

Abstract
Glial reactivity is implicated in CNS repair and regenerative responses. Microglia, the cells responding earliest to axonal injury, produce tumor necrosis factor-α (TNFα), a cytokine with both cytopathic and neuroprotective effects. We have studied activation of hippocampal microglia to produce TNFα in response to transection of perforant path axons in SJL/J mice. TNFα mRNA was produced in a transient manner, peaking at 2 d and falling again by 5 d after lesioning. This was unlike other markers of glial reactivity, such as Mac-1 upregulation, which were sustained over longer time periods. Message for the immune cytokine interferon-γ (IFNγ) was undetectable, and glial reactivity to axonal lesions occurred as normal in IFNγ-deficient mice. Microglial responses to lesion-induced neuronal injury were markedly enhanced in myelin basic protein promoter-driven transgenic mice, in which IFNγ was endogenously produced in hippocampus. The kinetics of TNFα downregulation 5 d after lesion was not affected by transgenic IFNγ, indicating that IFNγ acts as an amplifier and not an inducer of response. These results are discussed in the context of a regenerative role for TNFα in the CNS, which is innately regulated and potentiated by IFNγ.
Keywords: fascia dentata, axonal lesioning, microglia, tumor necrosis factor-α, transgenic mice, oligodendrocytes
Microglia and astrocytes react to CNS injury with cascades of cellular reactivity that include secretion of soluble mediators, leading to promotion of regeneration and repair. Frequently, this involves interaction with cells of the immune system (Wekerle et al., 1986; Raivich et al., 1998). Intercellular interactions are mediated at least in part by cytokines, prominent among which are the immune cytokine interferon-γ (IFNγ) and the more widely produced tumor necrosis factor-α (TNFα). IFNγ orchestrates glial reactivity, in particular through induction of TNFα (Renno et al., 1995). Stimulation of glial cells in vitro by IFNγ induces major histocompatibility complex (MHC) and adhesion molecule upregulation, proliferation (astrocytes), and production of cytokines and other soluble mediators, including TNFα (Fontana et al., 1984; Frei et al., 1987; Hayes et al., 1987; Yong et al., 1991; Aloisi et al., 1992a; Merrill et al., 1993; Sebire et al., 1993; Merrill and Benveniste 1996). Transgenic overexpression of IFNγ can induce demyelinating pathology and gliosis (Corbin et al., 1996;Horwitz et al., 1997; Renno et al., 1998). However, gliosis is also inducible in IFNγ-deficient animals (Rostworowski et al., 1997), and experimental autoimmune encephalomyelitis (EAE) can be induced, with production of TNFα, in mice lacking IFNγ (Krakowski and Owens, 1996). TNFα is associated with inflammation and neurotoxicity (Selmaj and Raine, 1988; Rothwell and Luheshi, 1996; Barone et al., 1997;Korner et al., 1997; Probert et al., 1997; Taupin et al., 1997;Stalder et al., 1998). TNFα also stimulates the production of growth factors in astroglial cells (Aloisi et al., 1992a,b; Lee et al., 1993;Shafit-Zagardo et al., 1993; Brodie, 1996) and may play a neuroprotective role. Ischemic neuronal degeneration was exacerbated in mice lacking TNFα receptors (Bruce et al., 1996), direct protective effects on neurons in culture have been described (Cheng et al., 1994), and in vivo administration of TNFα ameliorates EAE (Liu et al., 1998). These observations highlight the need to understand the relative roles of IFNγ and TNFα.
We have examined these issues by studying microglial response at a site of anterograde axonal and terminal degeneration in the CNS. Transection of perforant path (PP) afferents from the entorhinal cortex induces degeneration of PP fibers and synaptic terminals in the molecular layer of the fascia dentata of the hippocampus (Matthews et al., 1976a). This induces glial activation (Fagan and Gage,, 1990, 1994; Jensen et al., 1994, 1997, 1999) and leads to reactive sprouting and synaptogenesis in the denervated zones (Matthews et al., 1976b; Steward and Vinsant, 1983; Fagan and Gage, 1990, 1994; Frotscher et al., 1997). Glial reactivity in the PP lesion model is immune-independent (Fagan and Gage, 1994; Finsen et al., 2000). We find that reactive microglia produce TNFα with a transient time course that is distinct from kinetics for other markers of reactivity. Microglial reactivity was normal in IFNγ-deficient mice, and IFNγ expression was not detected by RT-PCR in hippocampus of normal mice. Nevertheless, microglial reactivity and TNFα production were significantly enhanced in transgenic mice that expressed IFNγ in hippocampus. Our data argue for an innately regulated program of TNFα production in the CNS that is subject to amplification by immune-derived IFNγ.
MATERIALS AND METHODS
Mice
The myelin basic protein (MBP)–IFNγ transgenic mice were homozygotes of the A519 line, backcrossed for six generations onto the SJL/J background, as described by Renno et al. (1998). In these mice the IFNγ transgene is constitutively expressed in the CNS under the control of a 1.3 kb MBP promoter. The transgenic mice develop and breed normally. Unmanipulated MBP–IFNγ transgenic mice show no spontaneous pathology, unlike other MBP promoter-driven IFNγ transgenic mice (Corbin et al., 1996; Horwitz et al., 1997). This has been attributed to thresholds for effect (Renno et al., 1998) and allows us to test the concomitant roles of IFNγ and other stimuli. There is expression of IFNγ mRNA in the spinal cord, and very low levels of TNFα mRNA are also detectable (Renno et al., 1998). BALB/c-backcrossed IFNγ-deficient GKO mice (Dalton et al., 1993) were originally obtained from Genentech (San Francisco, CA) and were maintained in our facility. SJL/J mice were purchased from Harlan Sprague Dawley (Indianapolis, IN) or Bomholtgaard (Skensved, Denmark). Animal breeding and experiments were conducted according to National Danish Animal Care Committee and Canadian Council on Animal Care guidelines, as administered by the McGill University Animal Care Committee.
Surgical procedures
Mice were subjected to wire knife-lesioning of the perforant path projection arising from the entorhinal cortex to terminate in the hippocampus. For lesioning the mice were anesthetized, and the perforant path was transected with a stereotaxically inserted wire knife as described by Jensen et al. (1999). Control mice were either unoperated or sham-operated, treated the same way as the lesioned animals except that the wire knife was not inserted into the brain. The contralateral, unoperated hippocampus also served as a control.
Histology
At survival times of 24 hr, 48 hr, 5 d, and 10 d after lesion, mice were anesthetized, and their brains were removed and snap-frozen in CO2 snow and processed histologically as described by Gregersen et al. (2000) and Jensen et al. (1999). Nonradioactive in situ hybridization (ISH) was used to visualize cellular TNFα mRNA expression (Gregersen et al., 2000). Parallel sections were stained with a modification of the Fink-Heimer silver impregnation method described by Hjorth-Simonsen (1970) to demonstrate argyrophilic, degenerating axons and terminals, or they were stained with toluidine blue as a general cell stain. Immunocytochemical staining for the microglial surface antigen complement receptor type 3 (Mac-1) (Perry et al., 1985) was performed as described previously (Jensen et al., 1999). Mice with inadequate lesions as shown by Fink-Heimer staining were excluded from further analysis. ISH for MBP mRNA was performed as described [Jensen et al. (2000), and see below]. Between 3 and 10 animals per group (e.g., at each time point per treatment) were processed, and every tenth section through the hippocampus was examined for each histological analysis.
In situ hybridization
Probes. TNFα mRNA was detected with a probe mixture composed of two alkaline phosphate (AP)-labeled probes (probe I: 5′-CTTCTCATCCCTTTGGGGACCGATCACC-3′; probe II: 5′-C GTA GTC GGG GCA GCC TTG TCC CTT GAA-3′) complementary to bases 305–332 and 570–597 of murine TNFα cDNA, respectively (Pennica et al., 1985). MBP mRNA was detected with an AP-labeled probe (5′-XTCT- CTGGGGCAGGGAGCCATAATGGGTAG T-3′) complementary to bases 56–85 of murine MBP cDNA (Takahashi et al., 1985). A probe specific for the “house-keeping” gene glyceraldehyde-3-phosphatedehydrogenase (GAPDH) (5′-XCCTGCTTCACCACCTTCTTGATGATGTCA-3′) complementary to bases 808–833 of murine GAPDH cDNA (Sabath et al., 1990) was used as positive control. All probes were purchased from DNA Technology (Aarhus, Denmark).
