

Abstract
The δ2 glutamate receptor (GluRδ2; GluD2), which is predominantly expressed on postsynaptic sites at parallel fiber (PF)–Purkinje cell synapses in the cerebellum, plays two crucial roles in the cerebellum: the formation of PF synapses and the regulation of long-term depression (LTD), a form of synaptic plasticity underlying motor learning. Although the induction of LTD and motor learning absolutely require signaling via the cytoplasmic C-terminal domain of GluD2, the mechanisms by which GluD2 regulates PF synaptogenesis have remained unclear. Here, we examined the role of the extracellular N-terminal domain (NTD) of GluD2 on PF synaptogenesis by injecting Sindbis virus carrying wild-type (GluD2wt) or mutant GluD2 into the subarachnoid supracerebellar space of GluD2-null mice. Remarkably, the expression of GluD2wt, but not of a mutant GluD2 lacking the NTD (GluD2ΔNTD), rapidly induced PF synapse formation and rescued gross motor dyscoordination in adult GluD2-null mice just 1 d after injection. In addition, although the kainate receptor GluR6 (GluK2) did not induce PF synaptogenesis, a chimeric GluK2 that contained the NTD of GluD2 (GluD2NTD–GluK2) did. Similarly, GluD2wt and GluD2NTD–GluK2, but not GluD2ΔNTD, induced synaptogenesis in heterologous cells in vitro. In contrast, LTD was restored in GluD2-null Purkinje cells expressing a mutant GluD2 lacking the NTD. These results indicate that the NTD of GluD2 is necessary and sufficient for the function of GluD2 in the regulation of PF–Purkinje cell synaptogenesis. Furthermore, our results suggest that GluD2 differently regulates PF synaptogenesis and cerebellar LTD through the extracellular NTD and the cytoplasmic C-terminal end, respectively.
Introduction
The δ2 glutamate receptor [GluRδ2; GluD2 (Collingridge et al., 2009)], which is highly and selectively expressed on the postsynaptic sites of parallel fiber (PF)–Purkinje cell synapses (Landsend et al., 1997; Zhao et al., 1997), is indispensable for cerebellar functions. Detailed analyses of GluD2-null mice [i.e., hotfoot (Lalouette et al., 1998, 2001; Motohashi et al., 2007)] and genetically engineered GluD2 knock-out mice (Kashiwabuchi et al., 1995; Kurihara et al., 1997) have revealed two crucial functions of GluD2: the formation or maintenance of PF–Purkinje cell synapses and the control of long-term depression (LTD), an elemental process underlying cerebellar motor learning such as delayed eye-blink conditioning (Ito, 1989). Nevertheless, the mechanisms by which GluD2 achieves these functions are not well understood.
As a first step toward identifying the mechanism of action of GluD2, we used a transgenic rescue approach by taking advantage of the fact that the domain structure and amino acid sequences within each domain are evolutionally well conserved in all ionotropic glutamate receptor (iGluR) family members, including GluD2 (Yuzaki, 2003). Because virally or genetically introduced mutant GluD2 transgenes, in which the glutamate-binding domain (Hirai et al., 2005a) or the ion selectivity filter domain (Kakegawa et al., 2007a,b) was mutated, rescued all abnormal phenotypes of GluD2-null mice, GluD2 was not thought to function as an ion channel gated by glutamate analogs. Instead, we noted that a mutant GluD2 lacking the most C-terminal domain (CTD), which serves as a binding site for several intracellular PDZ (PSD-95/Dlg/ZO-1) proteins, could not rescue abrogated LTD and eye-blink conditioning (Kohda et al., 2007; Kakegawa et al., 2008), indicating that the interaction of PDZ proteins with the CTD of GluD2 is essential for these functions. Unexpectedly, a mutant GluD2 lacking the CTD almost completely restored abnormal PF synapse formation in GluD2-null cerebellum (Uemura et al., 2007; Kakegawa et al., 2008). Therefore, although the induction of LTD and motor learning absolutely require signaling via the CTD of GluD2, morphological integrity at PF–Purkinje cell synapses does not; how the latter function is achieved by GluD2 has remained unclear.
GluD2 is required not only for the normal development of cerebellar circuits but also for the maintenance of PF–Purkinje cell synapses in adult cerebellum: ablation of the GluD2 gene in adult cerebellum using an inducible recombinase resulted in the gradual loss of normal PF synapses over 8 weeks (Takeuchi et al., 2005). Very recently, the N-terminal domain (NTD) of GluD2 was shown to induce synapse formation in heterologous cells in vitro (Uemura and Mishina, 2008). However, whether uninnervated spines in mature GluD2-null Purkinje cells in vivo could be restored by the expression of GluD2 remained unknown. Here, we show that a virally introduced wild-type GluD2 transgene rapidly induces PF synapse formation and rescues ataxia in adult GluD2-null mice 1 d after infection. Furthermore, we demonstrate that the NTD of GluD2 is necessary and sufficient for synaptogenesis both in vitro and in vivo. Therefore, we propose that, in addition to the CTD-based signaling, the NTD of GluD2 independently regulates morphological integrity at PF–Purkinje cell synapses in the cerebellum.
Materials and Methods
All procedures related to animal care and treatment were approved by the Animal Resource Committee of the School of Medicine, Keio University.
cDNA constructs.
The NTD region immediately after the signal sequence (amino acids 24–415) (see Fig. 1 A) was deleted using a standard PCR-based mutagenesis method with Pyrobest PCR polymerase (Takara) to produce a mutant GluD2 lacking the NTD (designated as GluD2ΔNTD) (see Fig. 1 A). Because the NTD is required for the assembly of the initial dimers of iGluR subunits (Ayalon and Stern-Bach, 2001) and an artificial tetramerization peptide GCN-LI (RMKQIEDKLEEILSKLYHIENELARIKKLLGER) was shown previously to replace this function of the NTD (Matsuda et al., 2005), we introduced cDNA encoding the tetramerization peptide GCN-LI to GluD2ΔNTD to produce GluD2tet-ΔNTD. However, because GluD2tet-ΔNTD and GluD2ΔNTD were similarly transported to the cell surface and synapses, GluD2tet-ΔNTD and GluD2ΔNTD were used interchangeably. The NTD of the kainate receptor GluR6 [GluK2 (Collingridge et al., 2009)] (amino acid 32–400) (see Fig. 6 A) was replaced with the NTD of GluD2 to produce a GluK2 chimera GluD2NTD–GluK2 (see Fig. 6 B). A cDNA encoding a hemagglutinin (HA) was added immediately upstream of the stop codon of GluR1 (GluA1), GluR2 (GluA2), GluR4 (GluA4), GluK2, and GluD2 cDNA (see Fig. 8). The valine at position 617 was replaced with arginine to produce GluD2V/R (see Fig. 7 A) (Kakegawa et al., 2007b), and the arginine at position 530 was replaced with lysine to produce GluD2R/K (see Fig. 7 B) (Hirai et al., 2005a). The nucleotide sequences of the amplified open reading frames were confirmed using bidirectional sequencing and were cloned into the expression vector pTracer–enhanced green fluorescent protein (GFP) (Invitrogen), pCAGGS (kindly provided by Dr. J. Miyazaki, Osaka University, Osaka, Japan), or the Sindbis virus vector (see below).
Figure 1.

Expression of GluD2wt, but not GluD2ΔNTD, rescued gross motor dyscoordination of mature GluD2-null mice. A, Schematic diagrams of GluD2wt and GluD2ΔNTD. The red line indicates the extracellular NTD (amino acids 28–427) of GluD2wt. The NTD was deleted from GluD2wt in GluD2ΔNTD. The open boxes at the C-terminal ends indicate the C-terminal PDZ ligand regions. TM, Transmembrane segment. B, Schema showing the subarachnoidal injection of the Sindbis virus into adult GluD2-null mice. The Sindbis virus was injected into the subarachnoidal supracerebellar space above the cerebellar vermis of GluD2-null mice aged P35–P45. C, Virally introduced GFP expression in GluD2-null cerebellum. One day after injection, GFP signals were observed in many Purkinje cells in almost all lobules, with a noticeable gradient from the injected site (C2–C4). A bright-field (BF) image is also shown in C1. D, Representative gait patterns of GluD2-null mice 1 d after introduction of various transgenes. The paws of the mice were marked with black paint. The irregular, shortened gait skips made by GluD2-null mice were markedly improved at 1 d after the injection of Sindbis viruses carrying GluD2wt, unlike those injected with viruses carrying GFP only (Vector) or GluD2ΔNTD. E, Results for the rotor-rod test. GluD2-null mice were placed on the rod rating at 20 rpm or before (Pre) and 1 d after (Post) receiving a subarachnoidal injection of Sin–GFP (Vector), Sin–GluD2wt–GFP (GluD2wt), or Sin–GluD2ΔNTD–GFP (GluD2ΔNTD). The average time was calculated from the results of six trials. *p < 0.05. ns, Not significant.
Figure 6.

