

Abstract
We have studied store-operated Ca2+ entry (SOCE) in Bergmann glia and granule cell layer astrocytes in acute brain slices of the rat cerebellum, using the Ca2+-sensitive fluorescent dye Fluo-4 and confocal laser scanning microscopy. Astrocytes were identified by their morphology, location, and their Ca2+ response in K+-free solution. Depletion of Ca2+ stores by cyclopiazonic acid (CPA) (20 μm) induced SOCE in both types of astrocyte. A similar Ca2+ influx was elicited by the calmodulin antagonist calmidazolium (CMZ) (1 μm). The SOCE channel blocker 2-aminoethoxy-diphenylborate (2-APB) (100 μm) and the Ca2+ release-activated channel blocker 3,5-bistrifluoromethyl pyrazole derivative (BTP2) (20 μm) suppressed the CPA- and the CMZ-induced Ca2+ influx. Pretreatment of acute slices with the specific Ca2+-independent phospholipase A2 (iPLA2) inhibitor bromoenol lactone (BEL) (25 μm) blocked the CPA- and the CMZ-induced Ca2+ influx. The lysophospholipid products of iPLA2, lysophosphatidylcholine (250 nm) and lysophosphatidylinositol (250 nm), but not lysophosphatidic acid (250 nm), induced a BTP2- and 2-APB-sensitive, but BEL-insensitive, Ca2+ influx. CPA or CMZ enhanced the BEL-sensitive enzymatic activity of iPLA2 in cerebellar astrocyte culture. Inhibition of iPLA2 expression by specific antisense oligodeoxynucleotide of iPLA2 reduced the SOCE and the Ca2+ store refilling in cultured astrocytes. Spontaneous Ca2+ oscillations in astrocytes in situ were reduced after inhibiting SOCE channels or iPLA2 activity. The results suggest that the depletion of Ca2+ stores activates iPLA2 to open Ca2+ channels in the plasma membrane by the formation of lysophospholipids in astrocytes, presumably to refill the stores and allow normal Ca2+ signaling.
Keywords: Fluo-4, cyclopiazonic acid, calmidazolium, lysophospholipids, calcium stores, Bergmann glia
Introduction
Depletion of intracellular Ca2+ stores can induce influx of Ca2+ from the extracellular space through channels in the plasma membrane, a mechanism known as store-operated Ca2+ entry (SOCE) or capacitative Ca2+ entry (Parekh and Putney, 2005). SOCE appears to be the predominant pathway of regulated Ca2+ influx in non-excitable cells but may also play a major role in some types of excitable cells, including neurons (Fagan et al., 2000; Emptage et al., 2001; Akbari et al., 2004; Verkhratsky, 2005). SOCE plays a major role not only in replenishing Ca2+ stores but also in various physiological processes ranging from short-term responses, such as exocytosis, contraction, and Ca2+ oscillations, to long-term control of gene transcription, cell cycle, and apoptosis (Berridge et al., 2000; Parekh and Putney, 2005).
A model proposed for SOCE channel activation implies the existence of a diffusible messenger termed Ca2+ influx factor (CIF), which can be released from the stores into the cytosol during depletion, thereby transmitting the signal to the plasmalemmal SOCE channels to trigger Ca2+ entry (Randriamampita and Tsien, 1993). Smani et al. (2004) have recently demonstrated that CIF activates SOCE channels in the plasma membrane through its interaction with Ca2+-independent phospholipase A2 (iPLA2). In this paradigm, CIF can displace inhibitory calmodulin (CaM) from iPLA2, resulting in activation of iPLA2 and formation of lysophospholipids, which in turn activates SOCE. During refilling of Ca2+ stores and termination of CIF production, calmodulin inhibits iPLA2 activity by binding to the enzyme, thereby suppressing SOCE. The EF hand protein stromal-interacting molecule (STIM1) was recently identified as a possible intracellular messenger, which migrates from the endoplasmic reticulum (ER) to the plasma membrane during depletion of Ca2+ stores, suggesting that STIM1 functions as the missing link between Ca2+ store depletion and SOCE activation (Liou et al., 2005; Roos et al., 2005; Zhang et al., 2005).
In glial cells, SOCE has been studied in some detail only in cell cultures. SOCE can be induced, e.g., in cultured rat cerebellar astrocytes (Jung et al., 2000; Lo et al., 2002) and C6 glioma cells (Wu et al., 1997), in which it is enhanced by either cAMP (Wu et al., 1999) or nitric oxide (Li et al., 2003), and reduced by arachidonic acid (AA) (Sergeeva et al., 2003; Yang et al., 2005). In glial cells in acute brain slices, however, mechanisms of regulating SOCE have not been investigated. The present study aims to elucidate the signaling pathway that activates SOCE in rat cerebellar astrocytes in situ. We investigated the SOCE in the two major types of astrocyte in the cerebellar cortex, the Bergmann glia and astrocytes in the granule cell layer (GCL), by imaging cytosolic Ca2+ with confocal microscopy. We found that SOCE exists in both types of astrocyte and is mediated by specific lysophospholipid products of iPLA2. Furthermore, we show that spontaneous Ca2+ oscillations of astrocytes in the cerebellum are reduced by inhibiting SOCE and by the iPLA2 inhibitor bromoenol lactone (BEL).
Materials and Methods
Slice preparation.
Cerebellar brain slices were prepared following the method of Edwards et al. (1989). In brief, a juvenile rat [postnatal day 8 (P8) to P15] was decapitated, and the cerebellum was isolated rapidly in an ice-cold, bicarbonate-buffered (5% CO2/95% O2, pH 7.4), Ca2+-reduced solution (0.5 mm; MgCl2 increased to 2.5 mm). Sagittal slices of the vermis (250 μm thick) were obtained using a vibratome (VT 1000; Leica, Darmstadt, Germany) and stored for 1 h in gassed Ca2+-reduced saline at 30°C before dye loading and later at room temperature (21–24°C). Cells of interest were Bergmann glial cells of the Purkinje cell layer and granule cell layer astrocytes of the cerebellar cortex. The cell types were identified as explained in Results.
Cell culture.
Astrocyte primary cultures were prepared from the cerebellar hemispheres of newborn rats (P1–P3) by a method similar to that described by Fischer (1984). Briefly, cerebella were rapidly excised and maintained in an ice-cold Ca2+-free isotonic salt solution containing the following (in mm): 120 NaCl, 5.4 KCl, 0.8 MgCl2, 25 Tris-HCl, and 15 glucose, pH 7.2. After removing the meninges, cerebella were digested with 0.1% trypsin for 5 min at room temperature, before trypsin was replaced with 10% fetal calf serum (FCS) in Eagle's Basal Medium (BME) (Sigma, St. Louis, MO). The tissue was triturated using successively decreasing bore size fire-polished Pasteur pipettes in the same medium. Cells were centrifuged at 600 rpm for 5 min and resuspended in DNase-containing solution. Centrifugation was repeated and the pellet was resuspended in DMEM (Sigma) (supplemented with 2.0 g of NaHCO3, 2.0 g of glucose, 1000 U/L penicillin, 1 g/L streptomycin, and 5% FCS). Cells were plated in poly-d-lysine (PDL)-coated (0.001%) bottles with DMEM and incubated at 37°C in a humidified atmosphere with 10% CO2. After the cells had reached confluence (∼10 d), the cells were dissociated by 0.1% of trypsin, and this step was stopped by fetal calf serum (10% in BME). Then the cells were incubated with DNase, centrifuged, resuspended in DMEM, plated on glass coverslips coated with PDL (0.001%), and incubated as described previously. The experiments were performed on secondary astrocyte cultures between 2 and 10 d after plating at room temperature (21–24°C). Cultures showed >95% staining for the astrocyte-specific marker glial fibrillary acidic protein (GFAP).
Transfection with oligodeoxynucleotides.
A 20-base-long antisense (AS) oligodeoxynucleotide (ODN) corresponding to the conserved nucleotides 59–78 in the murine group IV iPLA2 sequence was used (AS-ODN, 5′-fluorescein-ACTCCTTCACCCGGAATGGGT-3′; MWG-Biotech, Ebersberg, Germany). The corresponding sense (S) oligodeoxynucleotide complement (S-ODN, 5′-fluorescein-CCCATTCCGGGTGAAGGAG-3′; MWG-Biotech) was used as a control. Both S-ODN and AS-ODN contained nuclease-resistant phosphorothioate linkages to limit their degradation and were labeled with fluorescein to detect transfected cells (see below, Calcium imaging). Secondary astrocytes at 60% confluence were transfected using Lipofectamine 2000 (LF) (Invitrogen, Karlsruhe, Germany) in the dark following the protocol of the manufacturer. The experiments were performed 36–48 h after transfection. Transfection efficiency was determined with fluorescein-labeled ODNs and amounted to 71% in the astrocyte cultures used.
Dye loading.
Cerebellar slices were loaded with a solution containing 3 μm Fluo-4 AM (Invitrogen, Carlsbad, CA), made from a 6 mm stock solution in dimethylsulfoxide (DMSO) with 20% pluronic acid, in a Petri dish inside a small oxygenated closed box for 60 min at 20–24°C. After dye loading, the slices were kept on a mesh of nylon in artificial CSF (ACSF) (see below). All salines for acute brain slices were gassed with 5% CO2/95% O2 to maintain pH and oxygen levels at physiological levels. Cerebellar astrocyte cultures were incubated in 2 μm Fluo-4 AM-containing solutions for 45 min in the dark at room temperature. ODN-transfected and nontransfected cultures were loaded in a 1 μm fura-2 AM-containing saline, made from a 4 mm stock solution in DMSO, for 30 min at room temperature.
Calcium imaging.
Experiments with acute brain slices and cultures were performed with a confocal laser scanning microscope (LSM 510; Zeiss, Oberkochen, Germany). The Ca2+-sensitive dye Fluo-4 was excited by the 488 nm line of an argon laser, and images were taken with a frequency of 0.3–1.0 Hz. Excitation and emission signals were separated by a dichroic mirror at 500 nm. The emission signal was truncated by a 505 nm optical long-pass filter. The sequence of fluorescence images was sampled in one focal plane for Ca2+ measurements at room temperature (21–24°C). Regions of interest (ROIs) were defined in the first image, and the relative fluorescence changes ΔF (percentage) were measured throughout the series, related to the basic, normalized fluorescence intensity at the beginning of the experiment (defined as 100%). Hence, Ca2+ changes were referred to as this percentage change of the Fluo-4 fluorescence. All settings of the laser, optical filters, and the microscope as well as the data acquisition were controlled by personal computer software (Zeiss). Additional details have been described previously (Beck et al., 2004).
Experiments on ODNs-transfected and nontransfected cell cultures were performed with an Olympus Optical (Tokyo, Japan) upright microscope (BX50W1) using a Ca2+ imaging system (T.I.L.L. Photonics, Munich-Martinsried, Germany). Monochromator setting and data acquisition were controlled by software for a personal computer system. The transfected cells were identified by imaging the fluorescein (excitation at 488 nm, emission at 510 nm). Fura-2-loaded cells were excited by monochromatic wavelengths of 340 and 380 nm. The fluorescence emissions of several ROIs (each covering one single cell body) were simultaneously recorded with the CCD camera IMAGO (T.I.L.L. Photonics), using a 440 nm long-pass filter. The signals were sampled at 0.2–0.5 Hz, computed into relative ratio units F340/F380. The decay of intracellular Ca2+ transients was fitted by an equation for monoexponential decay using Origin software (Microcal Software, Northampton, MA), resulting in a time constant τ.
Antibody staining.