ISH procedure. Cryostat sections (16 μm thick) mounted on RNase-free glass slides were immersed in 96% ethanol for 18–24 hr and subsequently left to air dry for 20 min. Five picomoles of the probe were dissolved in 1 ml hybridization buffer consisting of 50% formamide, 20% 20× SSC, 2.5% 40× Denhardt's solution, 10% dextran, and 1% single-stranded DNA and hybridized overnight at 37°C. For posthybridization the sections were rinsed in 1× SSC, 3 × 30 min at 55°C, then transferred to Tris-HCl, pH 9.5, for 2 × 10 min at room temperature. Thereafter the sections were rinsed for 2 × 15 min in Tris-HCl, pH 9.5, and subjected to AP development. Substrates for the color reaction were nitro-blue tetrazolium (Sigma, Poole, UK) and 5-bromo-4-chloro-3-indoyl phosphate (Sigma). Development was performed in the dark at room temperature for up to 50 hr and arrested by immersing the sections in water. The sections were coverslipped with Aquamount (Merck, Darmstadt, Germany) and stored in the dark. Some sections were counterstained with hematoxylin before coverslipping.
Double labeling for TNFα mRNA and astroglial glial fibrillary acidic protein (GFAP) was performed as described by Gregersen et al. (2000).
Control reactions. Sections hybridized with TNFα probe I or II alone displayed identical but weaker hybridization signal compared with sections hybridized with the TNFα probe mixture. For additional controls, sections were incubated with the hybridization buffer alone, an excess (×100) of unlabeled TNF probe mixture, or hybridized subsequent to treatment with ribonuclease A (50 μg/ml; Pharmacia). None of these controls displayed specific hybridization signal (see Fig. 2G). Sections hybridized with the MBP or GAPDH probe showed a distinctly different hybridization signal. Specificity of MBP ISH was confirmed similarly.
Fig. 2.

Time course and cellular distribution of TNFα mRNA expression in PP-lesioned hippocampus. In situhybridization was used to show induction of TNFα mRNA in fascia dentata at various times after PP lesioning.A–G, SJL/J;H–J, A519 MBP–IFNγ transgenic.A, Low-power photomicrograph showing induction of TNFα mRNA in scattered cells (arrows) in the denervated molecular layer. B, C, Higher-power photomicrographs from the same section as shown in A. The TNFα mRNA-expressing cells (arrows inB) have small, round to oval microglia-like nuclei and occasionally elaborate process-like extensions.Arrowheads in C indicate a closely apposed cell “doublet.” D–F, Time course of TNFα induction. D–F show granule cells (left) and molecular layer (right) from mice lesioned 24 hr, 48 hr, and 5 d previously. G, Control showing a section equivalent to that in E but pretreated with RNase A before ISH.H, Fascia dentata from a PP-lesioned transgenic mouse (same magnification as A); I andJ show granule cells and molecular layer (as inD–G) from mice lesioned 48 hr and 5 d previously (same magnifications asD–G). Arrows indicate TNFα mRNA-expressing cells. g, Granule cell layer;mol, molecular layer. Scale bars: A, H, 100 μm; B, 125 μm; C, 250 μm; D–G,I, J, 50 μm.
RT-PCR analysis. Mice (10–13 per group) were transcardially perfused with ice-cold PBS, the brains were removed, and the hippocampi were dissected out under a dissecting microscope. The hippocampi were immediately snap-frozen in liquid nitrogen and stored at −80°C until further processing. Total RNA was purified from the dissected hippocampi using Trizol RNA isolation reagent (Life Technologies, Burlington, Ontario, Canada) according to the manufacturer's protocol. For PCR analysis, equivalent amounts of total hippocampal RNA were subjected to an RT protocol. Briefly, 3 μg mRNA was added to a tube containing 400 U Moloney murine leukemia virus-RT (Life Technologies), 10 mm of each dNTP (Pharmacia Biotech, Montreal, Quebec, Canada), 50 pmol random hexamer primer (Boehringer Mannheim, Laval, QC, Canada), 23 U RNA guard RNase inhibitor (Pharmacia Biotech), and a 5× first-strand buffer containing 250 mmTris-HCl, pH 8.3, 375 mm KCl, and 15 mmMgCl2 (Life Technologies).
The PCR conditions that were used were previously optimized for linear amplification to allow direct comparison between samples. Equal amounts of cDNA were amplified using 2.5 U Taq DNA polymerase (Life Technologies), 10 mm of each dNTP (Pharmacia Biotech), 50 pmol of each primer, and a 10× PCR buffer mixture containing 500 mm KCl, 100 mm Tris-HCl, pH 8.3, 15 mmMgCl, and 0.1% gelatin (Life Technologies). The primers that were used were as described by Renno et al. (1995), except for IFNγ: IFNγ sense primer 5′-ACACTGCATCTTGGCTTTGC-3′, IFNγ antisense primer 5′-CGACTCCTTTTCCGCTTCCT-3′, TNFα sense primer 5′-AGCACAGAAAGCATGATCCG-3′, TNFα antisense primer 5′-CAGAGCAATGACTCCAAAGT-3′. The primers for β-actin as an internal control were sense 5′-TGGGTCAGAAGGACTCCTATC-3′ and antisense 5′-CAGGCAGCTCATAGCTCTTCT-3′ (Renno et al., 1995). PCR was performed in a Programmable Thermal Controller-100 (MJ Research) for 28 cycles (IFNγ) (denaturation 1 min at 94°C, annealing 1 min at 60°C, extension 1 min at 72°C) or 34 cycles (TNFα) (denaturation 1 min at 95°C, annealing 1 min at 60°C, extension 1 min at 72°C). Fifty microliters per sample of PCR amplification products (450 bp for IFNγ, 289 bp for TNFα, 650 bp for β-actin) were run in 1.5% agarose gels in Tris-acetate-EDTA buffer and visualized by ethidium bromide or SYBR Green (Molecular Probes, Eugene, OR) staining. For quantitation, gels were visualized by Fluorimager (Molecular Dynamics, Sunnyvale, CA) and subsequently analyzed using ImageQuant v. 1.1 for Apple Macintosh. After background correction, the signals for IFNγ mRNA and TNFα mRNA were normalized against β-actin mRNA, from the same PCR run. Results are presented as ratios between ipsilateral (lesioned) and contralateral (control, unlesioned) RNA levels from the same mouse.
Densitometry and cell counting. TNFα mRNA-expressing cells in the molecular layer of the dorsotemporal part of the fascia dentata in SJL/J mice that were PP-lesioned 2 d previously were counted systematically using a 100× oil objective and the Cast-Grid microscope system (Olympus). Counting was performed in parallel ISH, blinded sections (n ≥ 8 per animal) with an intersectional distance of 160 μm. Counting was performed in the entire molecular layer because it was impossible to make a clear distinction between the PP-denervated perforant path zones and the inner nondenervated commissural–associational zone in ISH sections. The counting unit was a small, oval to elongated microglial-like nucleus, surrounded by ISH product (see Fig. 2C). Recounts showed <10% variability. An estimate of the cell density was obtained by dividing the total number of counted cells by the total sampling area. Density estimates (cells per millimeters squared) were plotted against the corresponding Fink-Heimer values measured in parallel sections, obtained as outlined by Jensen et al. (1999). For quantitative measurements of the level of microglial Mac-1 immunoreactivity and the extent and size of lesion, Mac-1-stained sections and corresponding Fink-Heimer-stained, blinded sections (n ≥ 6 for each animal) were analyzed as described by Jensen et al. (1999).
Statistical analysis. Wilcoxon matched pairs signed rank sum test was used to determine whether the Mac-1 and Fink-Heimer values for the medial perforant path (MPP) zone were different from the values for the lateral perforant path (LPP) zone. The relation between the Mac-1 and Fink-Heimer values was thereafter described by linear regression, as was the relation between the density of TNF mRNA-expressing cells and Fink-Heimer values, and the slopes of the two Mac-1 regression lines were compared by Student's t test. Comparison of IFNγ transgene expression at days 2 and 5, and comparison of TNFα levels in transgenic and nontransgenic mice at day 2, was performed by the Wilcoxon–Mann–Whitney test for comparison of unpaired samples.