The N-terminal domain of GluD2 is sufficient for the functional restoration of PF–Purkinje cell synapses in GluD2-null mice. A, B, Schematic diagrams and PF–EPSC traces recorded from GluD2-null Purkinje cells expressing GFP plus GluK2 (GluK2, A) or GFP plus GluD2NTD–GluK2 (GluD2NTD-GluK2, B). PF–EPSCs were recorded in the presence of NASP (100 μm), a selective blocker against Ca2+-permeable AMPA/kainate receptors, to exclude GluK2-mediated current responses. C, Averaged input--output (I-O) relationship of the PF–EPSCs. The error bars indicate the SEM. *p < 0.05.
Figure 8.

Expression of GluD2 in heterologous cells, with specific accumulation noted at the excitatory synaptic terminals in cerebellar cultures. A, Representative images of presynaptic terminals on HEK293 cells expressing HA-tagged GluD2, GluA1, GluA2, GluA4, or GluK2. The cells were plated on cerebellar neuron cultures and immunostained with HA (green) and a presynaptic marker, synaptophysin (red). B, Quantification of the signal intensity of synaptophysin on HA-immunopositive regions. The error bars indicate the SEM (n = 9 images from 3 independent experiments). C, D, GluD2 specifically accumulated at the VGluT1-positive excitatory terminals (red, D), not the VGAT-positive inhibitory terminals (red, C), on HEK293 cells expressing HA–GluD2 (blue). E, Representative images of presynaptic terminals on HEK293 cells expressing GluD2wt, GluD2ΔNTD, GluK2, or GluK2NTD–GluD2 with GFP. The cells were plated on cerebellar neuron cultures and immunostained with GFP (green) and synaptophysin (red). F, Quantification of the signal intensity of synaptophysin in the GFP-immunopositive regions. The error bars indicate the SEM (n = 9 cells from 2 independent experiments). **p < 0.01.
Figure 7.

GluD2 channel function and ligand binding ability are not required for the recovery of impaired PF–Purkinje cell synapses in GluD2-null mice. A, B, Schematic diagrams of mutant GluD2s and representative PF–EPSCs recorded from GluD2-null Purkinje cells expressing GFP plus GluD2V/R (GluD2V/R, A) or GFP plus GluD2R/K (GluD2R/K, B). The valine at position 617 in the selectivity filter domain (indicated by a star in A) was replaced with arginine and the arginine at position 530 in the putative ligand binding site (indicated by × in B) was replaced with lysine to produce the “pore-dead mutant” and “ligand binding-dead mutant,” respectively. C, Averaged input–output (I-O) relationship of the PF–EPSCs. The PF–EPSC amplitudes from GluD2-null Purkinje cells expressing GFP (Vector), GFP plus GluD2wt (GluD2wt), GFP plus GluD2V/R (GluD2V/R), or GFP plus GluD2R/K (GluD2R/K) were averaged. The error bars indicate the SEM. D, Results for the rotor-rod test. Mice were placed on the rotor-rod at 20 rpm before (Pre) and 1 d after (Post) receiving a subarachnoidal injection of Sin–GFP (Vector), Sin–GluD2wt–GFP (GluD2wt), Sin–GluD2V/R–GFP (GluD2V/R), or Sin–GluD2R/K–GFP (GluD2R/K). The average time was calculated from the results of six trials. For comparison, the data for Sin–GFP and Sin–GluD2wt–GFP in C and D were taken from Figures 5 and 1, respectively. *p < 0.05, **p < 0.01. ns, Not significant.
Recombinant Sindbis virus and in vivo injection.
The recombinant Sindbis virus for the expression of each GluR in combination with a GFP was constructed as described previously (Matsuda et al., 2003). Under deep anesthesia with an intraperitoneal injection of ketamine/xylazine (80/20 mg/kg; Sigma), the solution containing the recombinant Sindbis virus (titer, 1.0 × 108–9 TU/ml) was injected into the subarachnoidal space above the cerebellar vermis in postnatal day 35 (P35) to P45 GluD2-null mice (ho5J mice; The Jackson Laboratory), as described previously (Hirai et al., 2003). Briefly, a microsyringe needle (33 gauge) was inserted into the surface of the cerebellar vermis, and a 5 μl viral solution was injected into the subarachnoidal space at a rate of 40 μl/h. To prevent the leakage of viral solution from the puncture, the injection site was sealed with a silicone elastomer (Kwik-Sil; World Precision Instruments) during the operation. In some experiments for electrophysiology and immunohistochemistry, the viral solution (2 μl) was directly injected into the vermis of cerebellar lobules V–VIII using a glass pipette (30 μm in diameter) and a microinjector (Nanoliter; World Precision Instruments).
Immunohistochemistry and electron microscopy.
Under deep anesthesia, virus-infected mice were fixed by cardiac perfusion with 0.1 m sodium phosphate buffer (PB), pH 7.4, containing 4% paraformaldehyde (4% PFA/PB); the cerebellum was then removed and soaked in 4% PFA/PB for 4 h. After rinsing the specimens with PBS, parasagittal slices (100 μm) were prepared using a microslicer (DTK-2000; D.S.K.) and were permeabilized with 0.2% Triton X-100 in PBS with 2% normal goat serum and 2% bovine serum albumin (BSA) for 6 h at 4°C. Immunohistochemical staining was performed using selective antibodies against GluD2 (1:500; Millipore) at 4°C, followed by incubation with Alexa546-conjugated secondary antibodies (1:1000; Invitrogen). The stained slices were viewed using a confocal laser-scanning microscope (Fluoview; Olympus). For double fluorescence, microslicer sections pretreated with 1 mg/ml pepsin in 0.2N HCl at 37°C for 3 min were incubated with rabbit GluD2C antibody [1 μg/ml (Araki et al., 1993)] and rabbit GFP antibody (1 μg/ml; Invitrogen), followed by incubation of cyanine 3 (Cy3)-conjugated secondary antibodies (1:200 for each; Jackson ImmunoResearch). GFP and Cy3 signals were scanned by a confocal laser-scanning microscope (FV1000; Olympus).
For the electron microscopy (EM) analysis, mice were fixed transcardially with 0.1 m sodium PB, pH 7.4, containing 4% PFA and 0.5% glutaraldehyde, and the cerebellum was further immersed in the same fixative for 4 h. Immunogold EM analyses was performed as described previously (Fukaya et al., 2003). To identify the infected Purkinje cells, virally expressed GFP was labeled by the silver-enhanced preembedding immunogold method using rabbit anti-GFP antibody (1 μg/ml; Invitrogen), 1.4 nm gold particle-conjugated anti-rabbit antibody (Nanogold; Nanoprobes), and silver enhancement kit (HQ silver; Nanoprobes). To evaluate synaptic expression of virally transfected GluD2 and GluD2ΔNTD, double postembedding immunogold was performed using ultrathin Lowicryl HM20 resin sections. After etching with a saturated solution of NaOH in absolute ethanol for a few seconds and blocking with 2% normal goat serum (Nichirei) in 10 mm Tris-buffered saline (TBS), pH 7.6, containing 0.1% Tween 20 (TBS-T), nickel grids were first immunoreacted with rabbit anti-GFP antibody (5 μg/ml) overnight and 20 nm colloidal gold-conjugated anti-rabbit IgG (1:100; British Bio Cell International) in 2% normal goat serum for 2 h. Then, grids were blocked with normal rabbit serum and immunoreacted overnight with 10 nm colloidal gold-conjugated rabbit GluD2C antibody (OD520, 0.2) in 2% normal rabbit serum according to the method described by Slot and Geuze (1985). After each step, girds were washed with TBS-T. Finally, grids were stained with 2% uranyl acetate for 5 min and mixed lead solution for 30 s. Electron micrographs were taken randomly by an H-7100 electron microscope (Hitachi High-Technologies). For quantitative analysis, the numbers of contacted synapses expressing GFP and immunogold particles were counted on electron micrographs using MetaMorph software (Molecular Devices).
Behavioral analysis.
A walking footprint pattern analysis and the rotor-rod test were performed as described previously (Hirai et al., 2005a; Kakegawa et al., 2007a). Briefly, the GluD2-null mice were subjected to a rotor-rod test before and 1 d after viral injection. Each test comprised six continuous trials performed at 30 s intervals using a rotor-rod speed of 20 rotations per minute (rpm); the time that the mouse stayed on the rod was measured (maximum score, 120 s).
Electrophysiology.
