. or 4 × 106 PFU LCMV Clone 13 i.v. LCMV viruses were a generous gift from Dr. E John Wherry (University of Pennsylvania, Perelman School of Medicine). Peripheral blood was obtained from the mice at day 7 post infection (p.i.) and lymphocytes were enriched using LSM density centrifugation. Cells were prestained with a near-IR fixable live/dead marker (Life Technologies, cat# L34976) and an APC- conjugated dextramer reagent for gp33 (Immudex, cat# A2160-APC) according to manufacturer recommendations. The cells were then stained with the following antibodies: FITC 2B4 (BioLegend, cat# 133504), PerCP-Cy5.5 CD44 (BioLegend, cat# 103032), PE KLRG1 (BioLegend, cat# 138408), PE-Cy7 PD1 (BioLegend, cat# 135215), BV421 CD127 (BioLegend, cat# 135024), BV510 CD8A (BioLegend, cat# 100752).
Human CMV experiments (Donor 1)
Blood samples were obtained from a donor with detectable NLV-specific CD8+ T cell response. Lymphocytes were enriched via Ficoll gradient and prestained with a near-IR fixable live/dead marker (Life Technologies, cat# L34976) and an APC-conjugated dextramer reagent (Immudex, cat# WB2132-APC). The cells were then stained with the following antibodies: FITC CD8A (BioLegend, cat# 300906), PerCP-Cy5.5 CCR7 (BioLegend, cat# 353220), PE CD3 (BioLegend, cat# 317308), BV605 CD45RA (BioLegend, cat# 304133).
Human YFV experiment (Donor 2)
A healthy volunteer was vaccinated with a single dose (0.5 ml containing at least 105 PFU) of 17D live-attenuated yellow fever vaccine strain administered subcutaneously. Seroconversion after vaccination was confirmed by assaying the neutralizing antibody titers for YF-17D (data not shown). A whole blood sample was obtained 9 months post-vaccination and lymphocytes were enriched from whole blood via Ficoll gradient centrifugation and a CD8 negative selection magnetic bead kit (Miltenyi Biotec). Cells were prestained with a live/dead marker (Life Technologies, cat# L34976) and an APC-labeled tetramer reagent (NS4B 214–222 LLWNGPMAV, kindly provided by Dr Rafi Ahmed). The cells were then stained with the following antibodies: FITC CD8A (BioLegend, cat# 300906), PE CXCR3 (BioLegend, cat# 353705), PE-Cy7 CCR7 (BioLegend, cat# 353226), BV421 IL2Rb (BioLegend, cat# 339009), BV510 CD3 (BioLegend, cat# 317332), BV605 CD95 (BioLegend, cat# 305627), BV780 CD45RA (BioLegend, cat# 304140).
Human hepatitis C experiment (Donor 3)
Patient 355 (59-year old Male, infected with genotype 1a HCV, baseline viral load 467 000 IU/ml) received a prime vaccination of ChAd3-NSmut (2.5 × 1010 viral particles) and an MVA-NSmut (2 × 108 plaque forming units) boost vaccination 8 weeks later. PBMC were collected 14 weeks post-boost vaccination for assessment of single cell gene expression (22). PBMC were thawed and prestained with a live/dead marker (Life Technologies, cat# L34976) and a PE-conjugated pentamer reagent (PE-labeled HCV NS31406–1415 (KLSALGINAV; HLA-A*0201)). The cells were then stained with the following antibodies: FITC 2B4 (BioLegend, cat# 329505), PerCP-eFluor 710 LAG3 (eBioscience, cat# 46–2239), PE-Cy7 CCR7 (BioLegend, cat# 329919), APC CD39 (BioLegend, cat# 328209), BV421 PD1 (BioLegend, cat# 329919), BV510 CD3 (BioLegend, cat# 317332), BV605 CD8A (BioLegend, cat# 301040), BV780 CD45RA (BioLegend, cat# 304140).
The relevant institutional review boards approved all human subject protocols, and all subjects provided written consent before enrollment.
Single cell sorts
All single cell sorts were performed on a BD Aria II with a 70um nozzle. Cells were sorted into 5 μl of Qiagen TCL Buffer plus 1% beta-mercaptoethanol (v/v). Immediately following sorting, plates were sealed, vortexed on high for 30 s, and spun at 400 g for 1 min prior to flash freezing on dry ice. Samples were stored at –80°C until library preparation.
RNA sequencing
Single cell lysates were converted to cDNA following capture with Agencourt RNA Clean beads using the SmartSeq2 protocol as previously described (23). The cDNA was amplified using 22–24 PCR enrichment cycles prior to quantification and dual-index barcoding with the Illumina Nextera XT kit. The libraries were enriched with 12 cycles of PCR, then combined in equal volumes prior to final bead cleanup and sequencing. All libraries were sequenced on an Illumina HiSeq 2500 or NextSeq by either single-end 150 bp reads or short paired-end reads using the following read lengths: mouse samples—30 bp, human donor 1—26 bp for read 1 and 25 bp for read 2, human donor 2—30bp, human donor 3—26 bp. Donor 1 and donor 2 were sequenced using two batches, where every batch had cells from all of the donor's population (i.e. donor 1 batch 1 had both naive and CMV-specific cells, same for batch 2. Donor 2 batch 1 had YFV-specific, naive and effector memory cells, same for batch 2). Donor 3′s entire sample was sequenced on a single batch, and the LCMV samples from both mice were combined and sequenced on a single batch (Supplementary Table S2).
Preprocessing and Normalization of scRNA-seq data
Low quality bases were trimmed with trimmomatic (24) using the following parameters: LEADING:15, TRAILING:15, SLIDINGWINDOW:4:15, MINLEN:16. Trimmed reads were then aligned to the genome (hg38 or mm10 for human or mouse samples, respectively) with TopHat2 (25) for TCR reconstruction, and aligned to the transcriptome with RSEM (14) for transcriptome quantification.
For transcriptome analysis of the human CMV and YFV donors (donors 1 and 2), low quality cells were filtered out prior to normalization. Cells were filtered out if their read depth was less than 1 million pairs or if the cell expressed less than 20% of all expressed transcript, where a transcript was considered expressed if it had a transcripts per million (TPM) value of >10 in at least 10% of cells, leaving 353 out of 378 cells for further analysis.