RESULTS
Stereotactic lesioning of perforant path axons in SJL/J mice results in zonally defined axonal and terminal degeneration in the hippocampus (Fig.1A,B). This is accompanied by glial reactivity 2 d after lesion in the perforant path zones of the fascia dentata (Jensen et al., 1999). Microglial reactivity can be visualized as morphological changes, increased Mac-1 staining (Fig. 1C) (Jensen et al., 1999), and cellular proliferation, and these cells also upregulate MHC I and CD45 in the rat (Jensen et al., 1997). Astroglial and oligodendroglial reactivity are detectable as hypertrophy and increased GFAP staining (Jensen et al., 1994) and increased oligodendroglial MBP gene expression (Jensen et al. 2000), with slightly delayed time profile, in the same region. There is a statistically significant linear correlation between the immunohistochemically quantitated microglial Mac-1 reactivity and the extent of neurodegeneration (measured as Fink-Heimer staining) (Jensen et al., 1999). Neuronal, microglial, and oligodendroglial development, morphology, and distribution in fascia dentata and adjacent regions in MBP–IFNγ transgenic mice were identical to those in nontransgenic SJL/J mice (Renno et al., 1998) (data not shown). Neurodegenerative pathology after lesioning in transgenic hippocampus was indistinguishable from that in SJL/J mice, and Nissl and Fink-Heimer staining showed the same pattern and zonal restriction of changes.
Fig. 1.

Neurodegeneration and microglial reactivity in the perforant path lesion model. A–C, Perforant path-lesioned SJL/J mouse, 5 d after lesioning.A, Nissl staining of hippocampus from a lesioned SJL/J mouse. B, Fink-Heimer-stained degenerating fibers and terminals in the perforant path (pp) zone of the molecular layer of the fascia dentata, the molecular layer of CA3 (m), and stratum lacunosum-moleculare of CA1 (lm) of hippocampus. Same mouse as shown inA but section from a more ventral level.C, Mac-1-stained reactive microglia in the PP zone of dentate molecular layer and denervated zones in CA3 and CA1. Section parallel to that shown in A.D–F, Perforant path-lesioned GKO IFNγ-deficient mouse, 5 d after lesioning. D, Fink-Heimer staining of an incompletely lesioned mouse.E, F, Mac-1 staining of parallel section showing ipsilateral (E) and contralateral (F) fascia dentata.Arrows in A and B indicate the wire knife transection site. The large arrow inD indicates MPP; the small arrowindicates LPP. FD, Fascia dentata; g, granule cell layer; p, pyramidal cells;pp, perforant path zones of molecular layer;ipsi, ipsilateral; con, contralateral. Scale bar: A, B, 500 μm; C, 350 μm; D, 400 μm;E, F, 250 μm.
Transient induction of TNFα mRNA in microglia in PP-lesioned hippocampus
We first characterized the time course and regional and cellular expression of TNFα mRNA in SJL/J mice, using ISH. We found that TNFα was induced transiently in the denervated zones at day 2 (Fig.2A,E) but was expressed at undetectable levels at both earlier and later time points (Fig. 2D,F). The hybridization signal was located in the perinuclear region of the cells and occasionally radiated out in process-like extensions (Fig.2B,C), reminiscent of the activated microglial cells observed in parallel sections, as shown in later figures. ISH-positive cells were occasionally seen as closely apposed doublets, suggestive of recent cell division (Fig. 2C). In double-labeling experiments, no GFAP+cells were ISH positive (Fig.3A), confirming that microglia were the major source of TNFα. The density of TNF mRNA-expressing cells in the denervated zones showed a statistically significant correlation to the extent of neurodegeneration [Y = −7.90 + 8.90 ×, residual SD, Sres = 1.30 (p < 0.05)] (Fig. 3B). TNFα mRNA was not detected around the retrogradely degenerating neurons in the entorhinal cortex or around the transection site where the wire knife entered (data not shown). TNFα mRNA-positive cells were not detected in the contralateral hippocampus (data not shown) or in unlesioned or control tissue (data not shown, and Fig. 2G).
Fig. 3.

TNFα expression by microglia. A(large panel), Photomicrograph showing induction of TNFα by ISH (dark purple) and GFAP staining (brown) in the molecular layer of the fascia dentata of an SJL/J mouse that was lesioned 2 d previously. Two closely apposed ISH-positive cells are indicated by arrows. GFAP+ astrocyte is located at bottom right. The cell body is indicated with a large arrowhead, and a GFAP+ process withsmall arrowheads. GFAP-stained cells did not show ISH product, neither did ISH-positive cells stain for GFAP. Small panel, Focal plane emphasizes astrocyte morphology and processes. Scale bar, 100 μm. B, Graphic illustration of the relationship between the density of Fink-Heimer-stained degenerating axons and terminals and the number of TNFα-expressing cells in the fascia dentata 2 d after perforant path lesioning in six SJL/J mice. A statistically significant linear relation between microglial TNFα expression and degeneration density is evident (p < 0.05, Student's ttest, one-tailed probability).
RT-PCR detection of TNFα in lesioned hippocampus
Results from ISH analysis showed that TNFα expression was transient. To confirm downregulation after peak expression, we used the more sensitive RT-PCR analysis. We microdissected hippocampi from perfused SJL/J mice that had been lesioned 2 and 5 d previously, isolated RNA, and analyzed cytokine mRNA levels by RT-PCR. INFγ message was undetectable in RNA from any CNS tissue in SJL/J, whether lesioned or not (Fig.4A,C). We confirmed the absence of message by 40-cycle PCRs (data not shown). Messenger RNA for TNFα was weakly detectable by 34-cycle RT-PCR in unlesioned hippocampi (Fig. 4A). Levels of TNFα message increased significantly by 2 d after lesioning (Fig.4A,B,D). Because TNFα mRNA was undetectable by ISH in contralateral hippocampus, and Mac-1 induction resulting from degeneration of the crossed tempero-ammonic projection is confined to contralateral CA1, we have previously used the contralateral fascia dentata as a control for normalization of data (Jensen et al., 1999). We confirmed that RT-PCR-detectable increases in TNFα mRNA in unlesioned, contralateral hippocampi were small relative to whole brain (three- to eightfold at 2 d) compared with those seen in lesioned hippocampi (19- to 31-fold at 2 d) (Fig. 4D). Although it introduced a slight underrepresentation of the effect, data from a large number of mice were calculated as ipsilateral/contralateral ratios to normalize the results. Figure 4B shows as much as five- to sevenfold increases in TNFα levels at 2 d after lesion. Guided by the quantitative data showing highest numbers of TNF mRNA-expressing cells in the best lesioned animals (Fig. 3B) and absence of TNF mRNA-expressing microglia at day 5 (Fig.2F,J), we considered animals with a fold increase of ≤2.5 to be suboptimally lesioned (day 2) or their microglia to have downregulated TNF gene expression. The median value attained 3.6 for well-lesioned mice (Fig. 4B). TNFα levels fell by 5 d after lesion to, or close to, baseline levels (fold increase ≤2.5) (Fig.4A,B).
Fig. 4.