Current responses were recorded from human embryonic kidney 293 (HEK293) (a kind gift from Dr. R. Horn, Thomas Jefferson University Medical School, Philadelphia, PA) or Purkinje cells using an Axopatch 200B amplifier (Molecular Devices) and the pClamp system (version 9.2; Molecular Devices), as described previously (Kohda et al., 2000; Kakegawa and Yuzaki, 2005; Kakegawa et al., 2008). Sagittal and coronal slices (200 μm thick) were prepared from virus-infected cerebella, and whole-cell patch-clamp recordings were performed from Purkinje cells that emitted GFP fluorescence. The resistances of the patch pipettes were 3–5 MΩ when filled with an internal solution of the following composition (in mm): 65 Cs-methanesulfonate, 65 K-gluconate, 20 HEPES, 10 KCl, 1 MgCl2, 4 Na2ATP, 1 Na2GTP, 5 sucrose, and 0.4 EGTA, pH 7.25 (295 mOsm/kg). The solution used for slice storage and recording was composed as follows (in mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 d-glucose. This solution was bubbled continuously with a mixture of 95% O2 and 5% CO2 at room temperature. Picrotoxin (100 μm; Sigma) was always present in the saline to block inhibitory synaptic transmission. In the recordings made from Purkinje cells virally expressing GluK2 or GluD2NTD–GluK2 (in which the Q/R site was unedited), 1-naphtylacetyl spermine (NASP) (100 μm), a selective blocker of Ca2+-permeable AMPA and kainate receptors (Koike et al., 1997), was added to the extracellular solution to remove GluK2-mediated current responses. To elicit PF-evoked EPSCs (PF–EPSCs), a stimulating glass pipette was placed on the molecular layer (square pulse, 10 μs, ∼200 μA), and the selective stimulation of PFs was confirmed by the paired-pulse facilitation of PF–EPSCs with a 50 ms stimulation interval. In experiments on the input–output relationship of PF–EPSCs, increasing the stimulus intensity sometimes resulted in contamination with climbing fiber (CF)-evoked EPSC and granule cell axon-evoked responses, which exhibited a faster rise time and a higher release probability than the PF–EPSCs (Sims and Hartell, 2005) in sagittal slices of GluD2-null cerebella, probably because of their thinner molecular layer and the abnormally expanded CF territory (Kurihara et al., 1997; Hashimoto et al., 2001; Ichikawa et al., 2002). Therefore, recordings were performed using coronal slices, and a stimulating electrode was set midway between the Purkinje cell layer and the pia mater to a depth of 50–100 μm at a position >200 μm lateral to the recorded Purkinje cell to stimulate pure PFs (Marcaggi and Attwell, 2005; Sims and Hartell, 2005).
In the LTD sessions, PF–EPSCs were recorded successively at a frequency of 0.1 Hz from Purkinje cells clamped at −80 mV (Kakegawa et al., 2007a). After a stable PF–EPSC amplitude had been observed for at least 10 min, a conjunctive stimulation (CJ-stim) consisting of 30 single PF stimuli together with a 200 ms depolarizing pulse from a holding potential of −60 to +20 mV was applied for LTD induction. Access resistances were monitored every 10 s by measuring the peak currents in response to hyperpolarizing steps (50 ms, 2 mV) throughout the experiments; the measurements were discarded if the resistance changed by >20% of its original value. The normalized EPSC amplitude on the ordinate represents the EPSC amplitude for the average of six traces for 1 min divided by that of the average of six traces for 1 min immediately before CJ-stim. Signals were filtered at 1 kHz and digitized at 4 kHz.
Synapse formation assay in HEK293 cells.
HEK293 cells were cultured in DMEM (Invitrogen) supplemented with 10% fetal calf serum and l-glutamine (1 mm) and were grown in 10% CO2 at 37°C. “Mixed” cerebellar cultures were prepared from embryonic day 17 to day-of-birth ICR mice, as described previously (Matsuda et al., 2006). Cells were plated at a density of 2 × 105 cells on plastic coverslips (diameter, 13.5 mm) and were maintained in a mixed culture medium composed of DMEM/F-12 (Invitrogen) containing 100 μm putrescine, 30 nm sodium selenite, 0.5 ng/ml tri-iodothyronine (Sigma), 3.9 mm glutamate (Invitrogen), 0.25 mg/ml BSA (Sigma), and N3 supplement (100 μg/ml apo-transferrin, 10 μg/ml insulin, and 20 nm progesterone; all from Sigma) in 5% CO2 at 37°C. After transfection with or without a GFP expression vector (pCAGGS–GFP) using CellPhect (GE Healthcare), the HEK293 cells were added to the cerebellar culture at 7 d in vitro and cocultured for 5 d in the presence of 5-fluoro-2′-deoxyuridine (1 μm; Sigma). Cocultured cells were fixed with 4% PFA/PBS for 15 min and subsequently with 100% methanol at −20°C for 5 min. After permeabilization with 0.4% Triton X-100 in PBS with 2% normal goat serum and 2% BSA for 1 h, the cells were stained with synaptophysin (1:1000; Sigma), vesicular glutamate transporter 1 (VGluT1) (1:1000), vesicular GABA transporter (VGAT) (1:2000), HA (1:1000; Covance Research Products), or GFP (1:2000; Millipore), followed by incubation with Alexa546, 488, and 305-conjugated secondary antibodies (1:2000; Invitrogen).
To quantify synaptophysin accumulation in the transfected HEK293 cells, at least eight images were randomly taken using fixed gains and exposures for each fluorescent channel per transfected cDNA. The images were analyzed using IP-lab software (version 3.61; BD Biosciences). HA- or GFP-immunopositive regions were selected using macro “auto-segmentation.” The intensity of synaptophysin immunoreactivity within the segmented area was averaged, and the background immunoreactivity was subtracted. Subtracted intensity values above the threshold were measured and normalized to the autosegmented area of the transfected HEK293 cells.
Data analysis and statistics.
Data are presented as the mean ± SEM, and statistical significance was defined as p < 0.05, as determined using the paired Student's t test, the Mann–Whitney U test, the Fisher's exact test, or an ANOVA, followed by the Bonferroni's test for multiple comparisons.
Results
Virally introduced GluD2 rapidly rescues the ataxic gait and motor dyscoordination
We showed previously that antibodies [anti-GluD2 antibody (Hirai et al., 2003)] or proteins [Cbln1 (Ito-Ishida et al., 2008)] injected into the subarachnoid supracerebellar space above the cerebellar vermis affected motor coordination and gait pattern in vivo. Thus, to examine whether the expression of GluD2 could restore cerebellar functions in mature GluD2-null mice in vivo, we injected Sindbis virus carrying a gene encoding GluD2wt and GFP (Sin–GluD2wt–GFP) into the subarachnoidal space of GluD2-null mice aged P35–P45 (Fig. 1 B). Sindbis virus was used because it can rapidly express large cDNAs in neurons (Ehrengruber, 2002). Indeed, 1 d after injection, we observed GFP signals in many Purkinje cells (85% of total Purkinje cells in the injected region) (supplemental Fig. S1, available at www.jneurosci.org as supplemental material); these signals were distributed widely throughout the cerebellar cortex of all the lobules with a noticeable gradient from the injected site (Fig. 1 C). Virally expressed GluD2wt exerted a remarkable effect on the locomotive performance of adult GluD2-null mice 1 d after injection (Fig. 1 D). To evaluate the level of ataxia quantitatively, the mice were placed on a rod that rotated at 20 rpm (Fig. 1 E) (supplemental movie, available at www.jneurosci.org as supplemental material). The average retention time on the rotating rod significantly improved within 1 d after the injection of Sin–GluD2wt–GFP (3.5 ± 0.4 s before infection and 51.3 ± 14.3 s after infection; n = 6; paired Student's t test, p = 0.021) but not after the injection of an empty vector (Sin–GFP; 4.0 ± 0.3 s before infection and 6.7 ± 1.7 s after infection; n = 8; paired Student's t test, p = 0.171). These findings indicate that, although the recovery was partial compared with the motor performance of the wild-type mice (day 1, 81 ± 5 s; day 2, 113 ± 4 s; n = 7), the expression of GluD2 rapidly restored motor performance in adult GluD2-null mice in vivo.
We next examined the role of the most NTD on synapse formation by preparing GluD2 lacking the NTD (GluD2ΔNTD) (Fig. 1 A). Unlike Sin–GluD2wt–GFP, a Sindbis virus carrying GluD2ΔNTD (Sin–GluD2ΔNTD–GFP) could not rescue ataxic gait (Fig. 1 D) or poor performance on the rotor-rod test (4.0 ± 0.4 s before infection and 9.7 ± 2.7 s after infection; n = 7; paired Student's t test, p = 0.090) (Fig. 1 E) (supplemental movie, available at www.jneurosci.org as supplemental material) of adult GluD2-null mice, indicating that the NTD of GluD2 plays an indispensable role in cerebellar functions in vivo.
NTD of GluD2 regulates the receptor trafficking, but it is dispensable for LTD induction
When the surface delivery of GluD2 was evaluated using the lurcher mutant GluD2 (GluD2Lc), which exhibits continuous channel activity on the cell surface (Kohda et al., 2000; Wollmuth et al., 2000), the current amplitude was significantly reduced by deleting the NTD from GluD2Lc (Fig. 2 A,B) (ANOVA followed by Bonferroni's test, F = 15.047, p = 0.002). An immunohistochemical analysis of GluD2-null Purkinje cells transduced with Sin–GluD2wt–GFP or Sin–GluD2ΔNTD–GFP also indicated that, although immunoreactivities of GluD2ΔNTD and GluD2wt were similarly distributed throughout Purkinje cells, GluD2ΔNTD immunoreactivity was weaker than GluD2wt immunoreactivity within spines (Fig. 2 C). Furthermore, double-labeling postembedding immunogold analysis revealed that GluD2-immunopositive gold particles were less frequently observed at the postsynaptic density (PSD) of Purkinje cells transduced with Sin–GluD2ΔNTD–GFP (0.03 particles/100 nm PSD; n = 82 spines) than those transduced with Sin–GluD2wt–GFP (1.2 particles/100 nm PSD; n = 95 spines) (Fig. 2 D). Similarly, the NTD is reportedly required for the assembly of the initial dimers of iGluR subunits (Ayalon and Stern-Bach, 2001) and the trafficking of iGluRs to the cell surface (Matsuda and Yuzaki, 2002; Wang et al., 2003; Matsuda et al., 2005; Motohashi et al., 2007). These findings indicate that one of the functions of the NTD of GluD2 is to stably express GluD2 proteins at the cell surface.