Normalization of TPM values was done with our newly developed normalization framework SCONE (https://niryosef.wordpress.com/tools/scone/). SCONE considers a large number of unsupervised normalization pipelines (i.e. without using any prior biological information about samples’ origin), applying different ways to scale the data (e.g. full quantile, upper quantile) and perform factor analysis to eliminate unwanted variation. SCONE then uses a number of quality metrics to choose the best normalization, which reduces technical variation and maintain prior biological knowledge. In our study, the chosen normalization first scaled each sample with the DEseq (26) scaling factor to account for differences in sequencing depth. Then, we ran RUVg (27) with k = 1. In order to run RUVg, a list of genes that are constant across conditions should be provided. To find constant genes across the specific conditions that were tested in this paper, we also sequenced bulk populations of naive CD8+ T cells from donor 1 and CMV-specific effector memory CD8+ T cells, as well as populations of 50 cells of naive CD8+ T cells from donor 2 and YFV-specific effector memory CD8+ T cells. We ran DESeq2 (28) on those samples and defined the set of constant genes as the genes that showed no change (FDR-adjusted P-value > 0.98 and absolute log fold change < 0.2) across all pairwise comparisons (naive vs. all effector memory cells, naive versus CMV-specific effector memory, naive vs. YFV-specific effector memory and CMV-specific effector memory versus YFV-specific effector memory), resulting in a total of 373 genes.
Dimensionality reduction with PCA on samples from each donor after normalization revealed that the normalization process maintain biological information, while reducing the correlation between the data and technical variables such as batch, number of expressed genes in each cell, and the values of the first PC of the quality matrix (where the quality matrix includes for each cell technical information as previously described (29) (Supplementary Figure S10)).
Reconstructing TCR sequence from long reads
Detection of CDR3 sequence using long (150 bp) reads was performed similar to Venturi et al. (7). In short, reads were aligned against the set of known V and J segment using blastn (30). Reads with V and J segments aligning to their edges were selected, extracting the CDR3 sequence in each read. In case where more than one productive CDR3 sequence was discovered in a cell, the sequence with the highest number of supporting reads was selected.
Reconstructing TCR sequence from short paired-end reads using Trinity
Trinity (20) was run on each cell with the following parameters: –max_memory 10G, –min_contig_length 50. In addition, using the –KMER_SIZE parameter Trinity was run with four different k-mer sizes—13, 15, 17 and 19. For each k-mer size we ran Trinity twice: once in single-end mode, using the set of reads used by TRAPeS for CDR3 reconstruction, and once in paired-end mode, taking all the mapped and unmapped reads along with their pairs. Then, for each k-mer we combined the final Trinity output from both runs (paired-end and single-end) for each cell. To determine whether or not a transcript is productive and to annotate the CDR3 sequence, all possible reconstructed transcripts were run through IMGT/HighV-QUEST (31,32). We considered each productive chain output by IMGT as a successful reconstruction.
Comparing TRAPeS to TraCeR
TraCeR was run using default parameters. To compare TRAPeS to TraCeR on the benchmark data used by TraCeR (15), raw single cell RNA-seq data was downloaded as fastq files from ArrayExpress (accession number E-MTAB-3857). While the original data consisted of 100 bp paired-end reads, we converted it to that equivalent of short-read sequences by trimming each fragment to leave only the outer 25 or 30 bp of each read. We also ran TRAPeS on the original 100 bp paired-end data with the following parameters: -score 80 -bases 150 -top 15 -byExp -oneSide.
Comparing TRAPeS to scTCRseq and VDJPuzzle
VDJPuzzle was run using the default parameters. For scTCRseq, since running the software with the default parameters resulted in no alignments for human TRBV segments, we ran the software using the parameters -e 1e-7 -c 2. In addition, since scTCRseq does not summarize the data, we collected the fasta sequences of scTCRseq final results (*.gapfilled.final.vdj.fa files) and ran them through IMGT to annotate the junction sequence in each cell, taking only productive CDR3 with a complete reconstruction (no missing amino acids) as successful reconstructions. To compare TRAPeS and scTCRseq on the benchmark data used by scTCRseq (33), raw single cell RNA-seq data was downloaded as fastq files from ArrayExpress (accession number E-MTAB-2512) and trimmed from 75 bp paired-end into 25 or 30 bp paired-end. We also ran TRAPeS on the original 75 bp paired-end data with the following parameters: -score 65 -bases 150 -top 10 -bases 100
Gini coefficient calculation:
For each population, cells were considered from the same clone if they had identical CDR3 sequences of both alpha and beta chains. Cells with only one reconstructed chain were excluded from this analysis. The number of cells for each clone was counted and the Gini coefficient was calculated by using the Gini command in R from the ‘ineq’ package.
Inference of cell clusters, visualization and differential expression analysis
For cluster inference in the YFV + CMV human data, we defined an expression matrix consisting of normalized TPM values of 353 cells by 10827 transcripts (expressed at a level of ≥5 TPM in at least 1% of cells; Supplementary Table S11). We applied the SC3 software (34) for clustering the cells in this matrix using default parameters.
To visualize the data, we first used the jackStraw package (35) to reduce the dimensionality of the data and retain only principal components (PC) that are statistically significant (P-value < 10−4) in terms of the respective percent of explained variance. This analysis retained the first three PCs. We then applied t-SNE (36) with default parameters and 2000 iterations to these significant PCs, further reducing the data for visualization in two dimensions.
We used the DESeq2 package (28) to identify genes that are differentially expressed (DE) between the different clusters. In this application, each cluster was compared to the other two clusters, looking for genes that are differentially expressed. Genes were called as differentially expressed using an FDR-adjusted P-value cutoff of 0.05. The heatmap in Figure 3B was populated with log2(TPM) values for genes identified as uniquely up- or down- regulated in each of three major phenotypic groups. We also see similar results of DE genes using the scRNA-seq analysis package Seurat (37,38). Enrichment of DE genes with respect to immunological pathways was determined using a Fisher exact test (FDR-adjusted P-value < 10−3) quantifying the significance of overlap between differential genes and signatures from the ImmuneSigDB database (39).
Figure 3.

Transcriptome analysis reveals distinct subpopulation of YFV-specific cells exhibiting a naive-like profile. (A) t-SNE projection of 353 CMV-specific, effector memory, YFV-specific, and Naïve cells, using normalized transcripts per million (TPM) values of 10,827 transcripts. Ellipses indicate three distinct spatial clusters. A discrete subset of YFV-specific cells cluster with Naive. (B) Genes differentially expressed between relevant phenotypic groups. YFV-specific cells were classified as effector memory-like or naive-like using SC3, a non-spatial consensus clustering approach (Figure S7). (C) t-SNE projections, each cell colored by relative signature score. Shown are two signatures from the ImmuneSigDB distinguishing CMV-specific from YFV-specific cells, and two signatures distinguishing Naïve or YFV-specific naive-like cells from effector memory, CMV-specific and YFV-specific effector memory-like populations. (D) FACS protein expression of CCR7 and CD45RA surface molecules from index sort of Effector Memory, YFV-specific effector memory-like, YFV-specific naive-like, and Naïve cells.