Cerebellar slices were fixed in 4% paraformaldehyde in PBS, pH 7.4, overnight at 4°C. Slices were rinsed with PBS and permeabilized in 0.5% Triton X-100 in PBS/10% normal goat serum (NGS), and the primary antibody (rabbit anti-glial fibrillary acidic protein, 1:1000; Z0334; DakoCytomation, Carpinteria, CA) in PBS containing 0.05% Triton X-100 was incubated overnight at 4°C. The slices were then rinsed with PBS and incubated in secondary antibody (Alexa 488 goat anti-rabbit IgG, 1:200; Invitrogen, Carlsbad, CA) and propidium iodide (5 μm) for 3 h at room temperature. After rinsing, slices were mounted on a coverslip and imaged using confocal microscopy. GFAP staining was visualized at 488 nm excitation and 510 nm emission, whereas the nuclei (labeled by propidium iodide) were visualized at 544 nm excitation and 620 nm emission.
ODNs-transfected cultures were washed with PBS and fixed in 4% paraformaldehyde in PBS for 15 min at room temperature. After washing, cells were permeabilized in 0.1% Triton X-100 in PBS/10% NGS for 1 h. Incubation with primary antibody [rabbit iPLA2 (type IV) polyclonal antiserum, 1:500 (Cayman Chemical, Ann Arbor, MI) was performed in PBS containing 0.05% Triton X-100 overnight at 4°C. Cells were then incubated with secondary antibody (Alexa 546 goat anti-rabbit IgG, 1:200; Invitrogen, Carlsbad, CA) for 1 h, washed, and mounted. Control staining was done in the same way but without the primary antibody. Cell cultures of all treatments, i.e., S-ODN, AS-ODN, and LF alone, were processed simultaneously with the same solutions and incubation times. The expression of iPLA2 in random fields was assessed by the iPLA2 staining using confocal microscopy. The transfected cells were identified based on the labeling of fluorescein, and iPLA2 staining was visualized at 543 nm excitation and with a 560 nm long-pass emission filter, whereas fluorescein was detected as described (see above, Calcium imaging). S-ODN-transfected cells, AS-ODN-transfected cells, and LF-treated cells were imaged with the same laser and microscope settings. Anti-iPLA2-dependent fluorescence was quantified to compare the amount of iPLA2 in the cells. Therefore, a stack of images was recorded, and the brightest image was chosen, in which the mean fluorescence intensities of the astrocyte somata were measured.
Solutions.
The standard saline for acute brain slices (ACSF) contained the following (in mm): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 25 d-glucose, 26 NaHCO3, 1.25 NaH2PO4, and 0.5 l-lactate (gassed with 5% CO2/95% O2 to adjust the pH to 7.4). In Ca2+-reduced (0.5 mm) saline, 1.5 mm Ca2+ was replaced by 1.5 mm MgCl2 and l-lactate was omitted. In salines with varying K+ concentrations, KCl was exchanged by or for NaCl. Ca2+-free saline was prepared by replacing CaCl2 by equimolar amounts of MgCl2 and by adding 1 mm EGTA. The standard saline for cultured astrocytes contained the following (in mm): 145 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 d-glucose, and 10 HEPES, pH set to 7.4. Ca2+-free saline was prepared by replacing CaCl2 by equimolar amounts of MgCl2 and by adding 0.5 mm EGTA. The experimental chamber containing either an acute slice or cell cultures was continuously perfused with the saline at room temperature. Cyclopiazonic acid (CPA) (Alexis Biochemicals, San Diego, CA), calmidazolium chloride (CMZ) (Sigma), BEL (Sigma), 1,1,1-trifluoro-2-heptadecanone (PACOCF3) (Tocris Cookson, Ballwin, MO), propranolol hydrochloride (Sigma), 2-aminoethoxydiphenylborate (2-APB) (Tocris Cookson), AA (Tocris Cookson), and N5-{4-[3,5-bis(trifluoromethyl)-1H-pyrazole-1-yl]phenyl}-4-methyl-1,2,3-thiadiazole-5-carboxamide] (BTP2) (Calbiochem, La Jolla, CA) were dissolved in DMSO as stock solutions. Lysophosphatidylcholine (LPC) (Sigma), lysophosphatidylinositol (LPI) (Sigma), ADP (Sigma), ATP (Sigma), and 9-(tetrahydro-2-furanyl)-9H-purin-6-amine (SQ 22536) (Alexis Biochemicals) were dissolved in deionized water and lysophosphatidic acid (LPA) (Sigma) was dissolved in an equal volume of ethanol/water to make a stock solution and used at concentrations as mentioned in Results. All drugs were added to the experimental saline immediately before use. The final concentration of DMSO never exceeded 0.1%.
iPLA2 activity assay.
iPLA2 activity was measured in cerebellar astrocyte culture using cPLA2 assay kit (Cayman Chemical) as described previously (Smani et al., 2003, 2004). In brief, cells were grown in six-well dishes until 80% confluency. The required wells of the cells were exposed to different treatments, as described in Results, and washed with PBS (in mm: 171 NaCl, 10 Na2HPO4, 3.4 KCl, and 1.8 KH2PO4, pH 7.4) before and after each treatment. All steps were performed on ice. The cells were collected with a scraper and homogenized using mechanical homogenizer in 50 μl (per well) of ice-cold buffer (in mm: 300 sucrose and 10 Tris-HCl, pH 7.0) with protease inhibitor cocktail (Roche, Indianapolis, IN). Cell homogenates were centrifuged at 20,000 × g for 20 min at 4°C, and the supernatant was collected. Protein content was measured using Bio-Rad (Hercules, CA) reagent, and the iPLA2 assay was done on the same day following the instructions of the manufacturer. Supernatants from control cultures and treated cultures, containing the same amount of protein (2–3 mg/ml) in a final volume of 10 μl, were added to microtiter plate wells consisting of 5 μl of a modified assay buffer (300 mm NaCl, 0.5% Triton X-100, 60% glycerol, 4 mm EGTA, 10 mm HEPES at pH 7.4, and 2 mg/ml BSA). The reaction was initiated by the addition of 200 μl of arachidonoyl thiophosphatidylcholine and incubated at 20°C for 60 min. The reaction was terminated by the addition of 10 μl of 5,5′-dithio-bis-2-nitrobenzoic acid for 5 min, and the absorbance was measured at 405 nm in a 96-well microplate reader (Multiscan EX; Thermo Labsystems, Dreieich, Germany). To determine the BEL-sensitive iPLA2 activity, the optical density obtained in the presence of BEL was subtracted from all readings, and the resulting optical density was expressed in percentage of respective positive control (see Results).
Statistics.
Measurements are expressed as mean values ± SEM; n indicates the number of cells (first value given) and the number of brain slices or culture plates (second value given), from which cells were analyzed (each experiment in acute brain slices was repeated in different animal preparations). All statistical tests were done with either SigmaPlot (Systat Software, Point Richmond, CA) or Origin 7.0 (Microcal Software). Statistical differences were tested using Student's t test in Origin 7.0. Means were defined as statistically different at an error probability of *p < 0.05, **p < 0.01, or ***p < 0.005.
Results
Identification of cell types in acute rat brain slices
We used acute slices of the rat cerebellum to monitor cytosolic Ca2+ in somata of Bergmann glia and of astrocytes in the GCL in situ. Bergmann glial cells are a special kind of cerebellar astrocytes with their cell bodies grouped around a Purkinje neuron and extend radial or Bergmann fibers toward the pial layer (Fig. 1A,B). In mammals, most of the radial glial cells disappear during development; only a vestige persists in some specialized areas, such as Bergmann glia in cerebellar cortex, in which they adapt to perform the local functional requirements (Rakic, 2003). Although Bergmann glial cells are classified as belonging to the astrocyte lineage (Campbell and Götz, 2002), we treated these cells as a distinct glial cell category because of their functional importance in the cerebellum. Bergmann glial cells were readily identified by their location, typical radial morphology, and their ability to generate Ca2+ transients during application of ATP or ADP (Kirischuk et al., 1995; Hammer, 2006). After dye loading, the cell body and the radial dendrites of Bergmann glia were clearly visible during their response to ATP (20 μm for 30 s) (Fig. 1B,D,E), whereas Purkinje neurons were hardly stained by the dye. Ca2+ responses to ATP indicated the functional status of the intracellular Ca2+ stores. Bergmann glial cells, however, often lacked the Ca2+ rise in response to K+-free saline (0 mm K+) typical for astrocytes, which was used to identify astrocytes in the GCL (Fig. 1C,F,G) (Dallwig and Deitmer, 2002; Beck et al., 2004).
Figure 1.
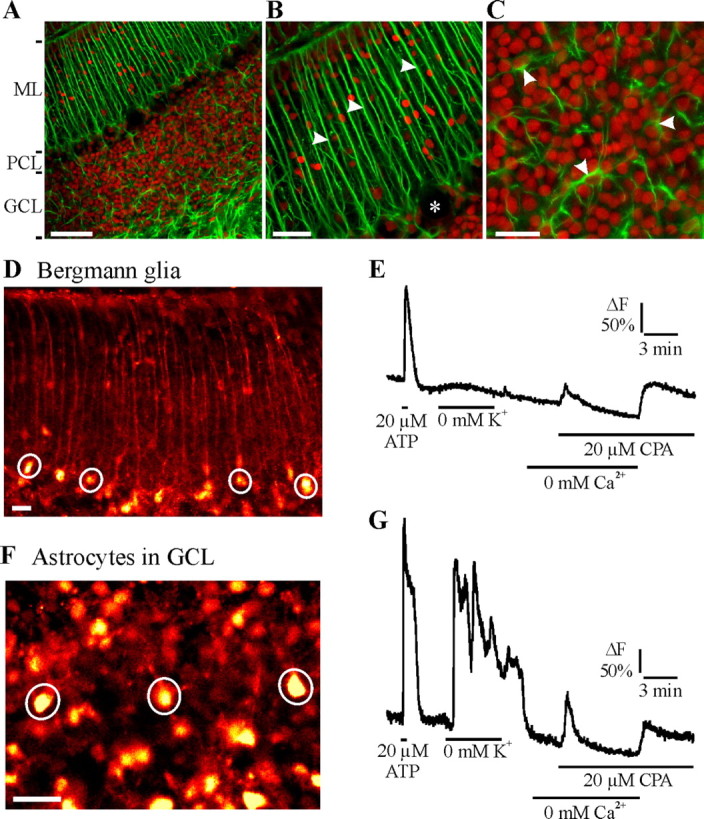
Identification of rat cerebellar astrocytes in situ. A, Labeling of astrocytes with anti-GFAP antibody (green) and cell nuclei with propidium iodide (red) in different cell layers of a cerebellar slice. ML, Molecular layer; PCL, Purkinje cell layer. B, At higher magnification, Bergmann glial cell processes in the molecular layer (arrowheads). Purkinje cell somata appear as black holes in the Purkinje cell layer (asterisk). C, Astrocytes (arrowheads) in the granule cell layer. D, F, Fluorescence images of Fluo-4-loaded Bergmann glia (D) and astrocytes in the GCL (F) during ATP application (20 μm for 30 s), when they respond with Ca2+ signals. Somata of Bergmann glial cells and GCL astrocytes are indicated by circles (regions of interest, in which cytosolic Ca2+ was recorded). E, G, The fluorescence recording, given as the relative change in fluorescence related to the value at the beginning of the experiments (ΔF), show typical Ca2+ responses of a Bergmann glial cell (D) and a GCL astrocyte (G) to ATP and 0 mm K+ and to CPA (20 μm) in the absence of extracellular Ca2+, followed by readdition of Ca2+. Scale bars: A, 100 μm; B, 50 μm; C, 30 μm; D, F, 20 μm.