Figure 2.

The role of the NTD in trafficking of GluD2. A, The current–voltage relationship of HEK293 cells expressing the lurcher mutant GluD2 (GluD2Lc; Lc), GluD2Lc lacking the NTD (Lc-ΔNTD), and a vector. B, Channel activity of mutant GluD2 in HEK293 cells expressing Lc and Lc-ΔNTD. The holding current at −80 mV was measured. The error bars indicate the SEM. *p < 0.05, **p < 0.01, ***p < 0.001. C, Confocal images of Purkinje cells transduced with GluD2wt and GluD2ΔNTD. The transduced GluD2 immunoreactivities were visualized using Alexa546 (red) and were overlaid with the GFP signals to indicate the infected Purkinje cells. Arrows indicate typical GluD2-immunopositive spines. D, Representative postembedding immunogold EM images showing synapses between PFs and Purkinje cells expressing GFP only (Vector), GFP plus GluD2wt (GluD2wt), and GFP plus GluD2ΔNTD (GluD2ΔNTD) in GluD2-null cerebella. Infected Purkinje cells were identified using anti-GFP antibodies and large gold particles (20 nm) and GluD2 was visualized using anti-GluD2 antibodies conjugated to small gold particles (10 nm). Arrows and arrowheads indicate GluD2-positive gold particles detected on the PSD and in the cytosol, respectively.
The induction of LTD and motor learning absolutely require signaling via the C-terminal end of GluD2 (Kohda et al., 2007; Kakegawa et al., 2008). However, the contribution of the NTD of GluD2 to LTD induction has remained unclear. Thus, we examined whether LTD could be restored in GluD2-null Purkinje cells transduced with Sin–GluD2ΔNTD–GFP. In GluD2-null Purkinje cells transduced with Sin–GluD2wt–GFP, a conjunctive stimulation, which consisted of 30 single PF stimuli together with a 200 ms depolarization of the Purkinje cells, successfully induced LTD in PF–EPSCs but did not induce LTD in Purkinje cells expressing GFP only (Fig. 3). The amplitude of the PF–EPSCs 25–30 min after conjunctive stimulation was 76 ± 5% (n = 7 from 5 mice) (Fig. 3) of the control responses in Purkinje cells transduced with Sin–GluD2wt–GFP, whereas that of cells treated with Sin–GFP was 96 ± 4% (n = 7 from 5 mice; ANOVA followed by Bonferroni's test, F = 4.508, p = 0.043). Interestingly, the conjunctive stimulation induced a robust LTD in GluD2-null Purkinje cells transduced with Sin–GluD2ΔNTD–GFP (Fig. 3). The amplitude of PF–EPSCs 25–30 min after conjunctive stimulation was 75 ± 7% (n = 8 from 5 mice) of the control responses (p = 0.041 vs Sin–GFP and p > 0.05 vs Sin–GluD2wt–GFP). These findings indicate that the NTD of GluD2 is dispensable for the induction of LTD. Previously, the motor performance and LTD were shown to be completely restored in transgenic mice when the expression levels of the GluD2wt transgene were as low as 10% of that in endogenous GluD2 in wild-type mice (Yuzaki, 2005). Therefore, a small amount of GluD2ΔNTD delivered to the PF synapses (Fig. 2 C,D) was likely sufficient to restore LTD in GluD2-null mice.
Figure 3.
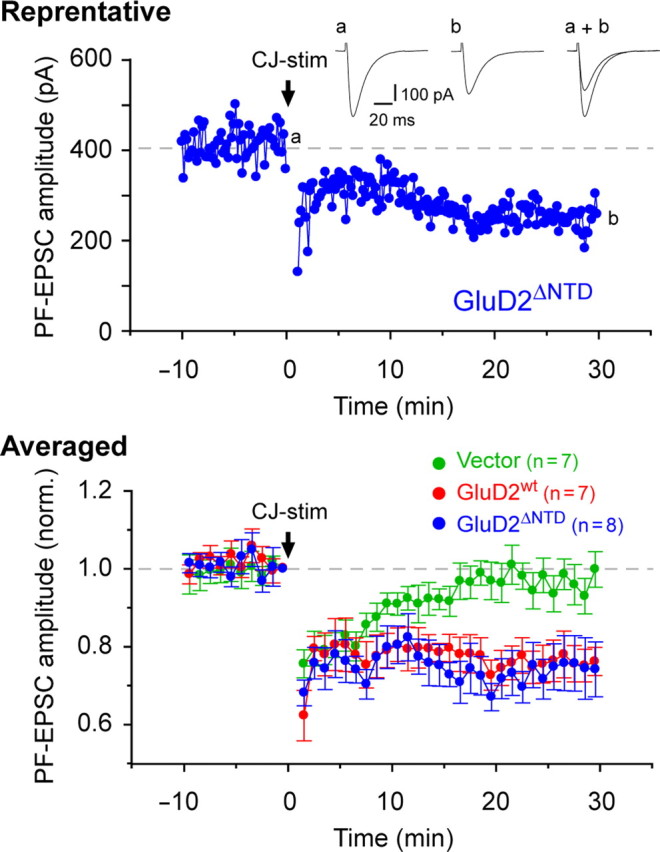
Recovery of abrogated LTD in GluD2-null Purkinje cells by viral expression of GluD2wt or GluD2ΔNTD. Representative (top) and averaged (bottom) data of cerebellar LTD recorded from GluD2-null Purkinje cells expressing GFP (Vector), GFP plus GluD2wt (GluD2wt), or GFP plus GluD2ΔNTD (GluD2ΔNTD). To induce LTD, CJ-stim [30 episodes of (Purkinje cell depolarization plus PF stimulation) at 1 Hz] was applied at time 0 (arrow). The inset traces show the PF–EPSCs just before (a) or 30 min after (b) CJ-stim and their superimposition (a + b).
NTD of GluD2 is necessary for PF–Purkinje cell synapse formation in vivo
A major morphological characteristic of GluD2-null mice is a marked reduction in PF–Purkinje cell synapses in the cerebellum; although the total spine densities are similar, the percentage of normal spines contacted by PF terminals is reduced in GluD2-null Purkinje cells to ∼50–60% of that in wild-type Purkinje cells (Kurihara et al., 1997; Lalouette et al., 2001). Thus, to examine the morphological basis of the behavioral recovery, we next performed an immunogold EM analysis on serial sections prepared from GluD2-null cerebellum 1 d after the injection of Sin–GluD2wt–GFP, Sin–GluD2ΔNTD–GFP, or Sin–GFP and determined the percentage of the contacted spines that were immunopositive for GFP (Fig. 4 A–C). Although we could not observe the longer-term effects because of the cytotoxicity of the Sindbis virus (Ehrengruber, 2002), the percentage of contacted spines was significantly higher (Fisher's exact test, p = 3.622 × 10−8) in Purkinje cells transduced with Sin–GluD2wt–GFP (87%; n = 101 spines) than in those transduced with Sin–GluD2ΔNTD–GFP (51%; n = 71 spines) or Sin–GFP (54%; n = 68 spines) (Fig. 4 D). These results indicate that, in mature GluD2-null cerebellum, the expression of GluD2wt rapidly induces synapse formation between PF and Purkinje cells as early as 1 d after infection and in a manner that is dependent on the NTD.
Figure 4.

Viral expression of GluD2wt, but not GluD2ΔNTD, rapidly induced PF–Purkinje cell synapses in mature GluD2-null mice. A–C, Representative immunogold EM images showing synapses between PFs and Purkinje cells expressing GFP (Vector, A1–A4), GFP plus GluD2wt (GluD2wt, B1–B4), or GFP plus GluD2ΔNTD (GluD2ΔNTD, C1–C4) in GluD2-null cerebella. Infected Purkinje cells were identified using anti-GFP antibodies and gold particle labels. F and C indicate free and contacted spines, respectively. To analyze the synapse ultrastructures in detail, serial sections were made (1 to 4 of A2–A4, B2–B4, and C2–C4). The arrows represent free spines, and the asterisks indicate Purkinje cell spines. D, Percentage of contacted synapses. The total numbers of the counted spines in GFP-expressing Purkinje cells are shown in the parentheses. **p = 3.622 × 10−8 (Fisher's exact test).
NTD of GluD2 is necessary and sufficient for functional PF synapse formation
To examine whether the new synapses induced by GluD2wt expression were functional, we next compared the input--output relationship of PF–EPSC amplitudes from GluD2-null Purkinje cells transduced with Sin–GluD2wt–GFP, Sin–GluD2ΔNTD–GFP, or Sin–GFP in acutely prepared cerebellar slices using the whole-cell patch-clamp method (Fig. 5 A–C). We found that GluD2-null Purkinje cells transduced with Sin–GluD2wt–GFP displayed significantly larger PF–EPSCs than those transduced with Sin–GluD2ΔNTD–GFP or Sin–GFP (n = 25 each; ANOVA followed by Bonferroni's test, F = 9.460, p = 0.005 vs Sin–GluD2ΔNTD–GFP, p < 0.001 vs Sin–GFP, and p > 0.05 in Sin–GluD2ΔNTD–GFP vs Sin–GFP) (Fig. 5 D). These results indicate that GluD2wt induced functional PF synapses in a manner dependent on the NTD and that the amplitudes of PF–EPSCs provide an alternative measure of the synaptic integrity of PF–Purkinje cell synapses.