Gene enrichment by signature analysis
We used FastProject (40) together with large collection of transcriptional signatures from ImmuneSigDB (39) to characterize the phenotype of our single cells. In short, each transcriptional signature is comprised of genes that are either over-expressed or under-expressed between two cell states of interest (e.g. using published bulk RNA-seq data from naive versus memory cells). For each single cell, the signature score is computed as:
 |
where  is the signature,
is the signature,  is the cell,
is the cell,  for genes under-expressed in this signature and +1 for over-expressed genes,
for genes under-expressed in this signature and +1 for over-expressed genes, 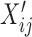 is the standardized (Z-normalized across all cells) log expression level of gene
is the standardized (Z-normalized across all cells) log expression level of gene  in cell
in cell  , and
, and 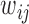 is the estimated false-negative weight for gene
is the estimated false-negative weight for gene 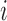 in cell
in cell  . To identify transcriptional signatures that are associated with an scRNA-seq data set of interest, FastProject looks for consistency between signatures and low-dimensional projections of the data. To this end, FastProject first computes a wide range of 2D projections (e.g. PCA, ICA, spectral embedding, tSNE), each capturing (possibly different) key axes of variation in the data. For each transcriptional signature and each projection it then computes a consistency score, which reflects the extent to which cells that have a similar signature score reside close to each other in the projection (thus extending our previous work (30) and facilitating the analysis of non-linear projections). The significance of the consistency score is evaluated by random shuffling.
. To identify transcriptional signatures that are associated with an scRNA-seq data set of interest, FastProject looks for consistency between signatures and low-dimensional projections of the data. To this end, FastProject first computes a wide range of 2D projections (e.g. PCA, ICA, spectral embedding, tSNE), each capturing (possibly different) key axes of variation in the data. For each transcriptional signature and each projection it then computes a consistency score, which reflects the extent to which cells that have a similar signature score reside close to each other in the projection (thus extending our previous work (30) and facilitating the analysis of non-linear projections). The significance of the consistency score is evaluated by random shuffling.
To include only relevant signatures, we analyzed only signatures with a significant consistency score (FDR-adjusted P-value < 0.05) in at least one projection. In addition, only signatures that include ‘CD8’ in their name were used for further analysis, leaving a total of 95 signatures for the YFV + CMV human data and 154 signatures for the YFV-specific analysis.
Characterization of TCR properties of YFV-specific cells
TCR expression
To compute the expression of each reconstructed TCR, we added the reconstructed sequences to the transcriptome and ran RSEM on the complete extended transcriptome, using the original sequencing results (the complete fastq files) as input. This was performed for each cell separately, i.e. for each cell only its TCR sequences were added to the transcriptome. In cases where a cell had more than one reconstructed alpha or beta chain (by having two productive chains or having one productive and one unproductive chain) they were both added to the transcriptome.
Germline score
Classification of each base in the CDR3 as germline (originating from the V, D, J regions) or added nucleotide was done by running the reconstructed TCR sequences thorough IMGT/V-Quest (41,42). The germline score was calculated by dividing the number of nucleotides encoded by V, D, J segments by the length of the CDR3 (16).
Comparing transcriptomic signatures with TCR length
Identification of gene signatures associated with TCR length was done with the PARIS algorithm (43), a module in GenePattern (44). PARIS describes the association between each signature score and TCR length by estimating their differential mutual information. For each signature, the mutual information is computed between the TCR length and the signature, and then normalized using the joint entropy. This score is rescaled with the mean of the score of the TCR length against itself and the score of the signature against itself, resulting in a rescaled normalized mutual information (RNMI) matching score. The significance of the score is evaluated by a permutation test (performed on the TCR length) and then FDR correction.
Hydrophobicity
The mean hydrophobicity of each CDR3 was computed using the Kyte-Doolittle (45) numeric hydrophobicity scale. In order to account for CDR3 length, we also computed mean hydrophobicity for each CDR3 using a sliding window (of both size 3 and 5), taking the mean across all windows. However, the sliding window also didn’t result in significant differences between YFV-specific naive-like and YFV-specific effector memory-like cells (K–S test P-value > 0.1, data not shown).
Normalized tetramer binding intensity
Normalized tetramer binding intensity was defined based on flow cytometry data acquired at the time of sorting. The tetramer binding was measured with the APC-labeled tetramer reagent. To correct for baseline expression of CD3, we divided the APC-labeled tetramer measurement by the expression of CD3 surface molecules.
RESULTS
TRAPeS reconstructs TCR sequences using short (25–30 bp) scRNA-seq
TRAPeS starts by recognizing putative pairs of V and J segments that flank the CDR3 region, using genome alignment (46) (Figure 1, top; see Materials and Methods for a complete description of the algorithm). It then identifies the set of unaligned reads that may have originated from the CDR3 region, taking the unmapped mates of reads aligned to the putative V-J segments or to the constant (C) segment (Figure 1, middle). Next, it uses an iterative dynamic programming scheme to piece together the putative CDR3 reads, gradually extending the CDR3 reconstruction on both ends (V and J) until convergence (Figure 1, bottom). Finally, after the TCR chain has been reconstructed, TRAPeS determines whether it is productive (i.e., has an in-frame CDR3 without a stop codon) and determines its exact CDR3 sequence, based on the criteria established by the international ImMunoGeneTics information system (IMGT) (47). For each cell, TRAPeS outputs a set of reconstructed TCR transcripts (from both chains), along with their complete sequence, an indication of whether or not they are productive, and the number of reads mapped to them. In some cases multiple reconstructions can be generated for the same cell. This may happen when more than one chain is produced in the cell (a phenomenon that have been previously reported (15,17,18)), or when sequence similarity between some V or J segments results in several possible V-J pairs with an identical CDR3 reconstruction. In such cases, we report all V-J pairs, while ranking the putative TCR transcripts in accordance to their estimated expression levels (Supplementary Table S1). The average running time of TRAPeS on a Human single cell library with an average two million reads per cell is less than two minutes per cell on a standard machine (Supplementary Figure S1).
Figure 1.