Application of CPA (20 μm), an inhibitor of the sarcoendoplasmic Ca2+-ATPase, in the absence of external Ca2+ evoked a transient cytosolic Ca2+ rise (Fig. 1E,G), indicative of Ca2+ leak from the intracellular Ca2+ stores. Readdition of Ca2+ in the presence of CPA resulted in another cytosolic Ca2+ rise (Fig. 1E,G), indicative of SOCE after the depletion of the intracellular Ca2+ stores. These Ca2+ responses were recorded in both Bergmann glial cells and astrocytes in the GCL.
Store-operated Ca2+ entry after Ca2+ store depletion by cyclopiazonic acid
First, we studied SOCE as induced by CPA in Bergmann glia and in astrocytes of the GCL with respect to its sensitivity to different drugs (Fig. 2). In Bergmann glia, CPA-induced depletion of Ca2+ stores resulted in Ca2+ transients during readdition of Ca2+ (SOCE), with a mean amplitude of 48 ± 8% ΔF (n = 92 cells/8 slices). This Ca2+ transient was significantly reduced to 21 ± 2% ΔF (p < 0.05; n = 42/3) by 2-APB (100 μm), an inhibitor of SOCE channels (Ma et al., 2000; Bakowski et al., 2001; Prakriya and Lewis, 2001; Voets et al., 2001). 2-APB was applied 5 min before the readdition of Ca2+ in the presence of CPA (Fig. 2A,B). Comparable results were obtained with BTP2 (20 μm) (Ishikawa et al., 2003; Zitt et al., 2004), a structurally distinct blocker of Ca2+ channels that are activated by store depletion. BTP2, preincubated for 15–20 min, significantly reduced SOCE to 12 ± 1.3% ΔF (p < 0.01; n = 46/3) (Fig. 2A,B). To test whether iPLA2 plays a role in SOCE in Bergmann glia, the specific, irreversible inhibitor of iPLA2, BEL (25 μm) (Ackermann et al., 1995; Winstead et al., 2000), was used. BEL requires the basal activity of iPLA2 and thus inhibits the enzyme more effectively at physiological temperature (37–40°) than at room temperature (20–22°), when the enzyme activity is low. Moreover, it permeates only slowly into intact cells and hence requires relatively long incubation times (Balsinde et al., 1995). Because the efficacy of BEL to block iPLA2 is temperature and time dependent, acute brain slices were preincubated in BEL for at least 30 min at 37°C. BEL was removed by washing for 15–20 min before starting the protocol of measuring cytosolic Ca2+ changes. In BEL-treated Bergmann glial cells, the mean amplitude of the SOCE was reduced to 4.5 ± 0.2% ΔF (n = 62/6; p < 0.01) (Fig. 2A,B). In addition to iPLA2 inhibition, BEL has been shown to inhibit another cellular phospholipase, the Mg2+-dependent phosphatidate phosphohydrolase (PAP-1) (Balsinde and Dennis, 1996; Fuentes et al., 2003), which is known to be involved in the synthesis of diacylglycerol. To check the possible role of PAP-1 in BEL-induced inhibition of SOCE, we used propranolol (50–100 μm) to inhibit PAP-1 (Fuentes et al., 2003). The acute slices were pretreated with propranolol for 30 min at room temperature. Propranolol did not inhibit the CPA-induced Ca2+ influx in either Bergmann glial cells or astrocytes of the GCL (data not shown), which indicates that PAP-1 is not involved in SOCE regulation in cerebellar astrocytes.
Figure 2.

SOCE in Bergmann glia and GCL astrocytes. A, C, Original recordings of the cytosolic Ca2+ concentration in somata of a single Bergmann glial cell (A) and in a single GCL astrocyte (C). Acute brain slices were exposed to 20 μm CPA (indicated by arrowhead) for 7 min in Ca2+-free condition before 2 mm Ca2+ was readded, and the resulting cytosolic Ca2+ rise was indicative of SOCE. Traces are for the cells treated with CPA alone, with CPA and drugs added, or preincubated: 100 μm 2-APB added (arrow), 20 μm BTP2 incubated for 15 min, 25 μm BEL preincubated for 30–40 min at 37°C, and 10 μm PACOCF3 added 15 min before the readdition of Ca2+. Cells only kept in Ca2+-free solution for 8–10 min served as control (without CPA, bottom trace). B, D, Bar plots showing the average, relative amplitude of the fluorescence signal (ΔF), indicative of cytosolic Ca2+ rise after readdition of Ca2+. The Ca2+ signals (SOCE) were reduced by the SOCE blockers 2-APB and BTP2 and by the iPLA2 inhibitors BEL and PACOF3 in Bergmann glia (B) and GCL astrocytes (D). The insets in B and D show Ca2+ responses to 20 μm ATP in a Bergmann glial cell (B) and a GLC astrocyte (D) after preincubation of slices in BEL to demonstrate functional status of the Ca2+ release capacity of the cells. *p < 0.05, **p < 0.01, ***p < 0.005 versus CPA-induced SOCE; n indicates the number of cells tested/number of slices.
BEL has also been reported to induce cell death by inhibiting endoplasmic iPLA2 (Cummings et al., 2002) or PAP-1 (Fuentes et al., 2003). After BEL treatment, Bergmann glial cells still generated Ca2+ transients in response to ATP (20 μm, 30 s), which evokes IP3-mediated Ca2+ release from ER stores (Shao and McCarthy, 1995) (Fig. 2B, inset). This indicates the functional status of the Ca2+ stores, ruling out the possibility that BEL, at the concentration used here, caused cell death in Bergmann glia. We also used a second iPLA2 blocker, PACOCF3 (10 μm), which belongs to a different chemical family than BEL and is known to inhibit iPLA2 with an IC50 value of 3.8 μm (Ackermann et al., 1995), to further confirm the involvement of iPLA2 in regulating SOCE. The Ca2+ transient during Ca2+ readdition in the presence of CPA was significantly reduced to 5.0 ± 0.8% ΔF (n = 73/6; p < 0.01) (Fig. 2A,B) by PACOCF3 (Fig. 2A,B). In the presence of BEL or PACOCF3, the Ca2+ transient during Ca2+ readdition was even smaller than in control cells (the mean amplitude being 10.2 ± 1% ΔF; p < 0.05; n = 52/5), which were not exposed to CPA but left for the same time period in Ca2+-free solution. This suggests that some iPLA2 activity is present even when Ca2+ stores are not depleted by CPA (Fig. 2B).
Like in Bergmann glial cells, readdition of Ca2+ induced an influx of Ca2+ in GCL astrocytes in the presence of CPA (Fig. 2C,D). During readdition of Ca2+ to the extracellular solution, cytosolic Ca2+ increased by 70 ± 8% ΔF (n = 62/8). This SOCE was significantly reduced by 2-APB (100 μm) to 32 ± 2.5% ΔF (p < 0.01; n = 31/4) and by BTP2 (20 μm) to 13 ± 1% ΔF (p < 0.005; n = 22/3). Preincubation with BEL or PACOCF3 strongly inhibited SOCE to 5.0 ± 0.5% ΔF (p < 0.005; n = 57/6) and to 8 ± 1% ΔF (p < 0.005; n = 53/6), respectively, indicating that iPLA2 is essential for the activation of SOCE (Fig. 2C,D). The PAP-1 inhibitor propranolol (50–100 μm) did not affect SOCE induced in the presence of CPA (data not shown). GCL astrocytes responded to ATP (20 μm, 30 s) with a Ca2+ transient after BEL treatment, which indicated the functional Ca2+ stores in the cells (Fig. 2D, inset). In control cells, readdition of external Ca2+ induced a Ca2+ transient of 22 ± 1.3% ΔF (p < 0.005; n = 59/5), which was still larger than in cells treated with BEL or PACOCF3 (p < 0.01) (Fig. 2C,D). These results suggest that iPLA2 activation by depletion of Ca2+ stores in both Bergmann glia and GCL astrocytes is required for the SOCE in these cells. Thus, similar results were obtained in experiments using either BEL or PACOCF3 to inhibit iPLA2 activity, and, for the following experiments, BEL was chosen as potent iPLA2 inhibitor.
Ca2+ influx induced by calmidazolium
After obtaining evidence for a role of iPLA2 in CPA-induced Ca2+ influx, we wanted to know whether the activation of iPLA2 itself, without previous Ca2+ store depletion, induces influx of Ca2+ in Bergmann glial cells and GCL astrocytes. The activity of iPLA2 is regulated by CaM, which binds to the C terminal of the enzyme and thereby inhibits its activity (Wolf and Gross, 1996a,b; Wolf et al., 1997). We used the CaM antagonist CMZ at a concentration of 1 μm to dissociate CaM from iPLA2. At this concentration, CMZ does not cause release of Ca2+ from stores itself (Smani et al., 2004), which was confirmed in the present study by comparing the cytosolic Ca2+ response induced by ADP (20 μm) in untreated cells and cells treated with 1 μm CMZ. No significant difference in the amplitude of the Ca2+ transients evoked by ADP was found in untreated (n = 24/3) and CMZ-treated (n = 29/3) cells (data not shown), indicating that CMZ does not affect the filling state of the Ca2+ stores.
CMZ was applied in the absence of external Ca2+, and a Ca2+ transient was recorded in Bergmann glial cells, when Ca2+ was readded to the extracellular solution (Fig. 3A). This Ca2+ transient was similar to that induced by the depletion of Ca2+ stores by CPA and had an amplitude of 39 ± 4% ΔF (n = 61/7) (Fig. 3B). To determine the mechanism by which CMZ activates Ca2+ influx in these cells, we examined the effect of SOCE channel inhibitors 2-APB (100 μm) and BTP2 (20 μm) on the Ca2+ rise in the presence of CMZ. In Bergmann glia, this Ca2+ rise was reduced to 13 ± 1.3% ΔF in the presence of 2-APB (p < 0.005; n = 40/3), and pretreatment of the slices with BTP2 for 15 min decreased this Ca2+ rise to 11 ± 1.2% ΔF (p < 0.005; n = 25/3). The iPLA2 inhibitor BEL reduced the Ca2+ rise in CMZ even to 1.0 ± 0.1% ΔF (p < 0.005; n = 39/3) compared with the untreated control slices, in which the Ca2+ rise was 10 ± 1% ΔF (p < 0.005; n = 52/5) (Fig. 3A,B).
Figure 3.

Calmodulin inhibitor calmidazolium evokes a Ca2+ influx in Bergmann glia and GCL astrocytes. A, C, Original recordings of the cytosolic Ca2+ concentration in somata of a single Bergmann glial cell (A) and a single GCL astrocyte (C). Acute slices were exposed to 1 μm CMZ (arrowhead) for 7 min in Ca2+-free saline before Ca2+ was readded (2 mm; filled bar). Traces are for cells treated with CPA alone, with CPA and 100 μm 2-APB added (arrow), with CPA and pretreated with 20 μm BTP2 for 15 min, and with CPA and pretreated with 25 μm BEL for 30–40 min at 37°C. Cells only kept in Ca2+-free solution for 8–10 min served as control (without CPA, bottom trace). B, D, Bar plots showing the average, relative amplitude of the fluorescence signal (ΔF), indicative of cytosolic Ca2+ rise after readdition of Ca2+. The Ca2+ signals (SOCE) were reduced by the SOCE blockers 2-APB and BTP2 and by the iPLA2 inhibitor BEL in Bergmann glia (B) and in GCL astrocytes (D). **p < 0.01, ***p < 0.005 versus CMZ-induced Ca2+ influx; n indicates the number of cells tested/number of slices.