Figure 5.

Functional restoration of PF–Purkinje cell synapses in GluD2-null mice by viral expression of GluD2wt but not GluD2ΔNTD. A–C, Schematic diagrams of GluD2 constructs and representative PF-evoked EPSC traces in GluD2-null Purkinje cells transduced with GFP (Vector, A), GFP plus GluD2wt (GluD2wt, B), or GFP plus GluD2ΔNTD (GluD2ΔNTD, C). D, Averaged input--output (I-O) relationship of PF–EPSCs in each construct. The error bars indicate the SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Next, to examine the specificity of the NTD of GluD2, we transduced GluD2-null Purkinje cells with the kainate receptor GluK2, which is not highly expressed in Purkinje cells (Wisden and Seeburg, 1993). To exclude the contamination of PF–EPSCs by currents directly mediated by homomeric GluK2 channels, we measured PF–EPSCs in the presence of NASP (100 μm), which blocks the channels formed by Ca2+-permeable AMPA or kainate receptors. We found that the expression of GluK2 did not increase PF–EPSCs in GluD2-null Purkinje cells (Fig. 6 A,C). In contrast, the expression of a chimeric GluK2 (GluD2NTD–GluK2), in which the NTD of GluD2 replaced that of GluK2, significantly increased PF–EPSCs in GluD2-null Purkinje cells (n = 25 each; Mann–Whitney U test, p = 0.025) (Fig. 6 C), although GluK2 was more efficiently trafficked to the cell surface than GluD2NTD–GluK2 (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Therefore, a small amount of the NTD of GluD2 was sufficient to induce functional PF–Purkinje cell synapses in mature GluD2-null Purkinje cells. In addition, these findings indicate that the NTD is not only involved in trafficking of GluD2 but also plays an essential role in PF synapse formation.
Channel activities and ligand binding are not required for the synapse-forming ability of GluD2
The replacement of valine with arginine in the putative channel pore of GluD2 (GluD2V/R) (Fig. 7 A) blocks the constitutive currents observed in GluD2Lc (Kakegawa et al., 2007b); a similar mutation is also known to disrupt the channel pores of AMPA and kainate receptors (Dingledine et al., 1992; Robert et al., 2002). Because the introduction of GluD2V/R completely rescued impaired LTD in GluD2-null Purkinje cells, we indicated previously that channel activity was not required for GluD2 to regulate LTD induction (Kakegawa et al., 2007b). However, whether another function of GluD2 (i.e., the formation of PF–Purkinje cell synapses) requires the channel activities of GluD2 has remained unclear. Thus, we examined PF–EPSCs from GluD2-null Purkinje cells transduced with Sin–GluD2wt–GFP, a Sindbis virus carrying GluD2V/R (Sin–GluD2V/R–GFP), or Sin–GFP in acutely prepared cerebellar slices. We found that Sin–GluD2wt–GFP and Sin–GluD2V/R–GFP similarly increased the amplitudes of PF–EPSCs (n = 25 each; ANOVA followed by a Bonferroni's test, F = 7.320, p > 0.05 vs Sin–GluD2wt–GFP and p = 0.001 vs Sin–GFP) (Fig. 7 C). Similarly, both Sin–GluD2wt–GFP and Sin–GluD2V/R–GFP rescued the poor performances on the rotor-rod test (4.3 ± 0.6 s before infection and 42.4 ± 12.7 s after infection, n = 5; paired Student's t test, p = 0.040) (Fig. 7 D) of adult GluD2-null mice.
Recent x-ray crystallography revealed that d-serine and glycine bound to the ligand-binding core of GluD2 by interacting with the conserved arginine residue (Naur et al., 2007). Thus, to address the question whether synapse-forming activity of GluD2 requires agonist binding, we virally expressed a GluD2 transgene (GluD2R/K) (Fig. 7 B) in which lysine replaced the conserved arginine in the ligand-binding core. We found that GluD2R/K rescued decreased PF-EPSC amplitudes (n = 25 each; ANOVA followed by a Bonferroni's test, F = 6.756, p = 0.714 vs Sin–GluD2wt–GFP and p = 0.045 vs Sin–GFP) (Fig. 7 C) and the poor performance on the rotor-rod test (3.9 ± 0.3 s before infection and 36.7 ± 8.1 s after infection; n = 6; paired Student's t test, p = 0.010) (Fig. 7 D) of adult GluD2-null mice. Similarly, a genetically introduced GluD2R/K completely rescued all abnormal phenotypes of GluD2-null mice, including LTD and hypoplasia of PF–Purkinje cell synapses (Hirai et al., 2005a). Together, these findings indicate that two major functions of GluD2, the control of LTD induction and the formation of PF synapses, do not require the channel activity and the ligand binding ability of GluD2.
GluD2 specifically induces artificial excitatory synapses in vitro
So far, only three cell adhesion molecules, neuroligins, synCAMs, and netrin-G ligands (NGLs), have been shown to serve as presynaptic organizers, which are sufficient to drive presynaptic contact formation on heterologous cells in coculture systems (Scheiffele et al., 2000; Biederer et al., 2002; Levinson and El-Husseini, 2005; Kim et al., 2006). Thus, to further clarify the nature of the synaptogenic capability of GluD2, we transfected HEK293 cells with cDNA encoding HA-tagged GluD2, GluA1, GluA2, GluA4, or GluK2, together with GFP cDNA, and then cocultured these cells with cerebellar neurons. Synapses formed on the transfected HEK293 cells were estimated using immunohistochemical analysis with antibodies against HA and synaptophysin, a marker for presynaptic terminals (Fig. 8 A). We found that GluD2 specifically recruited presynaptic terminals around transfected HEK293 cells; such effects were never observed for other iGluRs (Fig. 8 B). Therefore, GluD2 is a unique iGluR member that can serve as a potent presynaptic organizer, and GluD2 is sufficient for the accumulation of presynaptic terminals on heterologous cells in vitro.
Unlike synCAMs and NGLs, neuroligins are involved in both excitatory and inhibitory synapses (Levinson and El-Husseini, 2005). Thus, we examined the identity of GluD2-induced presynaptic terminals by immunostaining against VGluT1, a specific marker for glutamatergic excitatory presynaptic terminals, and VGAT, a marker of glycine- and GABA-containing inhibitory terminals. We found that, unlike the immunoreactivity for VGAT (Fig. 8 C), the immunoreactivity for VGluT1 accumulated in the vicinity of cells expressing GluD2 and was colocalized with synaptophysin immunoreactivity (Fig. 8 D). These findings indicate that GluD2 specifically induced excitatory synapses in the current coculture system.
Finally, we examined the role of the NTD in the formation of artificial synapses by expressing GluD2wt or GluD2ΔNTD together with GFP in HEK293 cells and coculturing the cells with cerebellar neurons (Fig. 8 E). Unlike GluD2wt, mutant GluD2 lacking the NTD could no longer recruit presynaptic terminals in the vicinity of the transfected HEK293 cells (Fig. 8 F). Furthermore, although GluK2 could not induce synaptogenesis, GluD2NTD–GluK2 was capable of causing the accumulation of presynaptic terminals (Fig. 8 E,F). These findings are consistent with in vivo data (Fig. 6) and indicate that the NTD of GluD2 is necessary and sufficient to induce artificial synapses onto heterologous cells.
Discussion
GluD2 plays two unique and crucial roles at PF–Purkinje cell synapses in the cerebellum: control of LTD induction and formation of PF synapses (Yuzaki, 2004). Although the former function is known to be achieved by signaling via the CTD of GluD2 (Kohda et al., 2007; Kakegawa et al., 2008), how the latter function is regulated has remained unclear. Here, we show that, unlike GluD2wt, GluD2 lacking the NTD (GluD2ΔNTD) could not recruit presynaptic terminals onto mature GluD2-null Purkinje cell dendrites in vivo (Figs. 1, 4, 5) nor could it recruit them onto heterologous cells in vitro (Fig. 8). Furthermore, although GluK2 did not increase the PF–EPSC amplitudes in GluD2-null Purkinje cells in vivo (Fig. 6) and did not induce synaptogenesis in heterologous cells in vitro (Fig. 8), chimeric GluK2 containing the NTD of GluD2 (GluD2NTD–GluK2) was capable of performing both of these activities (Figs. 6, 8). These data indicate that the NTD of GluD2 is necessary and sufficient for the role of GluD2 in the regulation of PF–Purkinje cell synaptogenesis.