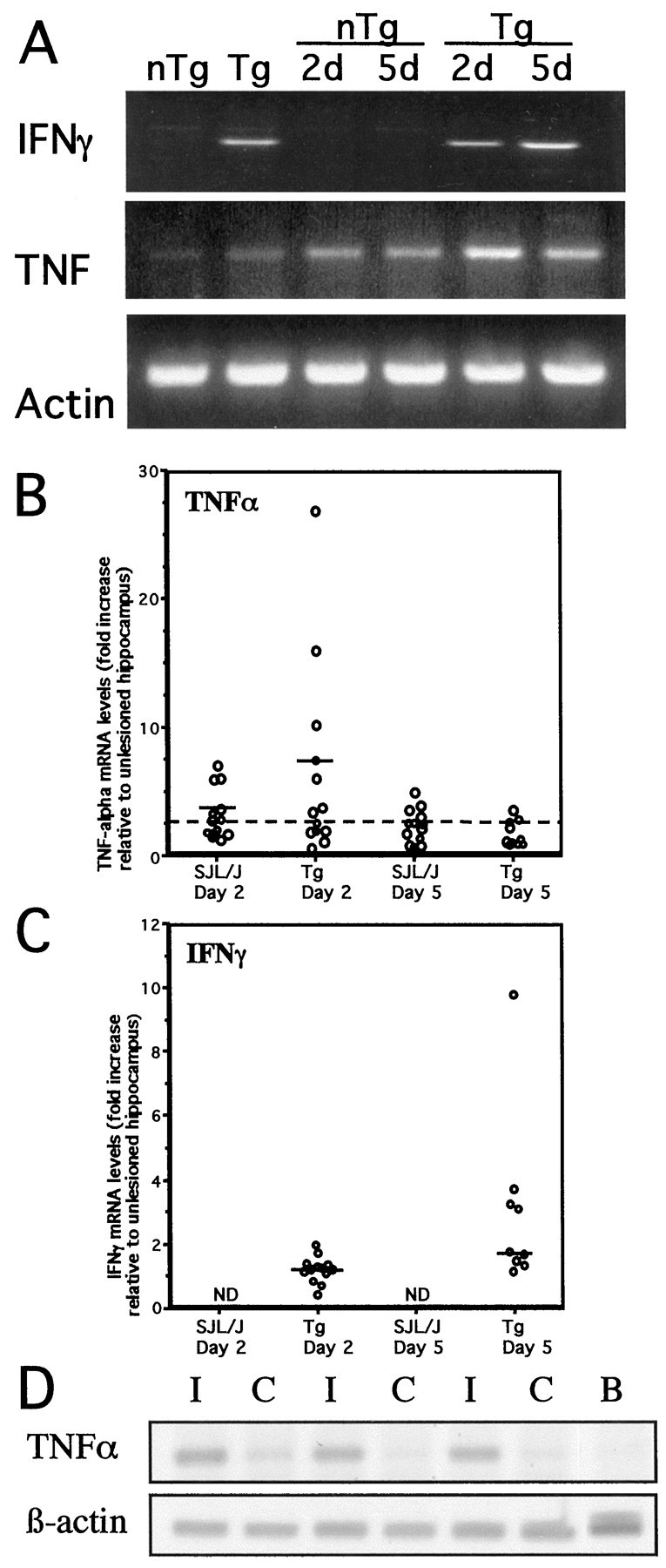
Cytokine levels in PP-lesioned hippocampus.A, Ethidium bromide-stained gels showing PCR-amplified IFNγ, TNFα, and β-actin cDNA from hippocampi of individual mice. IFNγ message was undetectable in both unlesioned and lesioned SJL/J mice (A, top gel, lanes 1,3, 4). IFNγ message was detected at roughly equivalent levels in both unlesioned and 2 day-lesioned MBP–IFNγ transgenic (Tg) mice (A,top gel, lanes 2, 5). TNFα mRNA was induced transiently in nontransgenic and MBP–IFNγ transgenic mice at day 2 (A, middle gel,lanes 3, 5). β-actin mRNA levels (bottom gel) were equivalent in all samples, indicating equal RNA input. B, C, Fluorimager quantitation of data from these and other experiments is shown as fold increase relative to unlesioned contralateral hippocampi for each mouse. Each point represents the relative cytokine level, normalized to β-actin, for one mouse. Data for IFNγ in C include samples independently analyzed for TNFα in B. Values for fold increase ≤2.5 are considered to be at or below baseline level, indicating either suboptimal lesioning (day 2) or downregulation to baseline levels (day 5). Medians for well-lesioned day 2 mice are indicated by a horizontal line. Wilcoxon–Mann–Whitney test shows induction of higher TNF levels in Tg than nTg mice at day 2 (p < 0.5, one-tailed probability). Wilcoxon–Mann–Whitney test also reveals a statistically significant increase in IFNγ levels from day 2 to day 5 after lesion in transgenic mice (p < 0.001, one-tailed probability), although some of the animals in the day 5 group appear to be suboptimally lesioned. The horizontal lines indicate median values. ND, None detected.D, Fluorimager image of a gel stained with SYBR Green, showing TNFα and β-actin amplimers after 30-cycle RT-PCR of RNA from paired ipsilateral (lesioned, I) and contralateral (unlesioned, C) hippocampi from three different SJL/J mice, 2 d after surgery. RNA from unlesioned brain was analyzed as a control (B).
Effect of IFNγ on TNFα expression
We then examined the induction and progression of the TNFα response in SJL/J-backcrossed MBP–IFNγ transgenic mice. Expression of IFNγ in the unlesioned hippocampus of transgenic mice was confirmed by RT-PCR (Fig. 4A). The time course of appearance and the cellular source of TNFα mRNA in hippocampus of lesioned MBP–IFNγ transgenic mice was determined by nonradioactive ISH and identical to SJL/J mice (Fig. 2, compareH--J, D–F). At 2 d after lesion, mRNA-expressing cells were morphologically identifiable as microglia in transgenic mice, similar to SJL/J (Fig. 2compare B, C; shown for SJL/J only). TNFα mRNA became undetectable at 5 d in both types of mice (Fig.2F,J).
RT-PCR-detectable TNFα mRNA levels were marginally higher in the hippocampus of transgenic mice than in nontransgenic mice, consistent with the original description of these mice (Fig.4A). This difference was slight and did not constitute a significant bias to lesion effects. Two days after axonal lesioning, an up to 10-fold or greater increase in TNFα mRNA was measured in the denervated hippocampus, levels that were never attained in nontransgenics (Fig. 4B). Wilcoxon–Mann–Whitney test on well-lesioned mice [fold increase >2.5; median of 7.3 for transgenics (n = 7) and of 3.6 for nontransgenic mice (n = 7), showed statistically higher TNF levels in transgenic than nontransgenic mice (p < 0.05, one-tailed probability)]. The elevated TNFα production at 2 d was transient and by 5 d levels had declined to, or close to, baseline levels (Fig. 4A,B). This decrease agreed with the ISH data in Figure 2, which showed that TNFα mRNA was induced transiently in microglial cells in the denervated areas in transgenic mice at day 2, becoming undetectable at day 5. Expression of IFNγ in the hippocampus therefore promoted a striking elevation of microglial TNFα production in response to anterograde axonal and terminal degeneration, but the transient nature of this TNFα production was unaffected by IFNγ.
Enhanced IFNγ expression in lesioned hippocampus of MBP–IFNγ transgenic mice
IFNγ was readily detectable by RT-PCR from uninjured hippocampi of transgenic mice (Fig. 4A). Levels of expression increased after PP lesion (Fig.4A,C). Strikingly, the kinetics of IFNγ upregulation were distinct from those for TNFα. IFNγ mRNA levels, in RNA samples for which elevated TNFα levels are shown in Figure 4B, barely increased over control at 2 d (median of 1.2, maximum of 1.95), but increased up to 9.7-fold in well lesioned mice at 5 d after the lesion (Fig. 4C) (Wilcoxon–Mann–Whitney test, p < 0.01, one-tailed probability).
Equivalent microglial reactivity in IFNγ-deficient mice
The complete absence of detectable IFNγ signal in SJL/J mice, even in mice with complete PP lesions (inferred from high TNFα levels), suggested that this cytokine might be dispensable for glial response. We confirmed this by stereotactically lesioning axons in BALB/c-backcrossed IFNγ-deficient GKO mice (Fig.1D,E). The PP lesion is functionally equivalent in BALB/c and SJL/J mice. Consistent with the lack of IFNγ in normal animals, both the extent of neurodegeneration (Fig. 1D) and the microglial reactivity in the hippocampus of lesioned GKO mice (Fig. 1E) were indistinguishable from responses in normal mice (Fig. 1, compareB, D, and C, E).
IFNγ enhances microglial reactivity in transgenic mice
IFNγ is therefore not required for glial reactivity to neurodegenerative injury and did not affect the time course of TNFα expression induced by axotomy. Nevertheless, this cytokine clearly affected the level of glial response. There was a striking difference in the strength of the denervation-induced microglial morphological changes between lesioned transgenic and nontransgenic mice (Fig.5, compare C, Ewith D, F). In transgenic mice, the overall glial reaction was more pronounced in all aspects, especially for 5 d post-lesion animals (Fig. 5F). The increase in Mac-1 immunoreactivity was higher compared with nontransgenic controls, being visible to the naked eye on some slides, microglial processes were more extensively branched, and the number of cells in the denervated zones was clearly elevated (Fig. 5). In the denervated zones, the microglial cells seemed to form a continuous conglomerate, and without counterstain of their nuclei the individual cells were hard to distinguish from each other (Fig.5F).
Fig. 5.

Mac-1 reactivity in SJL/J and MBP–IFNγ transgenic mice. Mac-1 staining of fascia dentata (granule cells and molecular layer) from mice before and after PP lesioning.A, C, E, Unlesioned SJL/J (A) and SJL/J at 2 d (C) and 5 d (E) after lesioning; B, D, F, unlesioned MBP–IFNγ transgenic (B), and MBP–IFNγ transgenic, at 2 d (D) and 5 d (F) after lesioning. ca, Commissural associational zone; g, granule cell layer;lpp, lateral perforant path; mpp, medial perforant path. Scale bar, 100 μm. G shows a graphic illustration of the relationship between the density of Fink-Heimer (FH)-stained degenerating axons and terminals and the density of microglial Mac-1 immunoreactivity in the fascia dentata 5 d after perforant path lesioning in MBP–IFNγ (•) transgenic and SJL/J (○) mice. The graphs portray statistically significant linear relations between microglial Mac-1 immunoreactivity and degeneration density (FH) in both transgenic and nontransgenic, in that the higher the degeneration density the higher the microglial reactivity. Comparison of the slopes of the regression lines shows that microglial Mac-1 reactivity is significantly higher in MBP–IFNγ transgenic than in SJL/J mice (p < 0.01, Student'st test, one-tailed probability).