TRAPeS—an algorithm for TCR reconstruction in single cell RNA-seq Illustration of the TRAPeS algorithm. First, the V and J segment are identified by searching for paired reads with one read mapping to the V segment and its mate mapping to the J segment. Then, a set of putative CDR3-originating reads is identified as the set of unmapped reads whose mates map to the V, J and C segments. Finally, an iterative dynamic programming algorithm is used to reconstruct the CDR3 region.
TRAPeS is accurate and more sensitive than previous methods using short reads and comparable to previous methods using long reads
We applied and tested TRAPeS to scRNA-seq data from a range of CD8+ T cell responses (Methods, Figure 2A). These data sets were selected to include both mouse and human CD8+ T cells as well as those expected to have a range of TCR complexities (Supplementary Figure S2). In mice, we used the lymphocytic choriomeningitis virus (LCMV) infection model, and profiled CD8+ T cells responding to either acute or chronic infection (using the Armstrong and Clone 13 strains of LCMV, respectively). In healthy human subjects we profiled naive CD8+ T cells, effector memory CD8+ T cells, and antigen-specific CD8+ T cells elicited by CMV infection; vaccination with the live attenuated yellow fever virus infection (YFV-17D) (48); or by vaccination with adenoviral and modified vaccinia Ankara vectors encoding HCV proteins (6,22). We sorted up to 128 single CD8+ T cells from each dataset to a total of 565 cells, and generated scRNA-seq libraries with short (25–30 bp) paired-end reads as previously described (23,49) and observed good quality metrics using previously used measures (29) (Supplementary Table S2, Materials and Methods). To test TRAPeS, we applied cell quality filtering scheme similar to the criteria used by others (15), removing samples with <2000 genes or with >10% of reads mapping to mitochondrial genes, resulting in a total of 513 high quality cells (Figure 2A). Importantly, our results below remain consistent also when cell filtering is not applied (Supplementary Figure S3).
Figure 2.

Validation of TRAPeS and comparison to other methods (A) Success rates for reconstruction of productive CDR3 in various CD8+ T cell data sets. Each line depicts the fraction of cells with a productive alpha or beta chain in a given data set with each one of the following methods—150 bp sequencing (black line), short paired-end data reconstructed using TRAPeS (red), TraCeR (turquoise), scTCRseq (gray), VDJPuzzle (dark blue) or Trinity (light blue). (B) Specificity of TRAPeS. Fraction of cells with identical CDR3 sequence between 150 bp data and the 25–30 bp data reconstructed either by TRAPeS, TraCeR, scTCRseq, VDJPuzzle or Trinity. This was calculated as the fraction out of cells with a productive chain in both 150 and 25–30 bp data. (C) Sensitivity of TRAPeS. Same as b, except the fraction of cells is calculated out of the total number of cells that had a successful reconstruction using 150 bp sequencing only. (D) Single cell RNA-sequencing captures a variety of clonal responses. Bars represent the Gini coefficient of each human CD8+ T cell data set. The Gini coefficient can range from zero (a complete heterogeneous population) to one (a complete homogenous population). Pie charts represent the distribution of clones in each population, n represents the number of cells with a successful reconstruction of both alpha and beta chains.
To evaluate the accuracy of TRAPeS, we compared its output with that of directly sequencing the TCR sequence using long reads (in which reconstruction is not required, Materials and Methods). To that end, we sequenced libraries of epitope-specific cells for Clone 13, Armstrong and CMV, and naive T cells from the CMV donor with both short (25–30 bp) paired-end and 150 bp single-end sequence reads (Figure 2A). TCR sequences identified by TRAPeS were almost perfectly consistent with those produced based on the long read data (Methods; Figure 2B and C), indicating a high level of specificity.
We compared TRAPeS to previously published methods for TCR reconstruction in single cells. First, we compared TRAPeS to TraCeR (15)—a TCR reconstruction software that is built upon Trinity (20), a de-novo transcriptome assembly tool. We found that the sensitivity of TRAPeS was markedly higher (Figure 2A–C). On average (across all data sets), TRAPeS successfully reconstructed productive alpha chains from 66% of the cells and productive beta chains from 80% of the cells, using the short (25–30 bp) libraries. In contrast, TraCeR resulted in no reconstruction for the 25 bp paired-end libraries, and was able, for the 30 bp libraries, to reconstruct CDR3 regions in an average of 43% and 15% of the cells for alpha and beta chains respectively.
Next, we considered two additional recently published methods—VDJPuzzle (18) and scTCRseq (17), both based on de-novo assembly algorithms (Trinity and GapFiller (19), respectively). As above, we observe substantially higher sensitivity with TRAPeS (Figure 2A–C, Materials and Methods). VDJPuzzle was also unable to reconstruct any productive chains in the 25 bp data and, for the 30 bp libraries, reconstructed CDR3 regions in an average of 40% and 63% of the cells for alpha and beta chains, respectively. scTCRseq, which is build upon GapFiller (19), managed to successfully reconstruct CDR3 regions in an average of 50% and 60% of the cells for alpha and beta chains, respectively. While scTCRseq achieves better results compared with Trinity-based methods, TRAPeS clearly outperforms all methods in terms of specificity and sensitivity (Figure 2A–C).
The low success rate of Trinity-based methods TraCeR and VDJPuzzle is likely due to its requirement for seed k-mer length (25nt) that is unsuitable for short reads. Thus, we also directly ran Trinity on our set of CDR3-originating reads, using a k-mer length of 13 (Materials and Methods). This resulted in an increased sensitivity for the 30 bp libraries compared to TraCeR and VDJPuzzle, but did not improve the reconstruction rates for 25 bp libraries (Figure 2A–C). Running Trinity with several other k-mer lengths (15, 17 and 19) did not significantly change the results (Supplementary Figure S4).
Notably, the average rate of successful reconstruction of TRAPeS in our mouse libraries is 93.7% (with 30 bp reads), which is higher than that achieved by TraCeR with the mouse libraries used by Stubbington et al. (86.3% with 100 bp reads) (15). To further substantiate this result, we applied TRAPeS and TraCeR on a trimmed version of this published data. We found that discarding 70–75% of the information (i.e. taking only 25 or 30 bp out of each 100 bp read) substantially hurts the performance of TraCeR, while TRAPeS is able to maintain rates of successful TCR reconstructions that are similar to those achieved in the original paper (15) (Supplementary Figure S5). Running TRAPeS on the original long read data is also comparable to the success rates obtained by TraCeR, demonstrating the ability of TRAPeS to be applied on long reads as well (Supplementary Figure S5). In addition, running TRAPeS on short or long reads is comparable to running scTCRseq using long reads, as evident by running TRAPeS on the original and a trimmed version of the data used to benchmark scTCRseq (17,33) (Supplementary Figure S6).