Dual functions differentially mediated by NTD and CTD of GluD2
How is NTD-mediated signaling related to CTD-mediated signaling? Synaptic adhesion molecules, such as EphB and the neural cell adhesion molecule, are not simple glues but act as signaling molecules to activate tyrosine kinase (Pasquale, 2008) or mitogen-activated protein kinase (Schmid et al., 1999; Kolkova et al., 2000), respectively. Thus, the NTD of GluD2, by interacting with a certain molecule expressed on PF terminals, may also modulate the CTD-based intracellular signaling involved in the LTD. However, LTD was restored in GluD2-null Purkinje cells expressing GluD2ΔNTD (Fig. 3), indicating that the CTD was sufficient for regulating LTD induction. Conversely, because GluD2NTD–GluK2, which contained the NTD but lacked the CTD of GluD2, could induce synapse formation in vitro and in vivo (Figs. 6, 8), the CTD of GluD2 was dispensable for the synaptogenic activity. Therefore, NTD-based signaling regulating PF synapse formation and CTD-based signaling controlling LTD induction are likely mediated by two independent mechanisms (supplemental Fig. S3, available at www.jneurosci.org as supplemental material).
The NTD of iGluRs is thought to have evolved from the bacterial periplasmic amino acid-binding protein LIVBP (leucine/isoleucine/valine-binding protein). The NTD of GluD2 shares a sequence similarity of ∼35% with the NTD of the AMPA or kainate receptor family. Like the NTD of iGluRs, the NTD of GluD2 was also involved in the trafficking of GluD2 to the cell surface (Fig. 2). Similarly, we showed previously that various mutations in the NTD of GluD2 impaired the assembly capabilities of GluD2, resulting in impaired trafficking of GluD2 to the cell surface and GluD2-null phenotypes in several kinds of hotfoot mutant mice (Matsuda and Yuzaki, 2002; Wang et al., 2003; Motohashi et al., 2007). In the present study, we have clarified another function of the NTD of GluD2. Interestingly, the NTD of the AMPA receptor subunit GluA2 has been shown to promote the formation and growth of dendritic spines in cultured hippocampal neurons by interacting directly with the cell adhesion molecule N-cadherin (Saglietti et al., 2007). Similarly, axonally derived neuronal pentraxin (NP) 1 and NP receptor have been shown recently to cluster the postsynaptic AMPA receptor GluA4 by interacting directly with the NTD of GluA4 (Sia et al., 2007). Thus, the NTDs of AMPA receptors and GluD2 share two common functions: the regulation of subunit assembly and the control of morphological integrity at the synapses. However, GluD2 is unique in that it serves as a presynaptic organizer; in contrast, GluA2 and GluA4 mainly mediate certain aspects of postsynaptic differentiation. Furthermore, among the numerous molecules involved in synapse formation, only synCAMs, NGLs, neuroligins (Scheiffele et al., 2000; Biederer et al., 2002; Levinson and El-Husseini, 2005; Kim et al., 2006), and now GluD2 are capable of driving presynaptic contact formation on heterologous cells in vitro (Fig. 8) (Uemura and Mishina, 2008).
Sindbis virus-mediated behavioral recovery of GluD2-null mice
Immature Purkinje cells are normally innervated by multiple CFs, but redundant CFs are gradually eliminated until a one-to-one relationship is established by the end of the third week of postnatal development (Crepel et al., 1980). In contrast, GluD2-null Purkinje cells remain innervated by supernumerary CFs even during adulthood (Hashimoto et al., 2001; Ichikawa et al., 2002). We reported previously that the expression of GluD2wt by a Sindbis virus could not rescue the sustained innervation by multiple CFs and ataxic gait in GluD2-null mice (Kohda et al., 2007). Similarly, in the present study, we observed that GluD2-null Purkinje cells remained innervated by supernumerary CFs after transduction with Sin–GluD2wt (data not shown). Nevertheless, ataxic gait and motor dyscoordination (as measured using the rotor-rod test) were significantly rescued by transduction with Sin–GluD2wt in the present study. The reason why motor performance was restored in the present study was probably because the Sindbis virus injected into the subarachnoidal space transduced more Purkinje cells than when it was injected into the cerebellar cortex.
It has not been completely clear whether motor coordination depends on an animal's ability to acquire discrete motor learning and cerebellar LTD. Analyses of various gene knock-out mice displaying cerebellar ataxia have indicated that motor coordination (as measured using the rotor-rod test and the footprint analysis) is not dependent on LTD but rather affected by the innervation pattern of Purkinje cells by CFs (Chen et al., 1995). However, the present results suggest that the CF innervation pattern may not play a major role. Because a genetically introduced GluD2 transgene lacking the CTD rescued the hypoplasia of PF–Purkinje cell synapses and the motor dyscoordination of GluD2-null mice without affecting the abrogated LTD (Kakegawa et al., 2008), LTD failure is also not responsible for motor dyscoordination. Together, these results suggest that the number of normal PF–Purkinje cell synapses likely affects gross motor performance, such as gait and coordination on a rotating rod.
GluD2 rapidly and dynamically regulates synaptogenesis in adult cerebellum
The development of the complete synaptic ultrastructure and its mature electrophysiological properties is thought to require several days (McAllister, 2007). Indeed, mature synapses in adult barrel cortex were shown recently to develop a few days after the appearance of a new spine (Knott et al., 2006). In contrast, we observed a significant increase in ultrastructurally and functionally normal synapses in adult GluD2-null cerebellum just 1 d after the injection of the Sindbis virus, which must have required at least 4–6 h to express the transgene (Ehrengruber, 2002). Thus, the time course of synaptogenesis induced by GluD2 in vivo was exceptionally rapid. The extremely high transgene expression levels achieved by the Sindbis virus may have contributed to the rapidity of this effect. In addition, the rapid synaptogenic effects of GluD2 may reflect its role in the assembly of existing presynaptic and postsynaptic structures rather than the induction of completely new synapses. Indeed, dendritic spines are maintained as “free spines” without any attached PF terminals in adult GluD2-null cerebellum (Kurihara et al., 1997; Lalouette et al., 2001); in contrast, the differentiation of dendritic spines and presynaptic boutons is highly correlated in the hippocampus and in the cortex (Friedman et al., 2000; Okabe et al., 2001). Because of the cytotoxicity of the Sindbis virus (Ehrengruber, 2002), we could not observe the longer-term effects of GluD2 transduction on PF innervation. However, because the ablation of the GluD2 gene in adult cerebellum using an inducible recombinase resulted in the gradual loss of normal PF–Purkinje cell synapses (Takeuchi et al., 2005), GluD2 is likely required for the maintenance of PF–Purkinje cell synapses in adult cerebellum. These findings have led us to suspect that GluD2 plays a crucial role in dynamic regulation of the contact state of PF–Purkinje cell synapses.
Cbln1, a member of the C1q/tumor necrosis factor superfamily, is secreted from cerebellar granule cells (Yuzaki, 2008). Interestingly, the behavioral, physiological, and anatomical phenotypes of cbln1-null mice precisely mimic those of GluD2-null mice (Hirai et al., 2005b). Recently, we found that a single application of recombinant Cbln1 rapidly, but transiently, induced new PF–Purkinje cell synapses in adult cbln1-null mice (Ito-Ishida et al., 2008). Cbln1 also rapidly induced functional PF synapses as early as 8 h after its application in cerebellar slices acutely prepared from young cbln1-null mice (Ito-Ishida et al., 2008). Thus, in addition to the striking phenotypic similarities shared by GluD2-null and cbln1-null mice, GluD2 and Cbln1 had very similar rapid synaptogenic effects in mature cerebellum. We postulate that Cbln1, which is released from granule cells, may directly or indirectly interact with the NTD of GluD2 to hold together presynaptic and postsynaptic elements at PF–Purkinje cell synapses in adult cerebellum. As other members of the Cbln family and GluRδ1 (GluD1; a relative of GluD2) are expressed in other brain regions (Wada and Ohtani, 1991; Safieddine and Wenthold, 1997; Pang et al., 2000; Magdaleno et al., 2006; Miura et al., 2006), this trans-synaptic process may represent a novel and more widespread mechanism controlling synaptic structure. Therefore, additional analysis of the detailed molecular mechanism underlying GluD2–Cbln1-based signaling pathways is warranted.
Footnotes
This work was supported by a Grant-in-Aid from MEXT (W.K., M.Y.), Takeda Science Foundation (M.Y.), Naito Foundation (K.M.), Sankyo Foundation of Life Science (M.Y.), Keio Gijuku Academic Development Funds (W.K.), and a Keio University Grant-in-Aid for the Encouragement of Young Medical Scientists (W.K.). We thank Sakae Narumi for the technical assistance.