We confirmed a statistically significant linear relation between the density of microglial Mac-1 immunoreactivity and Fink-Heimer staining in both transgenic mice [Y = −1.50 + 2.70 ×, residual SD, Sres = 0.13 for MPP (p < 0.001) and nontransgenic SJL/J mice (Y = 0.17 + 0.87 ×,Sres=0.16 for MPP (p < 0.001)] (Jensen et al., 1999) (Fig.5G). Notably, the slope of the regression line was significantly steeper for transgenic than for nontransgenic SJL/J mice (p ≪ 0.001, one-sided Student's t test) (Fig. 5G). No difference in microglial reactivity in the MPP and the LPP zones was observed in transgenic or nontransgenic mice (p ≫ 0.1; Wilcoxon matched pairs signed rank sum test, two-tailed probability; data not shown).
Increased MBP expression in lesioned hippocampus
The glial response to axotomy extends at later time points to oligodendrocytes. The oligodendrocyte response to axonal lesioning and terminal degeneration that was previously shown by ISH for MBP mRNA in C57Bl/6 mice (Jensen et al., 2000) was confirmed to take place, with similar kinetics, in both transgenic and nontransgenic SJL/J mice. MBP transcription was not discernible 2 d after the lesioning, whereas at 5 d (Fig. 6) the staining density of individual cells was higher than in nonlesioned animals or on the contralateral side, and the number of MBP mRNA-expressing cells in the denervated perforant path zones had significantly increased. Upregulation of MBP was detectable in transgenic and nontransgenic mice (Fig. 6). The kinetics of MBP and IFNγ upregulation were therefore similar, and upregulated IFNγ levels in transgenic mice were likely caused by lesion-induced transcription of the MBP promoter-driven transgene.
Fig. 6.

Axotomy-induced increase in MBP mRNA expression.In situ hybridization for MBP mRNA in the molecular layer of SJL/J (A, C, E) and MBP–IFNγ transgenic (TG) mice (B,D, F). A,B, Unlesioned fascia dentata; C,D, 2 d after lesion; E,F, 5 d after lesion. MBP mRNA is upregulated in oligodendrocytes located within the denervated PP zones of the fascia dentata molecular layer at day 5 in both types of mice. MBP RNA-expressing cells are indicated by arrowheads.lpp, Lateral perforant path; mpp, medial perforant path; ca, commissural associational zone;g, granule cell layer. Scale bar, 50 μm.
DISCUSSION
Our results show that perforant path lesioning in mice leads to transient induction of TNFα mRNA in reactive microglia. Microglial reactivity also included morphological changes and increased Mac-1 expression. Although independent of IFNγ, all of these were enhanced by the presence of this cytokine in MBP–IFNγ transgenic mice. Oligodendroglial reactivity was shown as increased MBP gene transcription in denervated areas in both MBP–IFNγ transgenic and nontransgenic SJL/J mice. The kinetics of TNFα production, whether enhanced by IFNγ or not, differed strikingly from those of microglial or oligodendroglial reactivity. These results suggest innate glial programs of response to injury, some of which are amplified by immune cytokines.
Microglia are the earliest responders to axotomy and are the principal source of TNFα in the CNS (Dopp et al., 1997; Finsen et al., 2000). The kinetics of glial reactivity after perforant path axonal lesioning include early retraction of processes by microglia, followed by proliferation at 1 d, coincident with upregulation of Mac-1/CR3 (Fagan and Gage, 1994). Astrocyte and oligodendroglial responses occur later (Steward et al., 1990; Fagan and Gage, 1994; Jensen et al., 1994,2000). Double-labeling controls failed to detect GFAP+TNFα mRNA-expressing cells, confirming our findings in ischemic brain (Gregersen et al. 2000). Our results therefore show induction of TNFα in microglial cells after axonal lesioning and a pronounced effect of IFNγ on this.
The levels of TNFα detected in reactive hippocampal microglia were relatively low, compared with levels seen in macrophage-like cells in ischemic brain lesions (Gregersen et al. 2000). Additionally, the number of Mac-1-reactive microglia was in all cases greater than the number of ISH-detectable TNFα-producing cells (Figs.2A,H, 5). It is likely that only strongly induced cytokine responses were detected by ISH and RT-PCR, so that only a subpopulation of activated microglia were high expressors of TNFα. Fink-Heimer staining for degeneration showed considerable interanimal variability (Fig. 5), and it was not possible to assess the extent of lesioning in hippocampi from which RNA was isolated for PCR. Nevertheless, TNFα induction was clearly demonstrated.
Transient TNFα transcription may reflect an inherent microglial program. Although Mac-1 expression remain elevated through and beyond day 5, TNFα mRNA levels drop from day 2 to day 5. Microglial reactivity may be induced in response to phagocytosis of debris, but degeneration of myelinated fibers persists for many days (Jensen et al., 1999), so this is unlikely to account for the transient TNFα response. Similarly, short-lived release of injury-related mediators from neurons would not account for continued Mac-1 reactivity. TNFα transcription by microglia/macrophages in EAE and ischemia is also transient (Renno et al., 1995; Gregersen et al. 2000), whereas Mac-1 elevation is more prolonged. Our results show this putative microglial TNFα program to be IFNγ independent.
TNFα has been implicated as both a neuroprotective and a neurotoxic cytokine in ischemia (Bruce et al., 1996; Rothwell and Luheshi 1996;Barone et al., 1997), and in EAE it has been implicated in demyelination and oligodendrocyte death and as a mediator of counter-inflammatory effects (Selmaj and Raine 1988; Korner et al., 1997; Taupin et al., 1997; Liu et al., 1998). In the case of the perforant path lesion, there is no infiltration of leukocytes (Fagan and Gage, 1994), so effects of TNFα in promoting leukocyte and endothelial reactivity are likely not relevant here. TNFα can act through two receptors, the p55 TNFRI and the p75 TNFRII (Hsu et al., 1995; Rao et al., 1995). Although TNFRI was originally thought to induce apoptosis, recent work suggests that this receptor may promote neuronal survival (Gary et al., 1998). Both receptors are expressed by neurons (Blotchkina et al., 1997; Cunningham et al., 1997), and it is possible that one of the roles of transient TNFα production is to promote the axonal sprouting that is a feature of the PP lesion (Fagan and Gage, 1990; Frotscher et al., 1997). The lack of leukocytic infiltrate excludes T cells or macrophages, which can contribute to regenerative responses in the CNS (Moalem et al., 1999), from playing such a role here. Finally, TNFα may act in either autocrine or paracrine mode to induce secondary signals such as other cytokines (Becher et al., 1996). Their kinetic advantage in the overall glial response makes it likely that microglia would influence other cell types. This could include induction of secondary mediators by astrocytes as well as microglia (Gómez-Pinilla et al., 1992; Théry et al., 1992; Shafit-Zagardo et al., 1993;Guthrie et al., 1995, 1997), which could then act on axons or oligodendrocytes.
Whether the increase in MBP-transcribing cells reflects oligodendrocyte proliferation, differentiation of precursors, or upregulation of established cells, it represents a myelinating response that occurs downstream of microglial signaling. We have found a close correlation between onset of sprouting and MBP transcription (Matthews et al., 1976b; Steward and Vinsant 1983; Jensen et al. 2000), which taken together with instances of zone-specific MBP transcription without microglial reactivity argued that sprouting is sufficient to induce MBP (Finsen et al., 2000; Jensen et al. 2000). This would not exclude a role for glial products in promoting myelination, and products of microglial response could directly induce oligodendroglial MBP transcription (Hetier et al., 1988; Fagan and Gage, 1990, 1994;Bartholdi and Schwab, 1998). Whether TNFα itself acts directly on oligodendrocytes may depend on whether these cells preferentially express p55 TNFα receptors (Dopp et al., 1997) or both p55 and p75 (Tchelingerian et al., 1995). Indirect action of microglia could involve induction or promotion of production of FGF-2, CSF-1, or CNTF by astrocytes (Gómez-Pinilla et al., 1992; Théry et al., 1992; Guthrie et al., 1995,1997; Oh and Yong, 1996). Similarly, TNFα, possibly in conjunction with IL-1, could cause the proliferation of astrocytes that is associated with anterograde axonal degeneration (Selmaj et al., 1990; Aloisi et al., 1992a; Fagan and Gage, 1994).