CMZ has been shown to inhibit the activity of adenylyl cyclase (AC), albeit at greater concentrations than those used in the present study (Haunso et al., 2003). We tested whether the CMZ-induced augmentation of the Ca2+ transient during Ca2+ readdition can be mimicked by the inhibition of AC activity. We used the AC inhibitor SQ 22536 (200 μm) in the absence of CMZ for 15 min before reading Ca2+. Ca2+ readdition resulted in a Ca2+ influx with a mean amplitude of 13 ± 0.9% ΔF (n = 42/5; data not shown), which was not significantly different from control slices (same treatment except without SQ 22536). Hence, inhibition of AC does not increase the Ca2+ rise after readdition of external Ca2+, as CMZ does.
In GCL astrocytes, the Ca2+ rise in the presence of CMZ decreased from 78 ± 8% ΔF (n = 61/5) to 35 ± 6.5% ΔF in the presence of 2-APB (p < 0.01; n = 36/4) and to 25 ± 2% ΔF in the presence of BTP2 (p < 0.005; n = 25/3). Preincubation of BEL almost completely blocked the Ca2+ rise in CMZ to a mean amplitude of 2.5 ± 0.4% ΔF (p < 0.005; n = 27/3). This was significantly smaller than in control cells (p < 0.005), in which the readdition of Ca2+ induced a Ca2+ rise of 22 ± 1.3% ΔF (p < 0.005; n = 59/5) (Fig. 3C,D). When the adenylate cyclase inhibitor SQ 22536 was present, Ca2+ readdition evoked a Ca2+ influx with a mean amplitude of 26 ± 1% ΔF (n = 44/5; data not shown), which was not significantly different from control cells. Our experiments show that inhibiting calmodulin with CMZ induces a Ca2+ influx that is dependent on the activation of iPLA2 and SOCE channels. Furthermore, these results indicate that CMZ activates a Ca2+ influx even when intracellular Ca2+ stores are not depleted.
Ca2+ influx induced by lysophospholipids
Because the activation of iPLA2 releases lysophospholipids and a free fatty acid (AA), we tested whether these products can lead to Ca2+ influx into astrocytes. We used three different lysophospholipids: LPI (250 nm), LPC (250 nm), and LPA (250 nm). In Ca2+-free saline, all three lysophospholipids were added 3 min before readdition of Ca2+ to the extracellular solution. LPC and LPI induced a larger Ca2+ rise during Ca2+ readdition in Bergmann glia (Fig. 4) and GCL astrocytes (Fig. 5), whereas Ca2+ rises in the presence of LPA were not significantly changed compared with those in control experiments in the absence of lysophospholipids (Figs. 4D, 5D). Notably, both LPI and LPC still induced augmented Ca2+ rises after incubation of the slices with BEL (Figs. 4A–C, 5A–C). Because LPI and LPC are products of iPLA2 activity and appear downstream of iPLA2 activity, this indicates that BEL does not suppress Ca2+ influx per se. Moreover, this suggests that iPLA2, by producing lysophospholipids, promotes Ca2+ influx.
Figure 4.

Lysophospholipids evoke Ca2+ influx in Bergmann glia. A, Original recordings of cytosolic Ca2+ concentration in soma of a single Bergmann glial cell. LPI at 250 nm was added in Ca2+-free saline (arrowhead) 3 min before Ca2+ (2 mm) was readded. Traces are for the cells exposed to LPI alone, to LPI and pretreated with 25 μm BEL for 30–40 min at 37°C, to LPI and 100 μm 2-APB added before (arrow), and to LPI and pretreated with 20 μm BTP2 for 15 min. Cells only kept in Ca2+-free solution for 8–10 min served as control (without LPI, bottom trace). B, Bar plot showing the average, relative amplitude of the Ca2+ rise after readdition of Ca2+ in Bergmann glial cells. The LPI-induced Ca2+ influx was not affected by the iPLA2 inhibitor BEL but suppressed by SOCE blockers 2-APB and BTP2. C, Bar plot showing the average, relative amplitude of the Ca2+ rise in the presence of 250 nm LPC after readdition of Ca2+; LPC-induced Ca2+ influx was not affected by the iPLA2 inhibitor BEL but reduced by the SOCE blockers 2-APB and BTP2. D, Comparison of the Ca2+ rise after readdition of Ca2+ in the presence of 250 nm LPI, LPC, or LPA and without lysophopholipids as control. LPI and LPC, but not LPA, induced a significant Ca2+ influx. *p < 0.05, **p < 0.01, ***p < 0.005 versus LPI/LPC-induced Ca2+ influx; n indicates the number of cells tested/number of slices.
Figure 5.

Lysophospholipids evoke Ca2+ influx in GCL astrocytes. A, Original recordings of cytosolic Ca2+ concentration in soma of a single GCL astrocyte. LPI at 250 nm was added in Ca2+-free saline (arrowhead) 3 min before Ca2+ (2 mm) was readded. Traces are for the cells exposed to LPI alone, to LPI and pretreated with 25 μm BEL for 30–40 min at 37°C, to LPI and 100 μm 2-APB added before (arrow), and to LPI and pretreated with 20 μm BTP2 for 15 min. Cells only kept in Ca2+-free solution for 8–10 min served as control (without LPI, bottom trace). B, Bar plot showing the average, relative amplitude of the Ca2+ rise after readdition of Ca2+ in GCL astrocytes. The LPI-induced Ca2+ influx was not affected by the iPLA2 inhibitor BEL but was suppressed by SOCE blockers 2-APB and BTP2. C, Bar plot showing the average, relative amplitude of the Ca2+ rise in the presence of 250 nm LPC after readdition of Ca2+; LPC-induced Ca2+ influx was not affected by iPLA2 inhibitor BEL but was reduced by SOCE blockers 2-APB and BTP2. D, Comparison of the Ca2+ rise after readdition of Ca2+ in the presence of 250 nm LPI, LPC, or LPA and without lysophospholipids as control. LPI and LPC, but not LPA, induced a significant Ca2+ influx. *p < 0.05, **p < 0.01, ***p < 0.005 versus LPI/LPC-induced Ca2+ influx; n indicates the number of cells tested/number of slices.
In Bergmann glia, the Ca2+ rise during Ca2+ readdition had an amplitude of 42 ± 6% ΔF (n = 54/5) in the presence of added LPI. This Ca2+ rise was reduced to 16 ± 2.7% ΔF (n = 51/4) and 13 ± 2.3% ΔF (n = 28/3) in the presence of 100 μm 2-APB and after preincubation of 20 μm BTP2, respectively (p < 0.05) (Fig. 4A,B). The mean amplitude of the Ca2+ rise in the presence of added LPC was 36 ± 3.7% ΔF (n = 89/7), which was reduced to 11 ± 1.3% ΔF (n = 72/4) and 12 ± 1.3% ΔF (n = 32/3) by 2-APB and BTP2, respectively (p < 0.01) (Fig. 4C). The Ca2+ rise in the presence of LPA was 15 ± 1.6% ΔF (n = 67/5) and was not significantly different from control cells without any added lysophospholipids, under which condition the Ca2+ rose by 9 ± 1% ΔF (n = 32/5) (Fig. 4D).
In GCL astrocytes, the Ca2+ rise during Ca2+ readdition in the presence of LPI had an amplitude of 58 ± 6.2% ΔF (n = 61/5). This Ca2+ rise was inhibited to 27 ± 4.4% ΔF by 2-APB (p < 0.05; n = 48/4) and 25 ± 2.1% ΔF after preincubation with BTP2 (p < 0.05; n = 31/3) (Fig. 5A,B). The mean amplitude of this Ca2+ rise in the presence of LPC was 53 ± 7% ΔF (n = 89/7) and was reduced to 26 ± 4 and 22 ± 2.5% ΔF by 2-APB (n = 67/4) and BTP2 (n = 35/3), respectively (p < 0.05) (Fig. 5C). In the presence of LPA, the Ca2+ rise had an amplitude of 24 ± 1.7% ΔF (n = 67/6), which was not significantly different from that in control cells, which was 23 ± 2.5% ΔF (n = 43/5) (Fig. 5D). These results show that LPI and LPC, but not LPA, induce a Ca2+ influx mediated by 2-APB- and BTP2-sensitive ion channels.
We then asked whether the Ca2+ influx after Ca2+ readdition uses the same pathway after depleting the stores with CPA or with added LPI and LPC. Therefore, we tested whether LPI and LPC led to an additional Ca2+ influx after inducing SOCE in CPA. After depletion of Ca2+ stores by CPA, readdition of external Ca2+ evoked a Ca2+ increase (Fig. 6A–D), as described above (Fig. 2). In the continued presence of CPA and external Ca2+, LPI and LPC, both at 250 nm, were added (Fig. 6A,C, arrows). We observed no additional Ca2+ rise by either lysophospholipid in Bergmann glia (Fig. 6A,B) or in GCL astrocytes (Fig. 6C,D), suggesting that CPA- and lysophospholipid-induced Ca2+ influx are not additive.
Figure 6.

Ca2+ influx induced by lysophospholipids (LPL) is not additive to SOCE in Bergmann glia and GCL astrocytes. A, C, Original recordings of the cytosolic Ca2+ in the soma of a single Bergmann glial cell (A) and in a single GCL astrocyte (C) as induced after depleting the intracellular Ca2+ stores with CPA (20 μm) and addition of lysophospholipids (LPL, arrow) after readdition of Ca2+ (2 mm). Traces are for the cells exposed to CPA and then LPI, to CPA and then LPC, and to CPA alone (bottom trace, control). B, D, Bar plots showing the average, relative amplitude of the Ca2+ rise just after lysophospholipids addition in 2 mm Ca2+ in Bergmann glia (B) and GCL astrocytes (D). The lysophospholipids did not evoke a Ca2+ influx additional to the SOCE in CPA. The number of cells tested/number of slices is indicated by n in the bars.
Ca2+ influx induced by arachidonic acid
Because iPLA2-mediated hydrolysis of membrane phospholipids also generates AA, we also examined the effect of AA, which has been demonstrated to activate Ca2+ influx through arachidonate-regulated channels in a variety of different cell types, which is clearly distinct from SOCE (Shuttleworth and Thompson, 1999). Acute slices were exposed to AA (10 μm) in Ca2+-free solution 3 min before external Ca2+ was readded (Fig. 7A). In GCL astrocytes, AA induced a significant increase of the Ca2+ rise after readdition of Ca2+, which was often oscillatory and had a mean maximal amplitude of 48 ± 5% ΔF (p < 0.05; n = 53/6). In comparison, the Ca2+ influx in control cells without added AA had an amplitude of 23.5 ± 2% ΔF (n = 38/5). The larger Ca2+ rise in the presence of AA was not significantly changed in the presence of 2-APB (n = 47/6) (Fig. 7A,B), indicating that AA activated Ca2+ channels, which were insensitive to 2-APB in contrast to those activated in the presence of CPA, CMZ, and lysophospholipids. The Ca2+ rise in Bergmann glia in the presence of AA was not significantly larger compared with control experiments without AA (Fig. 7C), although their mean amplitude was 17 ± 3.3% ΔF (n = 36/4) compared with 8 ± 1.2% ΔF (n = 23/3) in control cells. 2-APB had no effect on the Ca2+ rise in the presence of AA.
Figure 7.