TRAPeS captures various clonality levels
We investigated the clonality of the TCR repertoire measured by TRAPeS among the human CD8+ T cells (Figure 2D, Supplementary Table S3), using the Gini Index, a clonality measure (50) ranging from zero (i.e. no two cells share the same TCR) to one (i.e. all cells are from the same clone; Methods). As expected, the naïve population had a Gini index of zero, indicating that each naive CD8+ T cell expressed a unique TCR. The CMV-specific CD8+ T cell population had a high Gini index (with 83% of CMV-specific CD8+ T cells with reconstructed alpha and beta chains originated from a single clone), indicating a high degree of oligoclonality as previously described (51, 52). In contrast, CD8+ T cells elicited by YFV or HCV vaccines showed much greater heterogeneity in TCR repertoire, consistent with a more limited, rather than persistent, exposure to antigen (6,53–56). This demonstrates the ability of TRAPeS to capture cells from the same clone even with relatively small number of antigen-specific cells, assuming a clonal response.
Single-cell transcriptome analysis detects subpopulations of YFV cells
In order to determine the relationship between TCR use and CD8+ T cell state, we focused on CD8+ T cells from two healthy donors (YFV and CMV peptide-specific, as well as naive and effector memory cells without sorting for peptide specificity; Methods) to avoid introducing additional complexity from chronic infection. To identify groups of cells with similar expression profiles, we used SC3 (34), a robust clustering method for sparse datasets, to identify subpopulations of cells (Supplementary Figure S7, Supplementary Table S4, Materials and Methods) which we then visualized using t-SNE (36) (Figure 3A). We found three clusters of cells: one that contained all CMV-specific cells (Figure 3A, purple symbols); one that contained all effector memory cells (blue symbols); and one that contained all naive CD8+ T cells (green symbols). In contrast to these discrete groupings, we observed that YFV-specific CD8+ T cells were split between two clusters: one containing effector memory CD8+ T cells and one containing naive CD8+ T cells.
Differential gene expression analysis between cell clusters revealed transcripts consistent with the known patterns of gene expression in antigen-experienced or naive CD8+ T cells (Supplementary Tables S5–S7, Figure 3B, Materials and Methods). CMV-specific CD8+ T cells expressed effector molecules and transcription factors characteristic of antigen experienced cells (e.g., Granzyme B, PRDM1), which were not detected in naive cells. Naive CD8+ T cells expressed canonical markers of the naive state (CCR7, SATB1, LEF1) that were absent in CMV-specific and effector memory CD8+ T cells. The expression of these genes in YFV-specific CD8+ T cells was consistent with the cluster in which they were associated, with those in the naive cluster expressing minimal Granzyme B or PRDM1, but showing robust expression of CCR7, SATB1, and LEF1 (Supplementary Figure S8).
To identify broader patterns of transcriptional signatures, we applied FastProject (40)—a software tool that enables the expression of gene sets of interest to be quantified in transcriptional profiles of single cells (Materials and Methods). We surveyed the enrichment of a collection of gene sets, from the C7 (ImmuneSigDB) (39) collection of MSigDB (57) corresponding to cell states and perturbations of CD8+ T cells. We found significant up-regulation of multiple gene sets corresponding to naive CD8+ T cells (K–S test FDR- adjusted P-value<0.01) in the naive cluster (cluster 3) compared to the other two clusters. Consistent with this, we found significantly greater up-regulation of effector signatures in clusters 1 and 2 compared with the other clusters (FDR-adjusted P- value < 0.01; Figure 3C and Supplementary Table S8).
To confirm these patterns of transcript abundance at the protein level, we compared flow cytometry data for a set of surface markers acquired at the time of sorting (Methods) with transcript abundance in the same cell (Figure 3D). Consistent with the gene expression profiles, we observed that YFV-specific CD8+ T cells in the naive-like cluster (open symbols) showed higher protein levels of CCR7 and CD45RA than those in the effector memory cluster (purple symbols). Thus, single-cell analysis shows that CD8+ T cells specific for the same peptide epitope from YFV are heterogeneous and includes both effector-memory and naive-like gene expression profiles, as has been reported previously for cells analyzed at the bulk level (4,5,58).
Combined TCR-transcriptome analysis reveals longer CDR3 regions for naive-like YFV-specific cells
We reasoned that differences in TCR might contribute to the heterogeneous differentiation of CD8+ T cells following YFV vaccination. To that end, we evaluated a number of properties to characterize each reconstructed TCR—CDR3 specific properties such as length, hydrophobicity and germline score as well as TCR expression. In addition, we measured the normalized tetramer staining intensity per cell (Supplementary Table S9, Methods). We then asked whether any of those properties differed between naive-like and effector memory-like YFV-specific CD8+ T cells. Naive-like and effector memory-like YFV-specific CD8+ T cells were indistinguishable (P-value>0.05, FDR-adjusted P-value > 0.1) in terms of TCR transcript expression, hydrophobicity of the CDR3 region and normalized tetramer staining intensity (Materials and Methods). However, we found that the CDR3 sequence was significantly longer in YFV-specific CD8+ T cells with a naive-like state compared with those with an effector memory profile for both alpha and beta chains (Figure 4A, K–S test P-value 0.038 and 0.027 for alpha and beta chains respectively, FDR-adjusted P-value = 0.084 for both alpha and beta chains).
Figure 4.

YFV-specific subpopulations display different TCR structure. (A) YFV-specific naive-like cells tend to have longer CDR3. Distribution of the YFV-specific effector memory-like and naive-like CDR3 lengths in both alpha (left) and beta (right) chains. P-values were calculated with K–S test. (B) Differences between naive-like and effector memory-like CDR3 lengths are due to added nucleotides. Distribution of the YFV-specific effector memory-like and naive-like CDR3 germline scores, defined as the number of nucleotides in the CDR3 encoded by the V, D or J segments divided by the total number of nucleotides in the CDR3, for both alpha (left) and beta (right) chains. P-values were calculated with K–S test. (C) Signature analysis reveals significant correlation between CDR3 length and cell state. The plot depicts the rescaled normalized mutual information score between CDR3 length and transcriptional signatures of CD8+ T cells from ImmuneSigDB. Signatures identified as statistically significant using a permutation test (FDR-adjusted P-value < 0.1) are highlighted in red. (D) YFV-specific cells with long CDR3 tend to have a higher transcriptomic naive signature than cells with short CDR3. Plot represents the score of each cell for a transcriptional signature of a naive versus effector CD8+ T cell state. A high signature score means that a cell has higher expression of naive signature genes compared to effector signature genes.