Potentially, effects of IFNγ include amplification of endogenous responses and induction of novel response. The inherent IFNγ independence of in vivo glial responses (Krakowski and Owens 1996; Rostworowski et al., 1997) suggests amplification rather than primary response induction. IFNγ can stimulate microglial cells to upregulate MHC class I and induce MHC class II expression in vitro (Otero and Merrill, 1994). Microglial cells are furthermore stimulated in vitro by IFNγ to produce cytokines (TNFα, IL-1, and IL-6) and other soluble mediators and to enhance phagocytic and cytopathic activity (Frei et al., 1987; Hetier et al., 1988;Merrill et al., 1993; Renno et al., 1995; Merrill and Benveniste, 1996). IFNγ, in addition to being a microglial activator, also induces an astroglial response, with cytokine production (CSF-1, IL-6, IL-8) (Aloisi et al., 1992b; Théry et al., 1992), and proliferation in vitro and reactive gliosis in vivo (Yong et al., 1991). Given the powerful potential of IFNγ to activate microglial cells in vitro, it is consistent that the microglial response to PP lesioning in vivo was much higher in transgenic mice, in which IFNγ was expressed in hippocampus. Similarly, the increased levels of TNFα in transgenic mice must reflect activity of IFNγ on microglia. Importantly, TNFα levels in transgenic mice downregulated with similar kinetics as in nontransgenic mice. So, whether IFNγ or other stimuli induce TNFα, a separate, distinct mechanism then overrides at later stages. The kinetics of IFNγ upregulation were also quite distinct from TNFα. This is more consistent with an amplifying role for IFNγ.
It is uncertain whether there are endogenous sources of IFNγ in the adult CNS. Motor and sensory neurons isolated from the CNS have been reported to express IFNγ immunoreactivity and mRNA, respectively (Olsson et al., 1989; Neumann et al., 1997), but there are no reports of expression in situ in adult animals. Nonetheless, induction of developmentally silenced events may occur during injury or inflammatory responses, so it cannot be excluded that neurons might upregulate IFNγ in response to certain stimuli in adult animals. However, our high-cycle RT-PCR analyses failed to detect it in lesioned hippocampus. It is more likely that IFNγ production within the adult CNS derives from extra CNS sources, probably immune leukocytes such as T and NK cells. The expression of IFNγ receptors by microglia and astrocytes therefore anticipates interaction with immune cells, and this probably reflects evolutionary selection for anti-viral responses (Griffin et al., 1992). Our results suggest that the IFNγ response may also optimize the opportunity for regenerative responses. Inflammation contributes to regeneration in the CNS (Tourbah et al., 1997; Moalem et al., 1999) and likely involves induction of TNFα in glial cells. We have shown that IFNγ amplifies these responses.
Our findings suggest a model for glial reaction to axonal injury and subsequent regenerative response by which TNFα and other cytokine production, amplified by IFNγ, are directed to regeneration. Involvement of these cytokines in inflammatory pathology may be seen as an inadvertent consequence of their overproduction or failure to remove inducing stimuli within a prescribed time.
Footnotes
This study was supported by grants to M.B.J. and B.F. from The Danish Medical Research Council, Retired President Leo Nielsen and Wife Karen Margrethe Nielsen's Foundation, Kong Christian d. X's Foundation, The Foundation to the Advance of Medical Science, The Danish Multiple Sclerosis Society, President Ejnar Jonassen's Foundation, Lily Benthine Lund's Foundation, The Munkemølle Foundation, A. J. Andersen's Foundation, and University of Southern Denmark/Odense University. Research at The Montreal Neurological Institute was supported by grants to T.O. from the Multiple Sclerosis Society of Canada and Medical Research Council–Canada. We thank Grethe Jensen, Dorete Jensen, Jacob Bang Jensen, and Margrethe Krogh Rasmussen (Odense University) for technical assistance, and Albert Meier (Aarhus University) for photography. We thank Lyne Bourbonnière, Grace Chan, and Elise Tran (Montreal Neurological Institute) for help with PCR and analysis.
Correspondence should be addressed to Dr. Bente Finsen, Department of Anatomy and Neurobiology, University of Southern Denmark/Odense University, Winsløwparken 21, Odense C, DK 5000 Denmark. E-mail:finsen@imbmed.usd.dk.
REFERENCES
- 1.Aloisi F, Borsellino G, Samoggia P, Testa U, Chelucci C, Russo G, Peschle C, Levi G. Astrocyte cultures from human embryonic brain: characterization and modulation of surface molecules by inflammatory cytokines. J Neurosci Res. 1992a;32:494–506. doi: 10.1002/jnr.490320405. [DOI] [PubMed] [Google Scholar]
- 2.Aloisi F, Care A, Borsellino G, Gallo P, Rosa S, Bassani A, Cabibbo A, Testa U, Levi G, Peschle C. Production of hemolymphopoietic cytokines (IL-6, IL-8, colony-stimulating factors) by normal human astrocytes in response to IL-1 beta and tumor necrosis factor-alpha. J Immunol. 1992b;149:2358–2366. [PubMed] [Google Scholar]
- 3.Barone FC, Arvin B, White RF, Miller A, Webb CL, Willette RN, Lysko PG, Feuerstein GZ. Tumor necrosis factor-α: a mediator of focal ischemic brain injury. Stroke. 1997;28:1233–1244. doi: 10.1161/01.str.28.6.1233. [DOI] [PubMed] [Google Scholar]
- 4.Bartholdi D, Schwab ME. Oligodendroglial reaction following spinal cord injury in rat: transient upregulation of MBP mRNA. Glia. 1998;23:278–284. doi: 10.1002/(sici)1098-1136(199807)23:3<278::aid-glia10>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 5.Becher B, Dodelet V, Fedorowicz V, Antel JP. Soluble tumor necrosis factor receptor inhibits interleukin 12 production by stimulated human microglial cells in vitro. J Clin Invest. 1996;98:1539–1543. doi: 10.1172/JCI118946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blotchkina GI, Meistrell ME, III, Blotchkina IL, Tracey KJ. Expression of TNF and TNF receptors (p55 and p75) in the rat brain after focal cerebral ischemia. Mol Med. 1997;3:765–781. [PMC free article] [PubMed] [Google Scholar]
- 7.Brodie C. Differential effects of Th1 and Th2 derived cytokines on NGF synthesis by mouse astrocytes. FEBS Lett. 1996;394:117–120. doi: 10.1016/0014-5793(96)00911-8. [DOI] [PubMed] [Google Scholar]
- 8.Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med. 1996;2:788–794. doi: 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- 9.Cheng B, Christakos S, Mattson MP. Tumor necrosis factors protect neurons against metabolic-excitotoxic insults and promote maintenance of calcium homeostasis. Neuron. 1994;12:139–153. doi: 10.1016/0896-6273(94)90159-7. [DOI] [PubMed] [Google Scholar]
- 10.Corbin JG, Kelly D, Rath EM, Baerwald KD, Suzuki K, Popko B. Targeted CNS expression of interferon-gamma in transgenic mice leads to hypomyelination, reactive gliosis, and abnormal cerebellar development. Mol Cell Neurosci. 1996;7:354–370. doi: 10.1006/mcne.1996.0026. [DOI] [PubMed] [Google Scholar]
- 11.Cunningham ET, Jr, Stalder AK, Sanna PP, Liu SS, Bloom FE, Howes EL, Jr, Campbell IL, Margolis TP. Distribution of tumor necrosis factor receptor messenger RNA in normal and herpes simplex virus infected trigeminal ganglia in the mouse. Brain Res. 1997;758:99–106. doi: 10.1016/s0006-8993(97)00169-8. [DOI] [PubMed] [Google Scholar]
- 12.Dalton DK, Pitts-Meek S, Keshav S, Figari IS, Bradley A, Stewart TA. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science. 1993;259:1739–1742. doi: 10.1126/science.8456300. [DOI] [PubMed] [Google Scholar]