AA evokes a 2-APB-insensitive Ca2+ influx in GCL astrocytes and Bergmann glia. A, Original recording of the cytosolic Ca2+ concentration in GCL astrocytes in response to AA. Acute slices were exposed to 10 μm AA (arrowhead) for 3 min in Ca2+-free solution before 2 mm Ca2+ was readded. Traces are for the cells exposed to AA alone with different Ca2+ response patterns (top 2 traces) and with AA together with 100 μm 2-APB added before (arrow). Cells only kept in Ca2+-free solution for 8–10 min served as control (without AA, bottom trace). B, C, Bar plots of the average, relative amplitude of the Ca2+ rise after Ca2+ readdition in the presence of AA alone, with 2-APB added and in control cells in GCL astrocytes (B) and in Bergmann glia (C). AA-induced Ca2+ influx was not blocked by 2-APB, suggesting that AA activated a SOCE-independent pathway for Ca2+ influx. *p < 0.05 versus AA-induced Ca2+ rise versus control; n indicates the number of cells tested/number of slices.
BEL-sensitive SOCE and iPLA2 activity in cultured astrocytes
To confirm the involvement of iPLA2 in SOCE in cerebellar astrocytes, we wanted to measure the activity of iPLA2 under store-depleted condition and after inhibition of CaM, in astrocyte culture. First, we established that the Ca2+ rises during readdition of external Ca2+ in the presence of CPA and CMZ were sensitive to the iPLA2 inhibitor BEL (Fig. 8A–C). Addition of CPA (10 μm) in Ca2+-free saline evoked a Ca2+ transient in all cells. During readdition of external Ca2+ after store depletion by CPA, a Ca2+ rise, indicative of SOCE, was measured with a mean amplitude of 75 ± 4% ΔF (n = 150/5). This Ca2+ rise was much larger than in control cells (12 ± 0.5% ΔF; p < 0.005; n = 163/5), in which the cells were not exposed to CPA. In astrocyte cultures pretreated with 25 μm BEL, the SOCE induced in the presence of CPA was reduced to 4 ± 0.9% ΔF (p < 0.005; n = 141/5) (Fig. 8A,C). In the presence of 1 μm CMZ, the cytosolic Ca2+ rose to 87 ± 7% ΔF (n = 132/5), which was reduced to 4 ± 0.8% ΔF (p < 0.005; n = 146/5) after preincubation with BEL (Fig. 8B,C). Again, the Ca2+ rise induced by CMZ was significantly smaller after BEL treatment than the Ca2+ rise in control cells without CMZ (p < 0.005).
Figure 8.

CPA- and CMZ-related Ca2+ influx and iPLA2 in cerebellar astrocyte culture. A, B, Original recordings of the cytosolic Ca2+ concentration in single, cultured astrocytes in the presence of CPA or CMZ. Astrocytes were exposed to either 10 μm CPA (A) or 1 μm CMZ (B) (indicated by arrowhead) for 7 min in Ca2+-free condition before 2 mm Ca2+ was readded. Traces are for the cells exposed to CPA or CMZ alone and to CPA or CMZ in cells pretreated with 25 μm BEL for 30 min at 37°C. Cells only kept in Ca2+-free solution for 8–10 min served as control (bottom trace). C, Bar plot of the average, relative amplitude of Ca2+ rise after readdition of Ca2+ as obtained from experiments as shown in A and B, showing that BEL suppressed the influx of Ca2+ in the presence of either CPA or CMZ. D, Bar plot showing the percentage of BEL-sensitive iPLA2 enzyme activity as induced by either CPA or CMZ relative to the value in cells treated with 10 mm EGTA (100%), as obtained in the in vitro enzyme assay. *p < 0.05 versus 10 mm EGTA; ***p < 0.005 versus CPA/CMZ-related Ca2+ influx; n indicates the number of astrocytes/number of culture plates.
We determined BEL-sensitive iPLA2 activity in cerebellar astrocyte cultures by absorbance measurements using an iPLA2 activity assay (see Materials and Methods). Maximal iPLA2 activity (100%) was measured in the presence of 10 mm EGTA to suppress all Ca2+/calmodulin-dependent iPLA2 inhibition. Basal iPLA2 activity was 4 ± 1.4% of the maximal iPLA2 activity as measured in cell homogenates from untreated cerebellar astrocytes (Fig. 8D). In astrocytes treated with 10 μm CPA (10 min at 37°C) or 1 μm CMZ (10 min at 37°C), the iPLA2 activity increased to 62 ± 1.2 and 55 ± 3.0% of the maximal iPLA2 activity, respectively. These results suggest that iPLA2 activity is increased under conditions when SOCE is activated in cerebellar astrocytes.
Antisense inhibition of iPLA2
In another approach to examine the role of iPLA2 in SOCE, we used antisense technology to knock down the expression of iPLA2. Cerebellar astrocyte cultures were transfected with S-ODN and AS-ODN of iPLA2-A isoforms using a Lipofectamine-assisted transfection protocol (Balsinde et al., 1997; Smani et al., 2003). Control cells were exposed to the transfection protocol without ODNs to check whether LF alone had any nonspecific effects. Cultured astrocytes were loaded with fura-2, and the fluorescence ratio at 340 and 380 nm excitation was measured, indicative of the Ca2+ concentration in the cells. First, we used ATP (20 μm, 20 s) to check the functional status of the Ca2+ stores. ATP induced a similar Ca2+ rise in LF-treated and S-ODN-transfected cells, with mean amplitudes of 0.55 ± 0.13 ratio units (n = 176/6) and 0.59 ± 0.12 ratio units (n = 142/8) (Fig. 9A,C), respectively. In AS-ODN-transfected cells, the Ca2+ transient induced by ATP was strongly reduced to 0.17 ± 0.08 ratio units (p < 0.001; n = 153/8) (Fig. 9B,C), possibly reflecting the impaired filling status of the intracellular Ca2+ stores, when iPLA2 expression was inhibited. CPA (10 μm) was then applied in the absence of extracellular Ca2+ for 7 min, which led to a slow transient Ca2+ rise in all cells. The absolute mean amplitude of Ca2+ transient after the addition of CPA in LF-treated cells was 0.16 ± 0.03 ratio units (n = 176/6), which was similar to that of S-ODN-treated cells, being 0.15 ± 0.03 ratio units (n = 142/8). The AS-ODN-transfected cells showed a reduced Ca2+ transient during CPA addition of 0.08 ± 0.04 ratio units (p < 0.001; n = 153/8) (Fig. 9B,C), suggesting reduced Ca2+ release from the stores, possibly attributable to an incomplete filling status of the Ca2+ stores after interference with iPLA2 expression.
Figure 9.

Antisense inhibition of iPLA2 reduced the CPA-related Ca2+ influx. A, B, Original recordings of the cytosolic Ca2+ concentration in single, cultured, transfected astrocytes in the presence of CPA. Astrocytes were briefly exposed to 20 μm ATP (20 s) to check functional status of Ca2+ stores, before 10 μm CPA was added for 7 min in Ca2+-free condition, followed by readdition of Ca2+ (2 mm). Traces in A are for the cells transfected with S-ODN of iPLA2 and LF alone. Traces in B show cells transfected with AS-ODN with two different response patterns. C, Bar plot of the average amplitude of Ca2+ transients evoked by ATP and CPA and of the Ca2+ rise after the readdition of Ca2+ as obtained from experiments as shown in A and B. D–G, Labeling of transfected astrocytes with anti-iPLA2 antibody, showing similar iPLA2 expression in LF-treated cells (D) and in cells treated with S-ODN (E). AS-ODN-treated cells showed reduced iPLA2 expression (F). G, Bar plot of the relative fluorescence in AU in transfected cells as shown in D–F. Control represents the background fluorescence from astrocytes labeled only with secondary antibody. ***p < 0.005; n indicates the number of transfected astrocytes investigated. Scale bars, 25 μm.
Because the Ca2+ store content appeared to be reduced by AS-ODN, we wanted to check whether this reduction might be attributable to dysregulated ER-Ca2+ homeostasis. The decay time constant τ of the Ca2+ transients evoked by added ATP was analyzed in the S-ODN- and AS-ODN-treated cells. In S-ODN-treated astrocytes, τ was 10 ± 3.8 s (n = 62), which was not significantly different from that in AS-ODN-treated cells, with a τ of 11 ± 5.7 s (n = 51; data not shown); this finding is in line with an unimpaired ER homeostasis in AS-ODN-treated cells. We also measured the basal Ca2+-dependent fura-2 ratio level in S-ODN- and AS-ODN-transfected cells to get an indication of a change in resting cytosolic Ca2+ level after inhibiting iPLA2. The basal ratio was 0.48 ± 0.03 (n = 142/8) in S-ODN transfected cells, which was indeed significantly different from the AS-ODN-transfected cells, with a ratio of 0.41 ± 0.03 (p < 0.001; n = 153/8; data not shown), suggesting that the reduced cytosolic Ca2+ level in the AS-ODN-transfected cells might be attributable to the impaired iPLA2 activity.
During readdition of Ca2+, the Ca2+ rise was similar in LF-treated cells and cells transfected with the S-ODN, with a mean amplitude of 0.30 ± 0.06 ratio units (n = 176/6) and 0.33 ± 0.09 ratio units (n = 142/8), respectively. There was a significant reduction, however, in the Ca2+ transient evoked by Ca2+ readdition in AS-ODN-transfected cells to 0.16 ± 0.07 ratio units (p < 0.001; n = 153/8) (Fig. 9B,C). Hence, only transfection with iPLA2 antisense-ODN, but not sense-ODN or LF alone, affected SOCE in cultured astrocytes, indicating a crucial role of iPLA2 for generating SOCE.
To ascertain the effective knockdown of iPLA2 expression in AS-ODN-transfected cells, we labeled the cells with anti-iPLA2 antibody and measured the relative fluorescence. Antibody staining of all transfected cells (LF, S-ODN, and AS-ODN) was performed 48 h after transfection. All cells were processed in parallel with the same protocol, and the anti-iPLA2 immunoreactivity was assessed under the same settings of the confocal microscope. The relative fluorescence intensity of iPLA2 expression was measured only in cells labeled with fluorescein, indicating successful transfection, and is given in arbitrary units (AU). In LF-treated and S-ODN-transfected cells, the mean fluorescence was 110 ± 1.5 AU (n = 324) (Fig. 9D,G) and 130 ± 1.6 AU (n = 330) (Fig. 9E,G), respectively. In AS-ODN-treated cells, the fluorescence intensity was significantly reduced to 69 ± 1.5 AU (p < 0.001; n = 509) (Fig. 9F,G), consistent with a reduction of iPLA2 expression in AS-ODN treated cells. The background fluorescence was measured to be 17 ± 0.8 AU (p < 0.001; n = 290) (Fig. 9G) in control cells, which were stained only with the secondary antibody.
Spontaneous Ca2+ oscillations in rat cerebellum
If SOCE was indeed instrumental for the refilling of intracellular Ca2+ stores, suppression of SOCE would be expected to affect spontaneous Ca2+ oscillations (Nett et al., 2002; Zur Nieden and Deitmer, 2006). We therefore examined the effects of two substances, known to inhibit SOCE, on spontaneous Ca2+ oscillations in astrocytes in situ. Bergmann glia and GCL astrocytes showed spontaneous activity in cerebellar brain slices (Fig. 10A,B), although Bergmann glial cells generated Ca2+ oscillations less frequently than GCL astrocytes. We analyzed the relative number of cells with spontaneous Ca2+ oscillations and the frequency of the Ca2+ oscillations. Over 20 min, spontaneous Ca2+ signals were recorded in the standard solution as control. The experimental protocol continued for another 20 min after the SOCE channel blocker 2-APB was applied. Ca2+ signals were analyzed for at least 15 min, leaving the first 5 min to allow the drugs to work. Spontaneous Ca2+ responses could be recorded in 28% (n = 87 of 313/31) of the cells identified as Bergmann glia and in 47% (n = 107 of 227/27) of the GCL astrocytes. The overall frequency of spontaneous Ca2+ transients averaged 0.17 ± 0.06 transients/min (n = 87/31) in Bergmann glial cells and 0.40 ± 0.07 transients/min (n = 107/27) in GCL astrocytes.