We next evaluated the germline score of CDR3 regions in YFV-specific CD8+ T cells, a measure of the contribution of germline nucleotides to the CDR3 region. The germline score is defined as the ratio between the number of nucleotides in the CDR3 that originate from the germline (V, D, J segments) to the total number of nucleotides in the CDR3 (16) (Materials and Methods). Consistent with the differences in the CDR3 length, we found that naive-like YFV-specific CD8+ T cells had a significantly lower germline score in both alpha and beta chains than did effector memory-like cells (Figure 4B, K–S test P-value of 0.034 and 0.029 for alpha and beta chains respectively, FDR-adjusted P-value 0.084 for both alpha and beta chains), suggesting that generating the CDR3 region of these TCRs involved a greater degree of nucleotide addition/subtraction.
To further characterize the relationship between CDR3 length and cellular state in YFV-specific CD8+ T cells, we identified CD8+ transcriptional signatures (extracted from ImmuneSigDB (39) and scored with FastProject (40), as above) that correlated with CDR3 length across all YFV-specific CD8+ T cells (Supplementary Table S10, Materials and Methods). Of all signatures evaluated, we found that only naive CD8+ T cell signatures showed a significant positive correlation with CDR3 length (FDR-adjusted P-value < 0.1; Figures 4C and D). Previous work has suggested that YFV-specific CD8+ T cells with a naive-like phenotype include those with a stem-cell memory (Tstem-memory) differentiation state. We found that Tstem-memory signatures were more enriched in naive-like YFV-specific CD8+ T cells than in effector memory YFV-specific CD8+ T cells (Supplementary Figure S9). However, the enrichment for these signatures was equivalent between naive-like YFV-specific and phenotypically naive CD8+ T cells, making it difficult to discern whether these cells manifest a specific stem-cell-like state. Our results, however, show that heterogeneity in the differentiation state of CD8+ T cells responding to a single epitope of YFV is strongly associated with the CDR3 length.
DISCUSSION
TRAPeS enables the analysis of TCR clonality in scRNA-seq profiles using short sequence reads. Other methods of direct TCR sequencing (7) or reconstruction (15,17,18) have lower rate of successful TCR reconstruction or requires long sequence reads, which substantially increase the per-cell cost of single cell profiling. As single-cell RNA-seq technologies move towards massively parallel scale, long-read sequencing is likely to become unfeasibly expensive, making approaches such as TRAPeS critical for studies of TCR use in single cells.
We applied TRAPeS to short-read sequencing data from human CD8+ T cells and were able to discover a new association between the differentiation state of CD8+ T cells specific to a single YFV antigen and the CDR3 length of the TCRs that they express. Long CDR3 lengths have been associated with private clonotypes, which in turn may reflect low precursor frequency within the naive T cell pool (7–10). We therefore speculate that within a population of naive T cells capable of recognizing a specific antigen, those that exist at low frequency may enter the T cell response later than more abundant precursors, resulting in an altered differentiation state compared to those that existed at a higher precursor frequency. Alternatively, a greater degree of cross-reactivity in T cells with short CDR3 regions may result in more repeated TCR stimulation, leading to the difference in T cell phenotype we observe. While in this case the phenotype could be validated with protein surface markers, this is not true for many other phenotypes, highlighting the importance of transcriptome analysis using scRNA-seq.
More generally, we anticipate that TRAPeS will facilitate broad efforts to determine the relationship between T cell state and TCR sequence in the immune response. TRAPeS can be applied to further basic biological understanding of the relationship between TCR avidity and T cell differentiation. Being able to identify alpha and beta chains allows cloning of TCRs into experimental systems to study their binding properties, which will help determine how TCR properties are related to TCR avidity and T cell biology. This is highly relevant for studying vaccine responses and for thymic development. Moreover, linking the CDR3 sequence to T cell transcriptome can help identify biological similarities in clonal populations of T cells. For instance, in tumors where the identities of T cells responding to the tumors are not known, identifying clonal expansion can be used to infer tumor-specificities both for analyzing gene expression profiles and cloning both alpha and beta chains of the same TCR for clinical use. Additionally, we recently applied TRAPeS to study the clonality of CD4+ and HLA class II-restricted CD8+ T cells in HIV-infected individuals (59), demonstrating the wide use for a combined analysis of transcriptome and TCR sequence at the same cell.
AVAILABILITY
TRPAeS is publicly available and can be found in the following link: https://github.com/YosefLab/TRAPeS.
ACCESSION NUMBER
scRNA-seq data have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE96993.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Rama Akondy and Rafi Ahmed for providing the YFV vaccine samples and related reagents; members of the Haining and Yosef lab for input; and subjects for their participation in the studies.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Medical Research Council UK (L.S. as an MRC CASE studentship) (to L.S and E.B); National Institute of Health [5U19AI090023-07 to N.Y]; US National Institute of Health [AI090023, AI057266 and AI082630]. Funding for open access charge: National Institute of Health [5U19AI090023-07].
Conflict of interest statement. None declared.