- 13.Dopp JM, Mackenzie-Graham A, Otero GC, Merrill JE. Differential expression, cytokine modulation, and specific functions of type-1 and type-2 tumor necrosis factor receptors in rat glia. J Neuroimmunol. 1997;75:104–112. doi: 10.1016/s0165-5728(97)00009-x. [DOI] [PubMed] [Google Scholar]
- 14.Fagan AM, Gage FH. Cholinergic sprouting in the hippocampus, a proposed role for IL-1. Exp Neurol. 1990;110:105–120. doi: 10.1016/0014-4886(90)90055-w. [DOI] [PubMed] [Google Scholar]
- 15.Fagan AM, Gage FH. Mechanisms of sprouting in the adult central nervous system: cellular responses in areas of terminal degeneration and reinnervation in the rat hippocampus. Neuroscience. 1994;58:705–725. doi: 10.1016/0306-4522(94)90449-9. [DOI] [PubMed] [Google Scholar]
- 16.Finsen B, Jensen MB, Lomholt ND, Hegelund IV, Poulsen FR, Owens T. Axotomy-induced glial reactions in normal and cytokine transgenic mice. In: Matsas R, Tsacopoulos M, editors. The functional roles of glial cells in health and disease. Dialogue between glia and neurons. Plenum; New York: 2000. pp. 157–171. [DOI] [PubMed] [Google Scholar]
- 17.Fontana A, Fierz W, Wekerle H. Astrocytes present myelin basic protein to encephalitogenic T-cell lines. Nature. 1984;307:273–276. doi: 10.1038/307273a0. [DOI] [PubMed] [Google Scholar]
- 18.Frei K, Seipl C, Groscurth P, Schwerdel C, Fontana A. Antigen presentation and tumour cytotoxicity by interferon-γ-treated microglial cells. Eur J Immunol. 1987;17:1271–1278. doi: 10.1002/eji.1830170909. [DOI] [PubMed] [Google Scholar]
- 19.Frotscher M, Heimrich B, Deller T. Sprouting in the hippocampus is layer specific. Trends Neurosci. 1997;20:218–223. doi: 10.1016/s0166-2236(96)01018-1. [DOI] [PubMed] [Google Scholar]
- 20.Gary DS, Bruce-Keller AJ, Kindy MS, Mattson MP. Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor. J Cereb Blood Flow Metab. 1998;18:1283–1287. doi: 10.1097/00004647-199812000-00001. [DOI] [PubMed] [Google Scholar]
- 21.Gómez-Pinilla F, Lee JW-K, Cotman CW. Basic FGF in adult rat brain: cellular distribution and response to entorhinal lesion and fimbria-fornix transection. J Neurosci. 1992;12:345–355. doi: 10.1523/JNEUROSCI.12-01-00345.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gregersen R, Lambertsen KL, Finsen B. Brain macrophages and microglia are major sources of tumor necrosis factor alpha (TNF-α) in a murine model of permanent middle cerebral occlusion. J Cereb Blood Flow Metab. 2000;20:53–65. doi: 10.1097/00004647-200001000-00009. [DOI] [PubMed] [Google Scholar]
- 23.Griffin DE, Levine B, Tyor WR, Irani DN. The immune response in viral encephalitis. Semin Immunol. 1992;4:111–119. [PubMed] [Google Scholar]
- 24.Guthrie KM, Nguyen T, Gall CM. Insulin-like growth factor-1 mRNA is increased in deafferented hippocampus: spatiotemporal correspondence of a trophic event with axon sprouting. J Comp Neurol. 1995;352:147–160. doi: 10.1002/cne.903520111. [DOI] [PubMed] [Google Scholar]
- 25.Guthrie KM, Woods AG, Nguyen T, Gall CM. Astroglial ciliary neurotrophic factor mRNA expression is increased in fields of axonal sprouting in deafferented hippocampus. J Comp Neurol. 1997;386:137–148. [PubMed] [Google Scholar]
- 26.Hayes GM, Woodroofe MN, Cuzner ML. Microglia are the major cell type expressing MHC class II in human white matter. J Neurol Sci. 1987;80:25–37. doi: 10.1016/0022-510x(87)90218-8. [DOI] [PubMed] [Google Scholar]
- 27.Hetier E, Ayala J, Denefle P, Bousseau A, Rouget P, Mallat M, Prochiantz A. Brain macrophages synthesize interleukin-1 and interleukin-1 mRNAs in vitro. J Neurosci Res. 1988;21:391–397. doi: 10.1002/jnr.490210230. [DOI] [PubMed] [Google Scholar]
- 28.Hjorth-Simonsen A. Fink-Heimer silver impregnation of degenerating axons and terminals in mounted cryostat sections of fresh and fixed brains. Stain Technol. 1970;45:199–204. doi: 10.3109/10520297009067479. [DOI] [PubMed] [Google Scholar]
- 29.Horwitz MS, Evans CF, McGavern DB, Rodriguez M, Oldstone MB. Primary demyelination in transgenic mice expressing interferon-gamma. Nat Med. 1997;3:1037–1041. doi: 10.1038/nm0997-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-κB activation. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 31.Jensen MB, González B, Castellano B, Zimmer J. Microglial and astroglial reactions to anterograde axonal degeneration: a histochemical and immunocytochemical study of the adult rat fascia dentata after entorhinal perforant path lesions. Exp Brain Res. 1994;98:245–260. doi: 10.1007/BF00228413. [DOI] [PubMed] [Google Scholar]
- 32.Jensen MB, Finsen B, Zimmer J. Morphological and microglial changes in the denervated fascia dentata: correlation with blood-brain-barrier damage and astroglial reactions. Exp Neurol. 1997;143:103–116. doi: 10.1006/exnr.1996.6337. [DOI] [PubMed] [Google Scholar]
- 33.Jensen MB, Hegelund IV, Poulsen FR, Owens T, Zimmer J, Finsen B. Microglial reactivity correlates to the density of the anterogradely degenerating axons and terminals following perforant path denervation of the mouse fascia dentata. Neuroscience. 1999;93:507–518. doi: 10.1016/s0306-4522(99)00139-6. [DOI] [PubMed] [Google Scholar]
- 34.Jensen MB, Poulsen FR, Finsen B. Oligodendrocytes upregulate myelin basic protein gene expression in fields of axonal sprouting in denervated mouse hippocampus. Int J Dev Neurosci. 2000;18:221–235. doi: 10.1016/s0736-5748(99)00091-x. [DOI] [PubMed] [Google Scholar]
- 35.Korner H, Riminton DS, Strickland DH, Lemckert FA, Pollard JD, Sedgwick JD. Critical points of tumor necrosis factor action in central nervous system autoimmune inflammation defined by gene targeting. J Exp Med. 1997;186:1585–1590. doi: 10.1084/jem.186.9.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krakowski ML, Owens T. Interferon-gamma confers resistance to experimental allergic encephalomyelitis. Eur J Immunol. 1996;26:1641–1646. doi: 10.1002/eji.1830260735. [DOI] [PubMed] [Google Scholar]
- 37.Lee SC, Liu W, Dickson DW, Brosnan CF, Berman JW. Cytokine production by human fetal microglia and astrocytes: differential induction by lipopolysaccharide and IL-1β. J Immunol. 1993;150:2659–2667. [PubMed] [Google Scholar]
- 38.Liu J, Marino MW, Wong G, Grail D, Dunn A, Bettadapura J, Slavin AJ, Old L, Bernard CC. TNF is a potent anti-inflammatory cytokine in autoimmune-mediated demyelination. Nat Med. 1998;4:78–83. doi: 10.1038/nm0198-078. [DOI] [PubMed] [Google Scholar]
- 39.Matthews DA, Cotman C, Lynch G. An electron microscopic study of lesion-induced synaptogenesis in the dentate gyrus of the adult rat. I. Magnitude and time course of degeneration. Brain Res. 1976a;115:1–21. doi: 10.1016/0006-8993(76)90819-2. [DOI] [PubMed] [Google Scholar]
- 40.Matthews DA, Cotman C, Lynch G. An electron microscopic study of lesion-induced synaptogenesis in the dentate gyrus of the adult rat. II. Reappearance of morphologically normal synaptic contacts. Brain Res. 1976b;115:23–41. doi: 10.1016/0006-8993(76)90820-9. [DOI] [PubMed] [Google Scholar]
- 41.Merrill JE, Benveniste EN. Cytokines in inflammatory brain lesions: helpful and harmful. Trends Neurosci. 1996;19:331–338. doi: 10.1016/0166-2236(96)10047-3. [DOI] [PubMed] [Google Scholar]
- 42.Merrill JE, Ignarro LJ, Sherman MP, Melinek J, Lane TE. Microglial cell cytotoxicity of oligodendrocytes is mediated through nitric oxide. J Immunol. 1993;151:2132–2141. [PubMed] [Google Scholar]
- 43.Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, Schwartz M. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med. 1999;5:49–55. doi: 10.1038/4734. [DOI] [PubMed] [Google Scholar]