Figure 10.

Spontaneous Ca2+ oscillations in acute rat cerebellar slices were reduced by suppressing SOCE and by inhibiting iPLA2 activity. A, Continuous, original recordings of spontaneous Ca2+ transients for 20 min in GCL astrocytes. Traces are for untreated slices (control, top trace) and for slices pretreated with BEL (25 μm for 30–40 min at 37°C; bottom trace). B, C, Bar plots showing the percentage of oscillating cells (B) and the relative frequency of Ca2+ transients (C) relative to control conditions (untreated slices, 100%) and for slices after treatment with 2-APB and with BEL in Bergmann glia and in GCL astrocytes.
Inhibiting iPLA2 by preincubation with BEL for 30–40 min, as described above, decreased the frequency of the Ca2+ signals in GCL astrocytes (Fig. 10A) on average to 40% of the control, whereas the number of spontaneously active cells decreased to 46% (Fig. 10B,C). In Bergmann glial cells, the number of active cells decreased to 28%, and the frequency of Ca2+ signals decreased to 39% of the control.
In the presence of 2-APB, Ca2+ signals were observed in only 58% of the Bergmann glial cells and 62% of the GCL astrocytes that exhibited spontaneous Ca2+ oscillations under control conditions (Fig. 10B). The frequency of the Ca2+ oscillations was reduced to 49% (n = 17/11) in Bergmann glia and to 55% (n = 24/11) in GCL astrocytes in the presence of 2-APB (Fig. 10C). These results suggest that the functional activity of iPLA2 and 2-APB-sensitive SOCE channels are required for maintaining spontaneous Ca2+ oscillations in Bergmann glia and GCL astrocytes at their normal level.
Discussion
The present study provides evidence that SOCE in rat cerebellar astrocytes in situ is mediated by the activation of iPLA2. We showed here the following: (1) depletion of intracellular Ca2+ stores by CPA activates Ca2+ influx (SOCE), which can be suppressed by inhibiting iPLA2 activity by BEL and PACOCF3; (2) inhibiting calmodulin by CMZ to relieve the calmodulin-dependent block of iPLA2 induces a Ca2+ influx, which shares the pharmacological profile of SOCE as obtained in CPA; (3) lysophospholipid products of iPLA2, LPI and LPC, but not LPA, evoke a Ca2+ influx, which could be suppressed by SOCE channel blockers; (4) depletion of Ca2+ stores by CPA and inhibition of calmodulin by CMZ increased BEL-sensitive iPLA2 enzyme activity in cultured cerebellar astrocytes; and (5) the specific antisense inhibition of iPLA2 reduced SOCE. Finally, the SOCE channel blocker 2-APB and the iPLA2 inhibitor BEL reduced spontaneous Ca2+ oscillations in astrocytes in situ, indicating functional significance of iPLA2-mediated SOCE for normal Ca2+ signaling in astrocytes. This is the first study demonstrating the involvement of iPLA2 activity for SOCE in acute tissue slices and in glial cells in situ.
CPA-induced SOCE requires the functional activity of iPLA2 in astrocytes
The possible link of iPLA2 activation to store depletion was first reported in rat smooth muscle cells (Wolf et al., 1997) and pancreatic islets (Nowatzke et al., 1998), in which the depletion of intracellular Ca2+ stores resulted in an increased iPLA2-mediated phospholipid hydrolysis and accumulation of free fatty acids. According to the proposed molecular mechanism for cultured smooth muscle cells, the generated lysophospholipids activate SOCE channels and capacitative Ca2+ influx, leading to the refilling of Ca2+ stores (Smani et al., 2004).
In rat brain, >70% of the PLA2 activity could be attributed to iPLA2 (Yang et al., 1999). iPLA2 was detected in the cerebellar cortex in all cell layers by means of in situ hybridization and antibody staining (Shirai and Ito, 2004). In our study, the activation of SOCE by store depletion appeared to be attributable to stimulation of iPLA2, because iPLA2 impairment by the iPLA2 inhibitors BEL and PACOCF3 completely prevented Ca2+ influx in response to CPA in acute brain slices and in culture (BEL), as outlined in the hypothetical diagram in supplemental Figure 1 (available at www.jneurosci.org as supplemental material). Moreover, iPLA2 activity increased when ER stores had been depleted in astrocyte culture. Importantly, the specific iPLA2 AS-ODN impaired SOCE, presumably by a reduction of iPLA2 expression in the cells. Notably, the CPA-mediated Ca2+ influx in the presence of BEL or PACOCF3 was even smaller than the Ca2+ influx in control cells, in which Ca2+ stores were not depleted by CPA. This suggests some constitutive activity of iPLA2, which might be independent of the filling state of intracellular Ca2+ stores. The Ca2+ influx evoked by lysophospholipids in BEL-treated brain slices indicated that BEL did not suppress Ca2+ influx per se and presumably did not affect SOCE directly. This drug has also been reported to inhibit PAP-1, but the CPA-mediated Ca2+ entry was not prevented by the PAP-1 inhibitor propranolol in our study, indicating that the BEL-induced inhibition of SOCE was not attributable to suppressing PAP-1 activity. Furthermore, ATP-induced Ca2+ transients in BEL-pretreated astrocytes demonstrated intact Ca2+ stores, suggesting that BEL did not induce conditions with impaired ER integrity or apoptosis, under our experimental conditions.
In cells not challenged with thapsigargin or CPA, CaM binds to iPLA2 and thereby suppresses the activity of this enzyme. After the dissociation of CaM, iPLA2 becomes functionally active (Wolf and Gross, 1996a,b). In cultured smooth muscle cells, the CaM inhibitor CMZ induces Ca2+ release as well as 2-APB-insensitive Ca2+ influx at concentrations larger than 1 μm, whereas lower doses of CMZ (≤1 μm) only evoke 2-APB-sensitive Ca2+ entry (Smani et al., 2004). In HeLa cells, CMZ at concentrations larger than 1 μm induced Ca2+ entry that involves a non-SOCE pathway and was sensitive to AA (Peppiatt et al., 2004). In the present study, we used 1 μm CMZ to evoke Ca2+ influx in acute brain slices and found that CMZ induced a Ca2+ influx without depleting the Ca2+ stores. This Ca2+ influx is sensitive to 2-APB and BTP2 and is suppressed after inhibiting iPLA2. Because the Ca2+ signals induced by CPA-mediated store depletion and by CMZ-mediated CaM inhibition exhibit the same pharmacological profile, we suggest a similar signaling pathway in glial cells as reported for smooth muscle cells (Smani et al., 2004) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Hence, store depletion as well as CMZ would release the inhibitory interaction of CaM on iPLA2 and then allow SOCE. This is supported by the enhanced enzyme activity of iPLA2 in cerebellar astrocyte culture after store depletion by CPA and after inhibiting CaM with CMZ.
Activation of SOCE channel appears to be mediated by the products of iPLA2 because both lysophospholipids LPC and LPI activated a SOCE-like Ca2+ influx. LPA did not generate a Ca2+ influx, which may be attributable to the lack of a large head group in LPA, in contrast to LPI and LPC. Suppression of iPLA2 activity by BEL did not affect the LPI- and LPC-induced Ca2+ influx, in line with the hypothesis that the lysophospholipid-mediated activation of Ca2+ entry is downstream to iPLA2 activation (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Moreover, our results suggest that LPI- and LPC-induced Ca2+ influx passes through 2-APB- and BTP2-sensitive channels. Lysophospholipid-activated Ca2+ entry in Bergmann glial cells and GCL astrocytes apparently uses a pathway similar, if not identical, to that triggered by store depletion. It is yet unknown, however, how the generation of lysophospholipids activates SOCE. Lysophospholipids may open the SOCE channels either indirectly by affecting the lipid environment of the channels or directly by interacting with the membrane channel protein. Notably, a direct interaction of lysophosphatidylcholine with the cation channel TRPC5 was recently suggested in transfected HEK293 cells and arterial smooth muscle cells (Flemming et al., 2006).
Ca2+ oscillations rely on SOCE in cerebellar astrocytes
Spontaneous Ca2+ oscillations have been reported in astrocytes in culture (Fatatis and Russell, 1992; Harris-White et al., 1998) and in situ (Parri et al., 2001; Aguado et al., 2002; Nett et al., 2002; Zur Nieden and Deitmer, 2006). Ca2+ signaling in the form of repetitive oscillations are associated with regulating a variety of physiological processes, including gene expression (Dolmetsch et al., 1998), K+ conductance (Chen et al., 1997), and secretion (Thomas et al., 1996). The number of spontaneously active astrocytes and the number of Ca2+ transients exhibited by each astrocyte increased by increasing the external Ca2+ (Parri and Crunelli, 2003) and was reduced by inhibiting metabotropic glutamate receptors of groups I and II (Zur Nieden and Deitmer, 2006). SOCE and Ca2+ store refilling are required to maintain both spontaneous Ca2+ oscillations (Nett et al., 2002; Zur Nieden and Deitmer, 2006) and Ca2+ oscillations evoked by stimulation of metabotropic glutamate receptors in type I cortical astrocytes (Pizzo et al., 2001). It has been shown in rat hippocampal astrocytes that Ca2+ oscillations were attributable to phospholipase C-mediated Ca2+ release from intracellular stores (Zur Nieden and Deitmer, 2006). 2-APB effectively inhibits SOCE channels in many cell types (Bakowski et al., 2001; Braun et al., 2001; Broad et al., 2001; Gregory et al., 2001; Kukkonen et al., 2001; Prakriya and Lewis, 2001) but can also be a membrane-permeable inhibitor of IP3 receptors (Wu et al., 2000). We observed that the number of spontaneously active astrocytes as well as the frequency of oscillations were reduced by blocking SOCE channels and by inhibiting iPLA2 activity. Our results provide evidence that iPLA2-mediated Ca2+ influx through SOCE channels is needed for the maintenance of Ca2+ signaling in astrocytes in situ, which requires continuous refilling of Ca2+ stores.
In summary, our results suggest that iPLA2 is a major regulator of SOCE in cerebellar astrocytes; during depletion of Ca2+ stores, iPLA2 could be activated to open the channels in plasma membrane by the formation of lysophospholipids (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). A CIF would transfer the signal from depleted stores to relieve the block of iPLA2 by CaM in the plasma membrane, consistent with studies of iPLA2-mediated SOCE in other cell types evoked by emptying stores by either inhibiting SERCA pumps (Smani et al., 2004; Van den Abeele et al., 2004) or activating translocon pores (Flourakis et al., 2006). CIF has been suggested to act through the activation of iPLA2 (Smani et al., 2004; Van den Abeele et al., 2004); however, it remains unresolved how the formation of CIF is triggered in response to depletion of Ca2+ stores. The STIM1 is likely the sensor (CIF) of depleted Ca2+ inside the ER Ca2+ stores, which translocates in response to store depletion from the ER to the plasma membrane (Roos et al., 2005; Zhang et al., 2005) and also controls the operation of the SOCE channels within the plasma membrane (Spassova et al., 2006). Recently, the plasma membrane four-transmembrane-spanning protein Orai1 or CRACM1 (Ca2+ release-activated channel blocker) has been reported to be necessary for SOCE and CRAC channel activity (Feske et al., 2006; Vig et al., 2006). Orail and STIM1 are likely to be functionally coupled and sufficient to mediate SOCE (Peinelt et al., 2006; Soboloff et al., 2006). Whether STIM1 is the first step in the molecular cascade to activate iPLA2- and LPL-induced Ca2+ entry to refill intracellular Ca2+ stores in astrocytes still needs experimental evidence.