REFERENCES
- 1. Appay V., Dunbar P.R., Callan M., Klenerman P., Gillespie G.M.A., Papagno L., Ogg G.S., King A., Lechner F., Spina C.A. et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat. Med. 2002; 8:379–385. [DOI] [PubMed] [Google Scholar]
- 2. Newell E.W., Sigal N., Bendall S.C., Nolan G.P., Davis M.M.. Cytometry by time-of-flight shows combinatorial cytokine expression and virus-specific cell niches within a continuum of CD8+ T cell phenotypes. Immunity. 2012; 36:142–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chattopadhyay P.K., Roederer M.. A mine is a terrible thing to waste: high content, single cell technologies for comprehensive immune analysis. Am. J. Transplant. 2015; 15:1155–1161. [DOI] [PubMed] [Google Scholar]
- 4. Fuertes Marraco S.A., Soneson C., Cagnon L., Gannon P.O., Allard M., Maillard S.A., Montandon N., Rufer N., Waldvogel S., Delorenzi M. et al. Long-lasting stem cell like memory CD8 T cells with a naïve-like profile upon yellow fever vaccination. Sci. Transl. Med. 2015; 7:282ra48. [DOI] [PubMed] [Google Scholar]
- 5. Pulko V., Davies J.S., Martinez C., Lanteri M.C., Busch M.P., Diamond M.S., Knox K., Bush E.C., Sims P.A., Sinari S. et al. Human memory T cells with a naive phenotype accumulate with aging and respond to persistent viruses. Nat. Immunol. 2016; 17:966–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Swadling L., Capone S., Antrobus R.D., Brown A., Richardson R., Newell E.W., Halliday J., Kelly C., Bowen D., Fergusson J. et al. A human vaccine strategy based on chimpanzee adenoviral and MVA vectors that primes, boosts, and sustains functional HCV-specific T cell memory. Sci. Transl. Med. 2014; 6:261ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Venturi V., Quigley M.F., Greenaway H.Y., Ng P.C., Ende Z.S., McIntosh T., Asher T.E., Almeida J.R., Levy S., Price D.A. et al. A mechanism for TCR sharing between T cell subsets and individuals revealed by pyrosequencing. J. Immunol. 2011; 186:4285–4294. [DOI] [PubMed] [Google Scholar]
- 8. Robins H.S., Srivastava S.K., Campregher P.V., Turtle C.J., Andriesen J., Riddell S.R., Carlson C.S., Warren E.H.. Overlap and effective size of the human CD8+ T cell receptor repertoire. Sci. Transl. Med. 2010; 2:47ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Venturi V., Price D.A., Douek D.C., Davenport M.P.. The molecular basis for public T-cell responses. Nat. Rev. Immunol. 2008; 8:231–238. [DOI] [PubMed] [Google Scholar]
- 10. Venturi V., Kedzierska K., Price D.A., Doherty P.C., Douek D.C., Turner S.J., Davenport M.P.. Sharing of T cell receptors in antigen-specific responses is driven by convergent recombination. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:18691–18696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ji X., Lyu S.-C., Spindler M., Bacchetta R., Goncharov I., Han A., Glanville J., Wang W., Roncarolo M., Meyer E. et al. Deep profiling of single T cell receptor repertoire and phenotype with targeted RNA-seq (TECH2P. 927). J. Immunol. 2015; 194:206–237. [Google Scholar]
- 12. Li B., Li T., Pignon J.-C., Wang B., Wang J., Shukla S.A., Dou R., Chen Q., Hodi F.S., Choueiri T.K. et al. Landscape of tumor-infiltrating T cell repertoire of human cancers. Nat. Genet. 2016; 48:725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Han A., Glanville J., Hansmann L., Davis M.M.. Linking T-cell receptor sequence to functional phenotype at the single-cell level. Nat. Biotechnol. 2014; 32:684–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li B., Dewey C.N.. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011; 12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stubbington M.J.T., Lönnberg T., Proserpio V., Clare S., Speak A.O., Dougan G., Teichmann S.A.. T cell fate and clonality inference from single-cell transcriptomes. Nat. Methods. 2016; 13:329–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yu X., Almeida J.R., Darko S., van der Burg M., DeRavin S.S., Malech H., Gennery A., Chinn I., Markert M.L., Douek D.C. et al. Human syndromes of immunodeficiency and dysregulation are characterized by distinct defects in T-cell receptor repertoire development. J. Allergy Clin. Immunol. 2014; 133:1109–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Redmond D., Poran A., Elemento O.. Single-cell TCRseq: paired recovery of entire T-cell alpha and beta chain transcripts in T-cell receptors from single-cell RNAseq. Genome Med. 2016; 8:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eltahla A.A., Rizzetto S., Pirozyan M.R., Betz-Stablein B.D., Venturi V., Kedzierska K., Lloyd A.R., Bull R.A., Luciani F.. Linking the T cell receptor to the single cell transcriptome in antigen-specific human T cells. Immunol. Cell Biol. 2016; 94:604–611. [DOI] [PubMed] [Google Scholar]
- 19. Boetzer M., Pirovano W.. Toward almost closed genomes with GapFiller. Genome Biol. 2012; 13:R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grabherr M.G., Haas B.J., Yassour M., Levin J.Z., Thompson D.A., Amit I., Adiconis X., Fan L., Raychowdhury R., Zeng Q. et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011; 29:644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Döring A., Weese D., Rausch T., Reinert K.. SeqAn an efficient, generic C++ library for sequence analysis. BMC Bioinformatics. 2008; 9:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Swadling L., Halliday J., Kelly C., Brown A., Capone S., Ansari M.A., Bonsall D., Richardson R., Hartnell F., Collier J. et al. Highly-immunogenic virally-vectored T-cell vaccines cannot overcome subversion of the T-cell response by HCV during chronic infection. Vaccines. 2016; 4:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Trombetta J.J., Gennert D., Lu D., Satija R., Shalek A.K., Regev A.. Preparation of single-cell RNA-Seq libraries for next generation sequencing. Curr. Protoc. Mol. Biol. 2014; 107:4.22.1–4.22.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bolger A.M., Lohse M., Usadel B.. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014; 30:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L.. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013; 14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Anders S., Huber W.. Differential expression analysis for sequence count data. Genome Biol. 2010; 11:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Risso D., Ngai J., Speed T.P., Dudoit S.. Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol. 2014; 32:896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Love M., Anders S., Huber W.. Differential analysis of count data–the DESeq2 package. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gaublomme J.T., Yosef N., Lee Y., Gertner R.S., Yang L.V., Wu C., Pandolfi P.P., Mak T., Satija R., Shalek A.K. et al. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell. 2015; 163:1400–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J.. Basic local alignment search tool. J. Mol. Biol. 1990; 215:403–410. [DOI] [PubMed] [Google Scholar]
- 31. Alamyar E., Giudicelli V., Li S., Duroux P., Lefranc M.-P.. IMGT/HighV-QUEST: the IMGT® web portal for immunoglobulin (IG) or antibody and T cell receptor (TR) analysis from NGS high throughput and deep sequencing. Immunome Res. 2012; 8:26. [Google Scholar]