References
- Araki K, Meguro H, Kushiya E, Takayama C, Inoue Y, Mishina M. Selective expression of the glutamate receptor channel delta 2 subunit in cerebellar Purkinje cells. Biochem Biophys Res Commun. 1993;197:1267–1276. doi: 10.1006/bbrc.1993.2614. [DOI] [PubMed] [Google Scholar]
- Ayalon G, Stern-Bach Y. Functional assembly of AMPA and kainate receptors is mediated by several discrete protein-protein interactions. Neuron. 2001;31:103–113. doi: 10.1016/s0896-6273(01)00333-6. [DOI] [PubMed] [Google Scholar]
- Biederer T, Sara Y, Mozhayeva M, Atasoy D, Liu X, Kavalali ET, Südhof TC. SynCAM, a synaptic adhesion molecule that drives synapse assembly. Science. 2002;297:1525–1531. doi: 10.1126/science.1072356. [DOI] [PubMed] [Google Scholar]
- Chen C, Kano M, Abeliovich A, Chen L, Bao S, Kim JJ, Hashimoto K, Thompson RF, Tonegawa S. Impaired motor coordination correlates with persistent multiple climbing fiber innervation in PKC gamma mutant mice. Cell. 1995;83:1233–1242. doi: 10.1016/0092-8674(95)90148-5. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Olsen RW, Peters J, Spedding M. A nomenclature for ligand-gated ion channels. Neuropharmacology. 2009;56:2–5. doi: 10.1016/j.neuropharm.2008.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crepel F, Delhaye-Bouchaud N, Guastavino JM, Sampaio I. Multiple innervation of cerebellar Purkinje cells by climbing fibres in staggerer mutant mouse. Nature. 1980;283:483–484. doi: 10.1038/283483a0. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Hume RI, Heinemann SF. Structural determinants of barium permeation and rectification in non-NMDA glutamate receptor channels. J Neurosci. 1992;12:4080–4087. doi: 10.1523/JNEUROSCI.12-10-04080.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrengruber MU. Alphaviral vectors for gene transfer into neurons. Mol Neurobiol. 2002;26:183–201. doi: 10.1385/MN:26:2-3:183. [DOI] [PubMed] [Google Scholar]
- Friedman HV, Bresler T, Garner CC, Ziv NE. Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron. 2000;27:57–69. doi: 10.1016/s0896-6273(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Fukaya M, Kato A, Lovett C, Tonegawa S, Watanabe M. Retention of NMDA receptor NR2 subunits in the lumen of endoplasmic reticulum in targeted NR1 knockout mice. Proc Natl Acad Sci USA. 2003;100:4855–4860. doi: 10.1073/pnas.0830996100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Ichikawa R, Takechi H, Inoue Y, Aiba A, Sakimura K, Mishina M, Hashikawa T, Konnerth A, Watanabe M, Kano M. Roles of glutamate receptor δ2 subunit (GluRδ2) and metabotropic glutamate receptor subtype 1 (mGluR1) in climbing fiber synapse elimination during postnatal cerebellar development. J Neurosci. 2001;21:9701–9712. doi: 10.1523/JNEUROSCI.21-24-09701.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H, Launey T, Mikawa S, Torashima T, Yanagihara D, Kasaura T, Miyamoto A, Yuzaki M. New role of δ2 glutamate receptors in AMPA receptor trafficking and cerebellar function. Nat Neurosci. 2003;6:869–876. doi: 10.1038/nn1086. [DOI] [PubMed] [Google Scholar]
- Hirai H, Miyazaki T, Kakegawa W, Matsuda S, Mishina M, Watanabe M, Yuzaki M. Rescue of abnormal phenotypes of the δ2 glutamate receptor-null mice by mutant δ2 transgenes. EMBO Rep. 2005a;6:90–95. doi: 10.1038/sj.embor.7400312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H, Pang Z, Bao D, Miyazaki T, Li L, Miura E, Parris J, Rong Y, Watanabe M, Yuzaki M, Morgan JI. Cbln1 is essential for synaptic integrity and plasticity in the cerebellum. Nat Neurosci. 2005b;8:1534–1541. doi: 10.1038/nn1576. [DOI] [PubMed] [Google Scholar]
- Ichikawa R, Miyazaki T, Kano M, Hashikawa T, Tatsumi H, Sakimura K, Mishina M, Inoue Y, Watanabe M. Distal extension of climbing fiber territory and multiple innervation caused by aberrant wiring to adjacent spiny branchlets in cerebellar Purkinje cells lacking glutamate receptor δ2. J Neurosci. 2002;22:8487–8503. doi: 10.1523/JNEUROSCI.22-19-08487.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M. Long-term depression. Annu Rev Neurosci. 1989;12:85–102. doi: 10.1146/annurev.ne.12.030189.000505. [DOI] [PubMed] [Google Scholar]
- Ito-Ishida A, Miura E, Emi K, Matsuda K, Iijima T, Kondo T, Kohda K, Watanabe M, Yuzaki M. Cbln1 regulates rapid formation and maintenance of excitatory synapses in mature cerebellar Purkinje cells in vitro and in vivo . J Neurosci. 2008;28:5920–5930. doi: 10.1523/JNEUROSCI.1030-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakegawa W, Yuzaki M. A mechanism underlying AMPA receptor trafficking during cerebellar long-term potentiation. Proc Natl Acad Sci USA. 2005;102:17846–17851. doi: 10.1073/pnas.0508910102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakegawa W, Miyazaki T, Hirai H, Motohashi J, Mishina M, Watanabe M, Yuzaki M. Ca2+ permeability of the channel pore is not essential for the δ2 glutamate receptor to regulate synaptic plasticity and motor coordination. J Physiol. 2007a;579:729–735. doi: 10.1113/jphysiol.2006.127100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakegawa W, Kohda K, Yuzaki M. The δ2 “ionotropic” glutamate receptor functions as a non-ionotropic receptor to control cerebellar synaptic plasticity. J Physiol. 2007b;584:89–96. doi: 10.1113/jphysiol.2007.141291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakegawa W, Miyazaki T, Emi K, Matsuda K, Kohda K, Motohashi J, Mishina M, Kawahara S, Watanabe M, Yuzaki M. Differential regulation of synaptic plasticity and cerebellar motor learning by the C-terminal PDZ-binding motif of GluRδ2. J Neurosci. 2008;28:1460–1468. doi: 10.1523/JNEUROSCI.2553-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwabuchi N, Ikeda K, Araki K, Hirano T, Shibuki K, Takayama C, Inoue Y, Kutsuwada T, Yagi T, Kang Y, Aizawa S, Mishina M. Impairment of motor coordination, Purkinje cell synapse formation, and cerebellar long-term depression in GluRδ2 mutant mice. Cell. 1995;81:245–252. doi: 10.1016/0092-8674(95)90334-8. [DOI] [PubMed] [Google Scholar]
- Kim S, Burette A, Chung HS, Kwon SK, Woo J, Lee HW, Kim K, Kim H, Weinberg RJ, Kim E. NGL family PSD-95-interacting adhesion molecules regulate excitatory synapse formation. Nat Neurosci. 2006;9:1294–1301. doi: 10.1038/nn1763. [DOI] [PubMed] [Google Scholar]
- Knott GW, Holtmaat A, Wilbrecht L, Welker E, Svoboda K. Spine growth precedes synapse formation in the adult neocortex in vivo . Nat Neurosci. 2006;9:1117–1124. doi: 10.1038/nn1747. [DOI] [PubMed] [Google Scholar]
- Kohda K, Wang Y, Yuzaki M. Mutation of a glutamate receptor motif reveals its role in gating and δ2 receptor channel properties. Nat Neurosci. 2000;3:315–322. doi: 10.1038/73877. [DOI] [PubMed] [Google Scholar]
- Kohda K, Kakegawa W, Matsuda S, Nakagami R, Kakiya N, Yuzaki M. The extreme C-terminus of GluRδ2 is essential for induction of long-term depression in cerebellar slices. Eur J Neurosci. 2007;25:1357–1362. doi: 10.1111/j.1460-9568.2007.05412.x. [DOI] [PubMed] [Google Scholar]
- Koike M, Iino M, Ozawa S. Blocking effect of 1-naphthyl acetyl spermine on Ca2+-permeable AMPA receptors in cultured rat hippocampal neurons. Neurosci Res. 1997;29:27–36. doi: 10.1016/s0168-0102(97)00067-9. [DOI] [PubMed] [Google Scholar]
- Kolkova K, Novitskaya V, Pedersen N, Berezin V, Bock E. Neural cell adhesion molecule-stimulated neurite outgrowth depends on activation of protein kinase C and the Ras-mitogen-activated protein kinase pathway. J Neurosci. 2000;20:2238–2246. doi: 10.1523/JNEUROSCI.20-06-02238.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurihara H, Hashimoto K, Kano M, Takayama C, Sakimura K, Mishina M, Inoue Y, Watanabe M. Impaired parallel fiber→Purkinje cell synapse stabilization during cerebellar development of mutant mice lacking the glutamate receptor δ2 subunit. J Neurosci. 1997;17:9613–9623. doi: 10.1523/JNEUROSCI.17-24-09613.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalouette A, Guénet JL, Vriz S. Hotfoot mouse mutations affect the δ2 glutamate receptor gene and are allelic to lurcher. Genomics. 1998;50:9–13. doi: 10.1006/geno.1998.5314. [DOI] [PubMed] [Google Scholar]
- Lalouette A, Lohof A, Sotelo C, Guénet J, Mariani J. Neurobiological effects of a null mutation depend on genetic context: comparison between two hotfoot alleles of the δ-2 ionotropic glutamate receptor. Neuroscience. 2001;105:443–455. doi: 10.1016/s0306-4522(01)00193-2. [DOI] [PubMed] [Google Scholar]