Footnotes
The financial support of the Deutsche Forschungsgemeinschaft is gratefully acknowledged (Sonderforschungsbereich 530, Teilprojekt B1, and Graduate Research School 845). We thank Dr. Sandra Vatter for helping with the enzyme assay, Dr. Markus Hoth and Dr. Anant Parekh for discussion, and Sandra Bergstein for technical assistance with the cell cultures.
References
- Ackermann et al., 1995.Ackermann EJ, Conde-Frieboes K, Dennis EA. Inhibition of macrophage Ca2+-independent phospholipase A2 by bromoenol lactone and trifluoromethyl ketones. J Biol Chem. 1995;270:445–450. doi: 10.1074/jbc.270.1.445. [DOI] [PubMed] [Google Scholar]
- Aguado et al., 2002.Aguado F, Espinosa-Parrilla JF, Carmona MA, Soriano E. Neuronal activity regulates correlated network properties of spontaneous calcium transients in astrocytes in situ. J Neurosi. 2002;22:9430–9444. doi: 10.1523/JNEUROSCI.22-21-09430.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbari et al., 2004.Akbari Y, Hitt BD, Murphy MP, Dagher NN, Tseng BP, Green KN, Golde TE, LaFerla FM. Presenilin regulates capacitative calcium entry dependently and independently of gamma-secretase activity. Biochem Biophys Res Commun. 2004;322:1145–1152. doi: 10.1016/j.bbrc.2004.07.136. [DOI] [PubMed] [Google Scholar]
- Bakowski et al., 2001.Bakowski D, Glitsch MD, Parekh AB. An examination of the secretion-like coupling model for the activation of the Ca2+ release-activated Ca2+ current ICRAC in RBL-1 cells. J Physiol (Lond) 2001;532:55–71. doi: 10.1111/j.1469-7793.2001.0055g.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsinde and Dennis, 1996.Balsinde J, Dennis EA. Bromoenol lactone inhibits magnesium-dependent phosphatidate phosphohydrolase and blocks triacylglycerol biosynthesis in mouse P388D1 macrophages. J Biol Chem. 1996;271:31937–31941. doi: 10.1074/jbc.271.50.31937. [DOI] [PubMed] [Google Scholar]
- Balsinde et al., 1995.Balsinde J, Bianco ID, Ackermann EJ, Conde-Frieboes K, Dennis EA. Inhibition of calcium-independent phospholipase A2 prevents arachidonic acid incorporation and phospholipid remodeling in P388D1 macrophages. Proc Natl Acad Sci USA. 1995;92:8527–8531. doi: 10.1073/pnas.92.18.8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balsinde et al., 1997.Balsinde J, Balboa MA, Dennis EA. Antisense inhibition of group VI Ca2+-independent phospholipase A2 blocks phospholipid fatty acid remodeling in murine P388D macrophages. J Biol Chem. 1997;272:29317–29321. doi: 10.1074/jbc.272.46.29317. [DOI] [PubMed] [Google Scholar]
- Beck et al., 2004.Beck A, Zur Nieden R, Schneider HP, Deitmer JW. Calcium release from intracellular stores in rodent astrocytes in situ. Cell Calcium. 2004;35:47–58. doi: 10.1016/s0143-4160(03)00171-4. [DOI] [PubMed] [Google Scholar]
- Berridge et al., 2000.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- Braun et al., 2001.Braun FJ, Broad LM, Armstrong DL, Putney JW., Jr Stable activation of single Ca2+ release-activated Ca2+ channels in divalent cation-free solutions. J Biol Chem. 2001;276:1063–1070. doi: 10.1074/jbc.M008348200. [DOI] [PubMed] [Google Scholar]
- Broad et al., 2001.Broad LM, Braun FJ, Lievremont JP, Bird GS, Kurosaki T, Putney JW., Jr Role of the phospholipase C-inositol 1,4,5-trisphosphate pathway in calcium release-activated calcium current and capacitative calcium entry. J Biol Chem. 2001;276:15945–15952. doi: 10.1074/jbc.M011571200. [DOI] [PubMed] [Google Scholar]
- Campbell and Götz, 2002.Campbell K, Götz M. Radial glia: multi-purpose cells for vertebrate brain development. Trends Neurosci. 2002;25:235–238. doi: 10.1016/s0166-2236(02)02156-2. [DOI] [PubMed] [Google Scholar]
- Chen et al., 1997.Chen J, Backus KH, Deitmer JW. Intracellular calcium transients and potassium current oscillations evoked by glutamate in cultured rat astrocytes. J Neurosci. 1997;17:7278–7287. doi: 10.1523/JNEUROSCI.17-19-07278.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings et al., 2002.Cummings BS, McHowat J, Schnellmann RG. Role of an endoplasmic reticulum Ca2+-independent phospholipase A2 in oxidant-induced renal cell death. Am J Physiol Renal Physiol. 2002;283:F492–F498. doi: 10.1152/ajprenal.00022.2002. [DOI] [PubMed] [Google Scholar]
- Dallwig and Deitmer, 2002.Dallwig R, Deitmer JW. Cell-type specific calcium responses in acute rat hippocampal slices. J Neurosci Methods. 2002;116:77–87. doi: 10.1016/s0165-0270(02)00030-4. [DOI] [PubMed] [Google Scholar]
- Dolmetsch et al., 1998.Dolmetsch RE, Xu K, Lewis RS. Calcium oscillations increase the efficiency and specificity of gene expression. Nature. 1998;392:933–936. doi: 10.1038/31960. [DOI] [PubMed] [Google Scholar]
- Edwards et al., 1989.Edwards FA, Konnerth A, Sakmann B, Takahashi T. A thin slice preparation for patch clamp recordings from neurones of the mammalian central nervous system. Pflügers Arch. 1989;414:600–612. doi: 10.1007/BF00580998. [DOI] [PubMed] [Google Scholar]
- Emptage et al., 2001.Emptage NJ, Reid CA, Fine A. Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron. 2001;29:197–208. doi: 10.1016/s0896-6273(01)00190-8. [DOI] [PubMed] [Google Scholar]
- Fagan et al., 2000.Fagan KA, Graf RA, Tolman S, Schaack J, Cooper DM. Regulation of a Ca2+-sensitive adenylyl cyclase in an excitable cell. Role of voltage-gated versus capacitative Ca2+ entry. J Biol Chem. 2000;275:40187–40194. doi: 10.1074/jbc.M006606200. [DOI] [PubMed] [Google Scholar]
- Fatatis and Russell, 1992.Fatatis A, Russell JT. Spontaneous changes in intracellular calcium concentration in type I astrocytes from rat cerebral cortex in primary culture. Glia. 1992;5:95–104. doi: 10.1002/glia.440050203. [DOI] [PubMed] [Google Scholar]
- Feske et al., 2006.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- Fischer, 1984.Fischer G. Growth requirements of immature astrocytes in serum-free hormonally defined media. J Neurosci Res. 1984;12:543–552. doi: 10.1002/jnr.490120403. [DOI] [PubMed] [Google Scholar]
- Flemming et al., 2006.Flemming PK, Dedman AM, Xu SZ, Li J, Zeng F, Naylor J, Benham CD, Bateson AN, Muraki K, Beech DJ. Sensing of lysophospholipids by TRPC5 calcium channel. J Biol Chem. 2006;281:4977–4982. doi: 10.1074/jbc.M510301200. [DOI] [PubMed] [Google Scholar]
- Flourakis et al., 2006.Flourakis M, Van Coppenolle F, Lehen’Kyi V, Beck B, Skryma R, Prevarskaya N. Passive calcium leak via translocon is a first step for iPLA2-pathway regulated store-operated channel activation. FASEB J. 2006;20:1215–1217. doi: 10.1096/fj.05-5254fje. [DOI] [PubMed] [Google Scholar]
- Fuentes et al., 2003.Fuentes L, Perez R, Nieto ML, Balsinde J, Balboa MA. Bromoenol lactone promotes cell death by a mechanism involving phosphatidate phosphohydrolase-1 rather than calcium-independent phospholipase A2. J Biol Chem. 2003;278:44683–44690. doi: 10.1074/jbc.M307209200. [DOI] [PubMed] [Google Scholar]
- Gregory et al., 2001.Gregory RB, Rychkov G, Barritt GJ. Evidence that 2-aminoethyl diphenylborate is a novel inhibitor of store-operated Ca2+ channels in liver cells, and acts through a mechanism which does not involve inositol trisphosphate receptors. Biochem J. 2001;354:285–890. doi: 10.1042/0264-6021:3540285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer 2006.Hammer K. Gliale Calciumsignale im Kleinhirn der Ratte: Charakterisierung metabotroper Purinozeptoren mit Konfokaler Laser-Raster-Mikroskopie. PhD, University of Kaiserslautern. 2006 [Google Scholar]
- Harris-White et al., 1998.Harris-White ME, Zanotti SA, Frautschy SA, Charles AC. Spiral intercellular calcium waves in hippocampal slice cultures. J Neurophysiol. 1998;79:1045–1052. doi: 10.1152/jn.1998.79.2.1045. [DOI] [PubMed] [Google Scholar]
- Haunso et al., 2003.Haunso A, Simpson J, Antoni FA. Small ligands modulating the activity of mammalian adenylyl cyclases: a novel mode of inhibition by calmidazolium. Mol Pharmacol. 2003;63:624–631. doi: 10.1124/mol.63.3.624. [DOI] [PubMed] [Google Scholar]
- Ishikawa et al., 2003.Ishikawa J, Ohga K, Yoshino T, Takezawa R, Ichikawa A, Kubota H, Yamada T. A pyrazole derivative, YM-58483, potently inhibits store-operated sustained Ca2+ influx and IL-2 production in T lymphocytes. J Immunol. 2003;170:4441–4449. doi: 10.4049/jimmunol.170.9.4441. [DOI] [PubMed] [Google Scholar]
- Jung et al., 2000.Jung S, Pfeiffer F, Deitmer JW. Histamine-induced calcium entry in rat cerebellar astrocytes: evidence for capacitative and non-capacitative mechanisms. J Physiol (Lond) 2000;527:549–561. doi: 10.1111/j.1469-7793.2000.00549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirischuk et al., 1995.Kirischuk S, Moller T, Voitenko N, Kettenmann H, Verkhratsky A. ATP-induced cytoplasmic calcium mobilization in Bergmann glial cells. J Neurosi. 1995;15:7861–7871. doi: 10.1523/JNEUROSCI.15-12-07861.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukkonen et al., 2001.Kukkonen JP, Lund PE, Akerman KE. 2-aminoethoxydiphenyl borate reveals heterogeneity in receptor-activated Ca2+ discharge and store-operated Ca2+ influx. Cell Calcium. 2001;30:117–129. doi: 10.1054/ceca.2001.0219. [DOI] [PubMed] [Google Scholar]
- Li et al., 2003.Li N, Sul JY, Haydon PG. A calcium-induced calcium influx factor, nitric oxide, modulates the refilling of calcium stores in astrocytes. J Neurosci. 2003;23:10302–10310. doi: 10.1523/JNEUROSCI.23-32-10302.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou et al., 2005.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Meyer T. STIM is a Ca2+ sensor essential for Ca2+ store depletion triggered Ca2+ influx. Curr Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo et al., 2002.Lo KJ, Luk HN, Chin TY, Chueh SH. Store depletion-induced calcium influx in rat cerebellar astrocytes. Br J Pharmacol. 2002;135:1383–1392. doi: 10.1038/sj.bjp.0704594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma et al., 2000.Ma HT, Patterson RL, Van Rossum DB, Birnbaumer L, Mikoshiba K, Gill DL. Requirement of the inositol triphosphate receptor for activation of store-operated Ca2+ channels. Science. 2000;287:1647–1651. doi: 10.1126/science.287.5458.1647. [DOI] [PubMed] [Google Scholar]
