

Abstract
Background
Pain management is a high priority for patients with rheumatoid arthritis (RA). Despite deficiencies in research data, neuromodulators have gained widespread clinical acceptance as adjuvants in the management of patients with chronic musculoskeletal pain.
Objectives
The aim of this review was to determine the efficacy and safety of neuromodulators in pain management in patients with RA. Neuromodulators included in this review were anticonvulsants (gabapentin, pregabalin, phenytoin, sodium valproate, lamotrigine, carbamazepine, levetiracetam, oxcarbazepine, tiagabine and topiramate), ketamine, bupropion, methylphenidate, nefopam, capsaicin and the cannabinoids.
Search methods
We performed a computer‐assisted search of the Cochrane Central Register of Controlled Trials (CENTRAL) (The Cochrane Library 2010, 4th quarter), MEDLINE (1950 to week 1 November 2010), EMBASE (Week 44, 2010) and PsycINFO (1806 to week 2 November 2010). We also searched the 2008 and 2009 American College of Rheumatology (ACR) and European League against Rheumatism (EULAR) conference abstracts and performed a handsearch of reference lists of articles.
Selection criteria
We included randomised controlled trials which compared any neuromodulator to another therapy (active or placebo, including non‐pharmacological therapies) in adult patients with RA that had at least one clinically relevant outcome measure.
Data collection and analysis
Two blinded review authors independently extracted data and assessed the risk of bias in the trials. Meta‐analyses were used to examine the efficacy of a neuromodulator on pain, depression and function as well as their safety.
Main results
Four trials with high risk of bias were included in this review. Two trials evaluated oral nefopam (52 participants) and one trial each evaluated topical capsaicin (31 participants) and oromucosal cannabis (58 participants).
The pooled analyses identified a significant reduction in pain levels favouring nefopam over placebo (weighted mean difference (WMD) ‐21.16, 95% CI ‐35.61 to ‐6.71; number needed to treat (NNT) 2, 95% CI 1.4 to 9.5) after two weeks. There were insufficient data to assess withdrawals due to adverse events. Nefopam was associated with significantly more adverse events (RR 4.11, 95% CI 1.58 to 10.69; NNTH 9, 95% CI 2 to 367), which were predominantly nausea and sweating.
In a mixed population trial, qualitative analysis of patients with RA showed a significantly greater reduction in pain favouring topical capsaicin over placebo at one and two weeks (MD ‐23.80, 95% CI ‐44.81 to ‐2.79; NNT 3, 95% CI 2 to 47; MD ‐34.40, 95% CI ‐54.66 to ‐14.14; NNT 2, 95% CI 1.4 to 6 respectively). No separate safety data were available for patients with RA, however 44% of patients developed burning at the site of application and 2% withdrew because of this.
One small, low quality trial assessed oromucosal cannabis against placebo and found a small, significant difference favouring cannabis in the verbal rating score 'pain at present' (MD ‐0.72, 95% CI ‐1.31 to ‐0.13) after five weeks. Patients receiving cannabis were significantly more likely to suffer an adverse event (risk ratio (RR) 1.82, 95% CI 1.10 to 3.00; NNTH 3, 95% CI 3 to 13). These were most commonly dizziness (26%), dry mouth (13%) and light headedness (10%).
Authors' conclusions
There is currently weak evidence that oral nefopam, topical capsaicin and oromucosal cannabis are all superior to placebo in reducing pain in patients with RA. However, each agent is associated with a significant side effect profile. The confidence in our estimates is not strong given the difficulties with blinding, the small numbers of participants evaluated and the lack of adverse event data. In some patients, however, even a small degree of pain relief may be considered worthwhile. Until further research is available, given the relatively mild nature of the adverse events, capsaicin could be considered as an add‐on therapy for patients with persistent local pain and inadequate response or intolerance to other treatments. Oral nefopam and oromucosal cannabis have more significant side effect profiles however and the potential harms seem to outweigh any modest benefit achieved.
Plain language summary
Neuromodulators for pain management in rheumatoid arthritis
This summary of a Cochrane review presents what we know from research about the effect of neuromodulators on pain in patients with rheumatoid arthritis.
The review shows that in people with rheumatoid arthritis
‐ Nefopam, topical capsaicin and oromucosal cannabis may improve pain levels
‐ Oromucosal cannabis may slightly improve sleep
‐ No trials were found that evaluated whether neuromodulators affect functional status, quality of life, withdrawals due to inadequate analgesia or depression
We also do not have precise information about serious side effects and complications. Possible side effects of nefopam that were found in the trials include nausea, dry mouth, sweating and feeling tired. However, rare complications have also been reported and include convulsions and cardiac arrhythmias.
Common side effects of topical capsaicin therapy include local skin irritation and burning. More serious allergic reactions are rare but have been reported.