- Landsend AS, Amiry-Moghaddam M, Matsubara A, Bergersen L, Usami S, Wenthold RJ, Ottersen OP. Differential localization of δ glutamate receptors in the rat cerebellum: coexpression with AMPA receptors in parallel fiber-spine synapses and absence from climbing fiber-spine synapses. J Neurosci. 1997;17:834–842. doi: 10.1523/JNEUROSCI.17-02-00834.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinson JN, El-Husseini A. Building excitatory and inhibitory synapses: balancing neuroligin partnerships. Neuron. 2005;48:171–174. doi: 10.1016/j.neuron.2005.09.017. [DOI] [PubMed] [Google Scholar]
- Magdaleno S, Jensen P, Brumwell CL, Seal A, Lehman K, Asbury A, Cheung T, Cornelius T, Batten DM, Eden C, Norland SM, Rice DS, Dosooye N, Shakya S, Mehta P, Curran T. BGEM: an in situ hybridization database of gene expression in the embryonic and adult mouse nervous system. PLoS Biol. 2006;4:e86. doi: 10.1371/journal.pbio.0040086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcaggi P, Attwell D. Endocannabinoid signaling depends on the spatial pattern of synapse activation. Nat Neurosci. 2005;8:776–781. doi: 10.1038/nn1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda K, Fletcher M, Kamiya Y, Yuzaki M. Specific assembly with the NMDA receptor 3B subunit controls surface expression and calcium permeability of NMDA receptors. J Neurosci. 2003;23:10064–10073. doi: 10.1523/JNEUROSCI.23-31-10064.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda K, Matsuda S, Gladding CM, Yuzaki M. Characterization of the delta2 glutamate receptor-binding protein delphilin: splicing variants with differential palmitoylation and an additional PDZ domain. J Biol Chem. 2006;281:25577–25587. doi: 10.1074/jbc.M602044200. [DOI] [PubMed] [Google Scholar]
- Matsuda S, Yuzaki M. Mutation in hotfoot-4J mice results in retention of δ2 glutamate receptors in ER. Eur J Neurosci. 2002;16:1507–1516. doi: 10.1046/j.1460-9568.2002.02219.x. [DOI] [PubMed] [Google Scholar]
- Matsuda S, Kamiya Y, Yuzaki M. Roles of the N-terminal domain on the function and quaternary structure of the ionotropic glutamate receptor. J Biol Chem. 2005;280:20021–20029. doi: 10.1074/jbc.M410513200. [DOI] [PubMed] [Google Scholar]
- McAllister AK. Dynamic aspects of CNS synapse formation. Annu Rev Neurosci. 2007;30:425–450. doi: 10.1146/annurev.neuro.29.051605.112830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura E, Iijima T, Yuzaki M, Watanabe M. Distinct expression of Cbln family mRNAs in developing and adult mouse brains. Eur J Neurosci. 2006;24:750–760. doi: 10.1111/j.1460-9568.2006.04950.x. [DOI] [PubMed] [Google Scholar]
- Motohashi J, Kakegawa W, Yuzaki M. Ho15J: a new hotfoot allele in a hot spot in the gene encoding the δ2 glutamate receptor. Brain Res. 2007;1140:153–160. doi: 10.1016/j.brainres.2006.03.068. [DOI] [PubMed] [Google Scholar]
- Naur P, Hansen KB, Kristensen AS, Dravid SM, Pickering DS, Olsen L, Vestergaard B, Egebjerg J, Gajhede M, Traynelis SF, Kastrup JS. Ionotropic glutamate-like receptor delta2 binds D-serine and glycine. Proc Natl Acad Sci USA. 2007;104:14116–14121. doi: 10.1073/pnas.0703718104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe S, Miwa A, Okado H. Spine formation and correlated assembly of presynaptic and postsynaptic molecules. J Neurosci. 2001;21:6105–6114. doi: 10.1523/JNEUROSCI.21-16-06105.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang Z, Zuo J, Morgan JI. Cbln3, a novel member of the precerebellin family that binds specifically to Cbln1. J Neurosci. 2000;20:6333–6339. doi: 10.1523/JNEUROSCI.20-17-06333.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquale EB. Eph-ephrin bidirectional signaling in physiology and disease. Cell. 2008;133:38–52. doi: 10.1016/j.cell.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Robert A, Hyde R, Hughes TE, Howe JR. The expression of dominant-negative subunits selectively suppresses neuronal AMPA and kainate receptors. Neuroscience. 2002;115:1199–1210. doi: 10.1016/s0306-4522(02)00534-1. [DOI] [PubMed] [Google Scholar]
- Safieddine S, Wenthold RJ. The glutamate receptor subunit δ1 is highly expressed in hair cells of the auditory and vestibular systems. J Neurosci. 1997;17:7523–7531. doi: 10.1523/JNEUROSCI.17-19-07523.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saglietti L, Dequidt C, Kamieniarz K, Rousset MC, Valnegri P, Thoumine O, Beretta F, Fagni L, Choquet D, Sala C, Sheng M, Passafaro M. Extracellular interactions between GluR2 and N-cadherin in spine regulation. Neuron. 2007;54:461–477. doi: 10.1016/j.neuron.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Scheiffele P, Fan J, Choih J, Fetter R, Serafini T. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell. 2000;101:657–669. doi: 10.1016/s0092-8674(00)80877-6. [DOI] [PubMed] [Google Scholar]
- Schmid RS, Graff RD, Schaller MD, Chen S, Schachner M, Hemperly JJ, Maness PF. NCAM stimulates the Ras-MAPK pathway and CREB phosphorylation in neuronal cells. J Neurobiol. 1999;38:542–558. [PubMed] [Google Scholar]
- Sia GM, Béïque JC, Rumbaugh G, Cho R, Worley PF, Huganir RL. Interaction of the N-terminal domain of the AMPA receptor GluR4 subunit with the neuronal pentraxin NP1 mediates GluR4 synaptic recruitment. Neuron. 2007;55:87–102. doi: 10.1016/j.neuron.2007.06.020. [DOI] [PubMed] [Google Scholar]
- Sims RE, Hartell NA. Differences in transmission properties and susceptibility to long-term depression reveal functional specialization of ascending axon and parallel fiber synapses to Purkinje cells. J Neurosci. 2005;25:3246–3257. doi: 10.1523/JNEUROSCI.0073-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slot JW, Geuze HJ. A new method of preparing gold probes for multiple-labeling cytochemistry. Eur J Cell Biol. 1985;38:87–93. [PubMed] [Google Scholar]
- Takeuchi T, Miyazaki T, Watanabe M, Mori H, Sakimura K, Mishina M. Control of synaptic connection by glutamate receptor δ2 in the adult cerebellum. J Neurosci. 2005;25:2146–2156. doi: 10.1523/JNEUROSCI.4740-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uemura T, Mishina M. The amino-terminal domain of glutamate receptor delta2 triggers presynaptic differentiation. Biochem Biophys Res Commun. 2008;377:1315–1319. doi: 10.1016/j.bbrc.2008.10.170. [DOI] [PubMed] [Google Scholar]
- Uemura T, Kakizawa S, Yamasaki M, Sakimura K, Watanabe M, Iino M, Mishina M. Regulation of long-term depression and climbing fiber territory by glutamate receptor δ2 at parallel fiber synapses through its C-terminal domain in cerebellar Purkinje cells. J Neurosci. 2007;27:12096–12108. doi: 10.1523/JNEUROSCI.2680-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada C, Ohtani H. Molecular cloning of rat cerebellin-like protein cDNA which encodes a novel membrane-associated glycoprotein. Brain Res Mol Brain Res. 1991;9:71–77. doi: 10.1016/0169-328x(91)90131-g. [DOI] [PubMed] [Google Scholar]
- Wang Y, Matsuda S, Drews V, Torashima T, Meisler MH, Yuzaki M. A hot spot for hotfoot mutations in the gene encoding the δ2 glutamate receptor. Eur J Neurosci. 2003;17:1581–1590. doi: 10.1046/j.1460-9568.2003.02595.x. [DOI] [PubMed] [Google Scholar]
- Wisden W, Seeburg PH. A complex mosaic of high-affinity kainate receptors in rat brain. J Neurosci. 1993;13:3582–3598. doi: 10.1523/JNEUROSCI.13-08-03582.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wollmuth LP, Kuner T, Jatzke C, Seeburg PH, Heintz N, Zuo J. The Lurcher mutation identifies δ2 as an AMPA/kainate receptor-like channel that is potentiated by Ca2+ . J Neurosci. 2000;20:5973–5980. doi: 10.1523/JNEUROSCI.20-16-05973.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzaki M. The δ2 glutamate receptor: 10 years later. Neurosci Res. 2003;46:11–22. doi: 10.1016/s0168-0102(03)00036-1. [DOI] [PubMed] [Google Scholar]
- Yuzaki M. The δ2 glutamate receptor: a key molecule controlling synaptic plasticity and structure in Purkinje cells. Cerebellum. 2004;3:89–93. doi: 10.1080/14734220410028921. [DOI] [PubMed] [Google Scholar]
- Yuzaki M. Transgenic rescue for characterizing orphan receptors: a review of delta2 glutamate receptor. Transgenic Res. 2005;14:117–121. doi: 10.1007/s11248-005-2685-6. [DOI] [PubMed] [Google Scholar]
- Yuzaki M. Cbln and C1q family proteins: new transneuronal cytokines. Cell Mol Life Sci. 2008;65:1698–1705. doi: 10.1007/s00018-008-7550-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao HM, Wenthold RJ, Wang YX, Petralia RS. δ-glutamate receptors are differentially distributed at parallel and climbing fiber synapses on Purkinje cells. J Neurochem. 1997;68:1041–1052. doi: 10.1046/j.1471-4159.1997.68031041.x. [DOI] [PubMed] [Google Scholar]