- 32. Li S., Lefranc M.-P., Miles J.J., Alamyar E., Giudicelli V., Duroux P., Freeman J.D., Corbin V.D.A., Scheerlinck J.-P., Frohman M.A. et al. IMGT/HighV QUEST paradigm for T cell receptor IMGT clonotype diversity and next generation repertoire immunoprofiling. Nat. Commun. 2013; 4:2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mahata B., Zhang X., Kolodziejczyk A.A., Proserpio V., Haim-Vilmovsky L., Taylor A.E., Hebenstreit D., Dingler F.A., Moignard V., Göttgens B. et al. Single-cell RNA sequencing reveals T helper cells synthesizing steroids de novo to contribute to immune homeostasis. Cell Rep. 2014; 7:1130–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kiselev V.Y., Kirschner K., Schaub M.T., Andrews T., Yiu A., Chandra T., Natarajan K.N., Reik W., Barahona M., Green A.R. et al. SC3: consensus clustering of single-cell RNA-seq data. Nat. Methods. 2017; 14:483–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chung N.C., Storey J.D.. Statistical significance of variables driving systematic variation in high-dimensional data. Bioinformatics. 2015; 31:545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Maaten L. van der, Hinton G.. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008; 9:2579–2605. [Google Scholar]
- 37. Mcdavid A., Finak G., Chattopadyay P.K., Dominguez M., Lamoreaux L., Ma S.S., Roederer M., Gottardo R.. Data exploration, quality control and testing in single-cell qPCR-based gene expression experiments. Bioinformatics. 2013; 29:461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Satija R., Farrell J.A., Gennert D., Schier A.F., Regev A.. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 2015; 33:495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Godec J., Tan Y., Liberzon A., Tamayo P., Bhattacharya S., Butte A.J., Mesirov J.P., Haining W.N.. Compendium of immune signatures identifies conserved and species-specific biology in response to inflammation. Immunity. 2016; 44:194–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. DeTomaso D., Yosef N.. FastProject: a tool for low-dimensional analysis of single-cell RNA-Seq data. BMC Bioinformatics. 2016; 17:315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Brochet X., Lefranc M.-P., Giudicelli V.. IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res. 2008; 36:W503–W508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Giudicelli V., Brochet X., Lefranc M.-P.. IMGT/V-QUEST: IMGT standardized analysis of the immunoglobulin (IG) and T cell receptor (TR) nucleotide sequences. Cold Spring Harb. Protoc. 2011; 2011, db.prot5633. [DOI] [PubMed] [Google Scholar]
- 43. Cowley G.S., Weir B.A., Vazquez F., Tamayo P., Scott J.A., Rusin S., East-Seletsky A., Ali L.D., Gerath W.F.J., Pantel S.E. et al. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci. Data. 2014; 1:140035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reich M., Liefeld T., Gould J., Lerner J., Tamayo P., Mesirov J.P.. GenePattern 2.0. Nat. Genet. 2006; 38:500–501. [DOI] [PubMed] [Google Scholar]
- 45. Kyte J., Doolittle R.F.. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982; 157:105–132. [DOI] [PubMed] [Google Scholar]
- 46. Trapnell C., Pachter L., Salzberg S.L.. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009; 25:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lefranc M.-P., Giudicelli V., Kaas Q., Duprat E., Jabado-Michaloud J., Scaviner D., Ginestoux C., Clément O., Chaume D., Lefranc G.. IMGT, the international ImMunoGeneTics information system®. Nucleic Acids Res. 2005; 33:D593–D597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Akondy R.S., Johnson P.L.F., Nakaya H.I., Edupuganti S., Mulligan M.J., Lawson B., Miller J.D., Pulendran B., Antia R., Ahmed R.. Initial viral load determines the magnitude of the human CD8 T cell response to yellow fever vaccination. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:3050–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Picelli S., Faridani O.R., Björklund A.K., Winberg G., Sagasser S., Sandberg R.. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014; 9:171–181. [DOI] [PubMed] [Google Scholar]
- 50. Qi Q., Liu Y., Cheng Y., Glanville J., Zhang D., Lee J.-Y., Olshen R.A., Weyand C.M., Boyd S.D., Goronzy J.J.. Diversity and clonal selection in the human T-cell repertoire. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:13139–13144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Weekes M.P., Wills M.R., Mynard K., Carmichael A.J., Sissons J.G.P.. The memory cytotoxic T-lymphocyte (CTL) response to human cytomegalovirus infection contains individual peptide-specific CTL clones that have undergone extensive expansion in vivo. J. Virol. 1999; 73:2099–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Trautmann L., Rimbert M., Echasserieau K., Saulquin X., Neveu B., Dechanet J., Cerundolo V., Bonneville M.. Selection of T cell clones expressing high-affinity public TCRs within human cytomegalovirus-specific CD8 T cell responses. J. Immunol. 2005; 175:6123–6132. [DOI] [PubMed] [Google Scholar]
- 53. DeWitt W.S., Emerson R.O., Lindau P., Vignali M., Snyder T.M., Desmarais C., Sanders C., Utsugi H., Warren E.H., McElrath J. et al. Dynamics of the cytotoxic T cell response to a model of acute viral infection. J. Virol. 2015; 89:4517–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Miles J.J., Thammanichanond D., Moneer S., Nivarthi U.K., Kjer-Nielsen L., Tracy S.L., Aitken C.K., Brennan R.M., Zeng W., Marquart L. et al. Antigen-driven patterns of TCR bias are shared across diverse outcomes of human hepatitis C virus infection. J. Immunol. 2011; 186:901–912. [DOI] [PubMed] [Google Scholar]
- 55. Bolinger B., Sims S., Swadling L., O’Hara G., de Lara C., Baban D., Saghal N., Lee L.N., Marchi E., Davis M. et al. Adenoviral vector vaccination induces a conserved program of CD8(+) T cell memory differentiation in mouse and man. Cell Rep. 2015; 13:1578–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Barnes E., Folgori A., Capone S., Swadling L., Aston S., Kurioka A., Meyer J., Huddart R., Smith K., Townsend R. et al. Novel adenovirus-based vaccines induce broad and sustained T cell responses to HCV in man. Sci. Transl. Med. 2012; 4:115ra1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Liberzon A., Subramanian A., Pinchback R., Thorvaldsdóttir H., Tamayo P., Mesirov J.P.. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011; 27:1739–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fuertes Marraco S.A., Soneson C., Delorenzi M., Speiser D.E.. Genome-wide RNA profiling of long-lasting stem cell-like memory CD8 T cells induced by Yellow Fever vaccination in humans. Genom Data. 2015; 5:297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ranasinghe S., Lamothe P.A., Soghoian D.Z., Kazer S.W., Cole M.B., Shalek A.K., Yosef N., Jones R.B., Donaghey F., Nwonu C. et al. Antiviral CD8(+) T cells restricted by human leukocyte antigen class II exist during natural HIV infection and exhibit clonal expansion. Immunity. 2016; 45:917–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.