- 44.Neumann H, Schmidt H, Wilharm E, Behrens L, Wekerle H. Interferon γ gene expression in sensory neurons: evidence for autocrine gene regulation. J Exp Med. 1997;186:2023–2031. doi: 10.1084/jem.186.12.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oh LY, Yong VW. Astrocytes promote process outgrowth by adult human oligodendrocytes in vitro through interaction between bFGF and astrocyte extracellular matrix. Glia. 1996;17:237–253. doi: 10.1002/(SICI)1098-1136(199607)17:3<237::AID-GLIA6>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 46.Olsson T, Kristensson K, Ljungdahl A, Maehlen J, Holmdahl R, Klareskog L. Gamma-interferon-like immunoreactivity in axotomized rat motor neurons. J Neurosci. 1989;9:3870–3875. doi: 10.1523/JNEUROSCI.09-11-03870.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Otero GC, Merrill JE. Cytokine receptors on glial cells. Glia. 1994;2:117–128. doi: 10.1002/glia.440110207. [DOI] [PubMed] [Google Scholar]
- 48.Pennica D, Hayflick JS, Bringman TS, Palladino MA, Goeddel DV. Cloning and expression in Escherichia coli of the cDNA for murine tumor necrosis factor. Proc Natl Acad Sci USA. 1985;82:6060–6064. doi: 10.1073/pnas.82.18.6060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perry VH, Hume DA, Gordon S. Immunohistochemical localization of macrophages and microglia in the adult and developing mouse brain. Neuroscience. 1985;15:313–326. doi: 10.1016/0306-4522(85)90215-5. [DOI] [PubMed] [Google Scholar]
- 50.Probert L, Akassoglou K, Kassiotis G, Pasparakis M, Alexopoulou L, Kollias G. TNF-α transgenic and knockout models of CNS inflammation and degeneration. J Neuroimmunol. 1997;72:137–141. doi: 10.1016/s0165-5728(96)00184-1. [DOI] [PubMed] [Google Scholar]
- 51.Raivich G, Jones LL, Kloss CUA, Werner A, Neumann H, Kreutzberg GW. Immune surveillance in the injured nervous system: T-lymphocytes invade the axotomized mouse facial motor nucleus and aggregate around sites of neuronal degeneration. J Neurosci. 1998;18:5804–5816. doi: 10.1523/JNEUROSCI.18-15-05804.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rao P, Hsu KC, Chao MV. Upregulation of NF-κB-dependent gene expression mediated by the p75 tumor necrosis factor receptor. J Interferon Cytokine Res. 1995;15:171–177. doi: 10.1089/jir.1995.15.171. [DOI] [PubMed] [Google Scholar]
- 53.Renno T, Krakowski M, Piccirillo C, Lin J-Y, Owens T. TNFα production by resident microglia and infiltrating leukocytes in the central nervous system of mice with experimental allergic encephalomyelitis. J Immunol. 1995;154:944–953. [PubMed] [Google Scholar]
- 54.Renno T, Taupin V, Bourbonnière L, Verge G, Tran E, De Simone R, Krakowski M, Rodriguez M, Peterson A, Owens T. Interferon-gamma in progression to persistent demyelination and neurological deficit following acute EAE. Mol Cell Neurosci. 1998;12:376–389. doi: 10.1006/mcne.1998.0725. [DOI] [PubMed] [Google Scholar]
- 55.Rostworowski M, Balasingam V, Chabot S, Owens T, Yong VW. Astrogliosis in the neonatal and adult murine brain post-trauma: elevation of inflammatory cytokines and the lack of requirement for endogenous interferon-gamma. J Neurosci. 1997;17:3664–3674. doi: 10.1523/JNEUROSCI.17-10-03664.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rothwell NJ, Luheshi GN. Brain TNF: damage limitation or damaged reputation? Nat Med. 1996;2:746–747. doi: 10.1038/nm0796-746. [DOI] [PubMed] [Google Scholar]
- 57.Sabath DE, Broome HE, Prystowsky MB. Glyceraldehyde-3-phosphate dehydrogenase mRNA is a major interleukin 2-induced transcript in a cloned T-helper lymphocyte. Gene. 1990;16:185–191. doi: 10.1016/0378-1119(90)90087-8. [DOI] [PubMed] [Google Scholar]
- 58.Sebire G, Hery C, Peudenier S, Tardieu M. Adhesion proteins on human microglial cells and modulation of their expression by IL-1 alpha and TNF alpha. Res Virol. 1993;144:47–52. doi: 10.1016/s0923-2516(06)80011-7. [DOI] [PubMed] [Google Scholar]
- 59.Selmaj KW, Raine CS. Tumor necrosis factor mediates myelin and oligodendrocyte damage in vitro. Ann Neurol. 1988;23:339–346. doi: 10.1002/ana.410230405. [DOI] [PubMed] [Google Scholar]
- 60.Selmaj KW, Farooq M, Norton WT, Raine CS, Brosnan CF. Proliferation of astrocytes in vitro in response to cytokines: a primary role for tumor necrosis factor. J Immunol. 1990;144:129–135. [PubMed] [Google Scholar]
- 61.Shafit-Zagardo B, Sharma N, Berman JW, Bornstein MB, Brosnan CF. CSF-1 expression is upregulated in astrocyte cultures by IL-1 and TNF and affects microglial proliferation and morphology in organotypic cultures. Int J Dev Neurosci. 1993;2:189–198. doi: 10.1016/0736-5748(93)90078-r. [DOI] [PubMed] [Google Scholar]
- 62.Stalder AK, Carson MJ, Pagenstecher A, Asensio VC, Kincaid C, Benedict M, Powell HC, Masliah E, Campbell IL. Late-onset chronic inflammatory encephalopathy in immune-competent and severe combined immune-deficient (SCID) mice with astrocyte-targeted expression of tumor necrosis factor. Am J Pathol. 1998;153:767–783. doi: 10.1016/S0002-9440(10)65620-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Steward O, Vinsant SL. The process of reinnervation in the dentate gyrus of the adult rat: a quantitative electron microscopic analysis of terminal proliferation and reactive synaptogenesis. J Comp Neurol. 1983;214:370–386. [Google Scholar]
- 64.Steward O, Torre ER, Phillips LL, Trimmer PA. The process of reinnervation in the dentate gyrus of adult rats: time course of increases in mRNA for glial fibrillary acidic protein. J Neurosci. 1990;10:2373–2384. doi: 10.1523/JNEUROSCI.10-07-02373.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Takahashi N, Roach A, Teplow DB, Prusiner SB, Hood L. Cloning and characterization of the myelin basic protein gene from mouse: one gene can encode both 14 kd and 18.5 kd MBPs by alternate use of exons. Cell. 1985;42:139–148. doi: 10.1016/s0092-8674(85)80109-4. [DOI] [PubMed] [Google Scholar]
- 66.Taupin V, Renno T, Bourbonniere L, Peterson AC, Rodriguez M, Owens T. Increased severity of experimental autoimmune encephalomyelitis, chronic macrophage/microglial reactivity, and demyelination in transgenic mice producing tumor necrosis factor-alpha in the central nervous system. Eur J Immunol. 1997;27:905–913. doi: 10.1002/eji.1830270416. [DOI] [PubMed] [Google Scholar]
- 67.Tchelingerian JL, Monge M, Le Saux F, Zalc B, Jacque C. Differential oligodendroglial expression of the tumor necrosis factor receptors in vivo and in vitro. J Neurochem. 1995;65:2377–2380. doi: 10.1046/j.1471-4159.1995.65052377.x. [DOI] [PubMed] [Google Scholar]
- 68.Théry C, Stanley ER, Mallat M. Interleukin 1 and tumor necrosis factor-alpha stimulate the production of colony-stimulating factor 1 by murine astrocytes. J Neurochem. 1992;59:1183–1186. doi: 10.1111/j.1471-4159.1992.tb08366.x. [DOI] [PubMed] [Google Scholar]
- 69.Tourbah A, Linnington C, Bachelin C, Avellana-Adalid V, Wekerle H, BaronVan Evercooren A. Inflammation promotes survival and migration of the CG4 oligodendrocyte progenitors transplanted in the spinal cord of both inflammatory and demyelinated rats. J Neurosci Res. 1997;50:853–861. doi: 10.1002/(SICI)1097-4547(19971201)50:5<853::AID-JNR21>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 70.Wekerle H, Linington C, Lassmann H, Meyermann R. Cellular immune reactivity within the CNS. Trends Neurosci. 1986;9:271–277. [Google Scholar]
- 71.Yong VW, Moumdjian R, Yong FP, Ruijs TC, Freedman MS, Cashman N, Antel JP. Gamma-interferon promotes proliferation of adult human astrocytes in vitro and reactive gliosis in the adult mouse brain in vivo. Proc Natl Acad Sci USA. 1991;88:7016–7020. doi: 10.1073/pnas.88.16.7016. [DOI] [PMC free article] [PubMed] [Google Scholar]