- Nett et al., 2002.Nett WJ, Oloff SH, McCarthy KD. Hippocampal astrocytes in situ exhibit calcium oscillations that occur independent of neuronal activity. J Neurophysiol. 2002;87:528–537. doi: 10.1152/jn.00268.2001. [DOI] [PubMed] [Google Scholar]
- Nowatzke et al., 1998.Nowatzke W, Ramanadham S, Ma Z, Hsu FF, Bohrer A, Turk J. Mass spectrometric evidence that agents that cause loss of Ca2+ from intracellular compartments induce hydrolysis of arachidonic acid from pancreatic islet membrane phospholipids by a mechanism that does not require a rise in cytosolic Ca2+ concentration. Endocrinology. 1998;139:4073–4085. doi: 10.1210/endo.139.10.6225. [DOI] [PubMed] [Google Scholar]
- Parekh and Putney, 2005.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- Parri and Crunelli, 2003.Parri HR, Crunelli V. The role of Ca2+ in the generation of spontaneous astrocytic Ca2+ oscillations. Neuroscience. 2003;120:979–992. doi: 10.1016/s0306-4522(03)00379-8. [DOI] [PubMed] [Google Scholar]
- Parri et al., 2001.Parri HR, Gould TM, Crunelli V. Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR-mediated neuronal excitation. Nat Neurosci. 2001;4:803–812. doi: 10.1038/90507. [DOI] [PubMed] [Google Scholar]
- Peinelt et al., 2006.Peinelt C, Vig M, Koomoa DL, Beck A, Nadler MJ, Koblan-Huberson M, Lis A, Fleig A, Penner R, Kinet JP. Amplification of CRAC current by STIM1 and CRACM1 (Orai1) Nat Cell Biol. 2006;8:771–773. doi: 10.1038/ncb1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppiatt et al., 2004.Peppiatt CM, Holmes AM, Seo JT, Bootman MD, Collins TJ, McDonald F, Roderick HL. Calmidazolium and arachidonate activate a calcium entry pathway that is distinct from store-operated calcium influx in HeLa cells. Biochem J. 2004;381:929–939. doi: 10.1042/BJ20040097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzo et al., 2001.Pizzo P, Burgo A, Pozzan T, Fasolato C. Role of capacitative calcium entry on glutamate-induced calcium influx in type-I rat cortical astrocytes. J Neurochem. 2001;9:98–109. doi: 10.1046/j.1471-4159.2001.00539.x. [DOI] [PubMed] [Google Scholar]
- Prakriya and Lewis, 2001.Prakriya M, Lewis RS. Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP3 receptors. J Physiol (Lond) 2001;536:3–19. doi: 10.1111/j.1469-7793.2001.t01-1-00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic, 2003.Rakic P. Developmental and evolutionary adaptations of cortical radial glia. Cereb Cortex. 2003;13:541–549. doi: 10.1093/cercor/13.6.541. [DOI] [PubMed] [Google Scholar]
- Randriamampita and Tsien, 1993.Randriamampita C, Tsien RY. Emptying of intracellular Ca2+ stores releases a novel small messenger that stimulates Ca2+ influx. Nature. 1993;364:809–814. doi: 10.1038/364809a0. [DOI] [PubMed] [Google Scholar]
- Roos et al., 2005.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G, Stauderman KA. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J Cell Biol. 2005;169:435–445. doi: 10.1083/jcb.200502019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeeva et al., 2003.Sergeeva M, Strokin M, Wang H, Ubl JJ, Reiser G. Arachidonic acid in astrocytes blocks Ca2+ oscillations by inhibiting store-operated Ca2+ entry, and causes delayed Ca2+ influx. Cell Calcium. 2003;33:283–292. doi: 10.1016/s0143-4160(03)00011-3. [DOI] [PubMed] [Google Scholar]
- Shao and McCarthy, 1995.Shao Y, McCarthy KD. Receptor-mediated calcium signals in astroglia: multiple receptors, common stores and all-or-nothing responses. Cell Calcium. 1995;17:187–196. doi: 10.1016/0143-4160(95)90033-0. [DOI] [PubMed] [Google Scholar]
- Shirai and Ito, 2004.Shirai Y, Ito M. Specific differential expression of phospholipase A2 subtypes in rat cerebellum. J Neurocytol. 2004;33:297–307. doi: 10.1023/B:NEUR.0000044191.83858.f7. [DOI] [PubMed] [Google Scholar]
- Shuttleworth and Thompson, 1999.Shuttleworth TJ, Thompson JL. Discriminating between capacitative and arachidonate-activated Ca2+ entry pathways in HEK293 cells. J Biol Chem. 1999;274:31174–31178. doi: 10.1074/jbc.274.44.31174. [DOI] [PubMed] [Google Scholar]
- Smani et al., 2003.Smani T, Zakharov SI, Leno E, Csutora P, Trepakova ES, Bolotina VM. Ca2+-independent phospholipase A2 is a novel determinant of store-operated Ca2+ entry. J Biol Chem. 2003;278:11909–11915. doi: 10.1074/jbc.M210878200. [DOI] [PubMed] [Google Scholar]
- Smani et al., 2004.Smani T, Zakharov SI, Csutora P, Leno E, Trepakova ES, Bolotina VM. A novel mechanism for the store-operated calcium influx pathway. Nat Cell Biol. 2004;6:113–120. doi: 10.1038/ncb1089. [DOI] [PubMed] [Google Scholar]
- Soboloff et al., 2006.Soboloff J, Spassova MA, Tang XD, Hewavitharana T, Xu W, Gill DL. Orail and STIM reconstitute store-operated calcium channel function. J Biol Chem. 2006;281:20661–20665. doi: 10.1074/jbc.C600126200. [DOI] [PubMed] [Google Scholar]
- Spassova et al., 2006.Spassova MA, Soboloff J, He LP, Xu W, Dziadek MA, Gill DL. STIM1 has a plasma membrane role in the activation of store-operated Ca2+ channels. Proc Natl Acad Sci USA. 2006;103:4040–4045. doi: 10.1073/pnas.0510050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas et al., 1996.Thomas AP, Bird GS, Hajnoczky G, Robb-Gaspers LD, Putney JW., Jr Spatial and temporal aspects of cellular calcium signaling. FASEB J. 1996;10:1505–1517. [PubMed] [Google Scholar]
- Van den Abeele et al., 2004.Van den Abeele F, Lemonnier L, Thebault S, Lepage G, Parys JB, Shuba Y, Skryma R, Prevarskaya N. Two types of store-operated Ca2+ channels with different activation modes and molecular origin in LNCaP human prostate cancer epithelial cells. J Biol Chem. 2004;279:30326–30337. doi: 10.1074/jbc.M400106200. [DOI] [PubMed] [Google Scholar]
- Verkhratsky, 2005.Verkhratsky A. Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol Rev. 2005;85:201–279. doi: 10.1152/physrev.00004.2004. [DOI] [PubMed] [Google Scholar]
- Vig et al., 2006.Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, Kinet JP. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006;312:1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets et al., 2001.Voets T, Prenen J, Fleig A, Vennekens R, Watanabe H, Hoenderop JGJ, Bindels RJM, Droogmans G, Penner R, Nilius B. CaT1 and the calcium release-activated calcium channel manifest distinct pore properties. J Biol Chem. 2001;276:47767–47770. doi: 10.1074/jbc.C100607200. [DOI] [PubMed] [Google Scholar]
- Winstead et al., 2000.Winstead MV, Balsinde J, Dennis EA. Calcium-independent phospholipase A2: structure and function. Biochem Biophys Acta. 2000;1488:28–39. doi: 10.1016/s1388-1981(00)00107-4. [DOI] [PubMed] [Google Scholar]
- Wolf and Gross, 1996a.Wolf MJ, Gross RW. The calcium-dependent association and functional coupling of calmodulin with myocardial phospholipase A2. Implications for cardiac cycle-dependent alterations in phospholipolysis. J Biol Chem. 1996a;271:20989–20992. doi: 10.1074/jbc.271.35.20989. [DOI] [PubMed] [Google Scholar]
- Wolf and Gross, 1996b.Wolf MJ, Gross RW. Expression, purification, and kinetic characterization of a recombinant 80-kDa intracellular calcium-independent phospholipase A2. J Biol Chem. 1996b;271:30879–30885. doi: 10.1074/jbc.271.48.30879. [DOI] [PubMed] [Google Scholar]
- Wolf et al., 1997.Wolf MJ, Wang J, Turk J, Gross RW. Depletion of intracellular calcium stores activates smooth muscle cell calcium-independent phospholipase A2. A novel mechanism underlying arachidonic acid mobilization. J Biol Chem. 1997;272:1522–1526. doi: 10.1074/jbc.272.3.1522. [DOI] [PubMed] [Google Scholar]
- Wu et al., 2000.Wu J, Kamimura N, Takeo T, Suga S, Wakui M, Maruyama T, Mikoshiba K. 2-Aminoethoxydiphenyl borate modulates kinetics of intracellular Ca2+ signals mediated by inositol 1,4,5-trisphosphate-sensitive Ca2+ stores in single pancreatic acinar cells of mouse. Mol Pharmacol. 2000;58:1368–1374. doi: 10.1124/mol.58.6.1368. [DOI] [PubMed] [Google Scholar]
- Wu et al., 1997.Wu ML, Kao EF, Liu IH, Wang BS, Lin-Shiau SY. Capacitative Ca2+ influx in glial cells is inhibited by glycolytic inhibitors. Glia. 1997;21:315–326. doi: 10.1002/(sici)1098-1136(199711)21:3<315::aid-glia6>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Wu et al., 1999.Wu ML, Chen WH, Liu IH, Tseng CD, Wang SM. A novel effect of cyclic AMP on capacitative Ca2+ entry in cultured rat cerebellar astrocytes. J Neurochem. 1999;73:1318–1328. doi: 10.1046/j.1471-4159.1999.0731318.x. [DOI] [PubMed] [Google Scholar]
- Yang et al., 1999.Yang HC, Mosior M, Johnson CA, Chen Y, Dennis EA. Group-specific assays that distinguish between the four major types of mammalian phospholipase A2. Anal Biochem. 1999;269:278–288. doi: 10.1006/abio.1999.4053. [DOI] [PubMed] [Google Scholar]
- Yang et al., 2005.Yang KT, Chen WP, Chang WL, Su MJ, Tsai KL. Arachidonic acid inhibits capacitative Ca2+ entry and activates non-capacitative Ca2+ entry in cultured astrocytes. Biochem Biophys Res Commun. 2005;331:603–613. doi: 10.1016/j.bbrc.2005.03.221. [DOI] [PubMed] [Google Scholar]
- Zhang et al., 2005.Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitt et al., 2004.Zitt C, Strauss B, Schwarz EC, Spaeth N, Rast G, Hatzelmann A, Hoth M. Potent inhibition of Ca2+ release-activated Ca2+ channels and T-lymphocyte activation by the pyrazole derivative BTP2. J Biol Chem. 2004;279:12427–12437. doi: 10.1074/jbc.M309297200. [DOI] [PubMed] [Google Scholar]
- Zur Nieden and Deitmer, 2006.Zur Nieden R, Deitmer JW. The role of metabotropic glutamate receptors for the generation of calcium oscillations in rat hippocampal astrocytes in situ. Cereb Cortex. 2006;16:676–687. doi: 10.1093/cercor/bhj013. [DOI] [PubMed] [Google Scholar]