Possible side effects of oromucosal cannabis found in this review were dizziness, fatigue and loss of balance. Readers should be aware that, although not seen in our review, rarer complications including psychosis and suicidal thoughts have also been reported.
What is rheumatoid arthritis and what are neuromodulators?
When you have rheumatoid arthritis your immune system, which normally fights infection, attacks the lining of your joints. This makes your joints swollen, stiff and painful. The small joints of your hands and feet are usually affected first. There is no cure for rheumatoid arthritis at present, so the treatments aim to relieve pain and stiffness and improve your ability to move.
Neuromodulators are broadly defined as substances which alter the way nerves communicate with each other and, consequently, the overall activity level of the brain. By acting on these nerve signals it is thought that these drugs can reduce the amount of pain felt by an individual. Neuromodulators sometimes used in pain management include the anticonvulsant agents (drugs used to prevent seizures); oral, intramuscular or intravenous ketamine; oral or intravenous nefopam; topical capsaicin, cannabis based medications (oral, oromucosal or inhaled); and more recently intra‐articular botulinum toxin.
Best estimate of what happens to people with rheumatoid arthritis who take neuromodulators
Oral nefopam
Pain (higher scores mean worse or more severe pain)
‐ People who took nefopam rated their pain 21 points lower on a scale of 0 to 100 after 2 weeks treatment with nefopam (21% absolute improvement)
‐ People who took nefopam rated their pain as 18 on a scale of 0 to 100 after 2 weeks
‐ People who took a placebo rated their pain as 39 on a scale of 0 to 100
Total adverse events
‐ 27 more people out of 100 experienced an adverse event after 4 weeks treatment with nefopam (absolute difference 27%). These were predominantly nausea (56%), sweating (44%), insomnia (11%), pruritus (11%) and malaise (11%). They completely settled once treatment was ceased
‐ 35 out of 100 people who took nefopam suffered an adverse event
‐ 8 out of 100 people who took a placebo suffered an adverse event
Topical capsaicin
Pain (higher scores mean worse or more severe pain)
‐ People who took capsaicin rated their pain 34 points lower on a scale of 0 to 100 after 2 weeks treatment (34% absolute improvement)
‐ People who took capsaicin rated their pain as 14 on a scale of 0 to 100 after 2 weeks
‐ People who took a placebo rated their pain as 48 on a scale of 0 to 100
Adverse events
No data
Oromucosal cannabis
Pain (higher scores mean worse or more severe pain)
‐ People who took oromucosal cannabis rated their pain 0.7 points lower on a scale of 0 to 5 after 5 weeks treatment
‐ People who took oromucosal cannabis rated their pain as 2.6 on a scale of 0 to 5 after 5 weeks
‐ People who took a placebo rated their pain as 3.3 on a scale of 0 to 5
Quality of sleep
‐ People who received oromucosal cannabis rated their sleep 1.2 points better on a scale of 0 to 10 after 5 weeks treatment (12% absolute improvement)
‐ People who received oromucosal cannabis rated their sleep as 4.6 on a scale of 0 to 10 after 5 weeks
‐ People who received a placebo rated their sleep as 3.4 on a scale of 0 to 10
Total adverse events
‐ 27 more people out of 100 experienced an adverse event after 4 weeks treatment with oromucosal cannabis (absolute difference 27%). These were most commonly dizziness (26%), light headedness (10%), dry mouth (13%), nausea (6%) and falls (6%); they completely resolved once treatment was ceased
‐ 35 out of 100 people who took oromucosal cannabis suffered an adverse event
‐ 8 out of 100 people who took a placebo suffered an adverse event
We looked at all the published scientific literature and identified four drug trials that evaluated different neuromodulators. Two small studies with a total of 52 patients tested the drug nefopam (which is only available in some parts of the world). One trial each tested capsaicin cream (31 participants) and a cannabis based mouth spray (58 participants). Note: use of medicinal cannabis is illegal and therefore unavailable in most countries.
When patients took nefopam they had a greater improvement in pain levels, on average 21 points on a 100 point scale, than those patients who were given a placebo (an inactive substance that has no treatment value). However, patients on nefopam also developed side effects, which mainly consisted of nausea and sweating. Many patients stopped taking the drug because the symptoms were so bad. These studies were performed in the 1980s when treatment for RA was very different to what it is now. Until further, larger studies are carried out to better assess nefopam, with many other effective pain relieving medications on the market, the risks of harm seem to outweigh the benefit arguing against its routine use.
In the one small study testing capsaicin cream (0.025%) in patients with persistent knee pain, patients also had better pain relief with capsaicin cream than for those given a placebo cream. On average, patients receiving the active treatment improved by 34 more points (out of 100) than the control group. The most common side effect was a local burning sensation at the site that the cream was applied. This was usually mild but was moderate to severe in a few patients. About 50% of patients who use capsaicin cream on their skin will develop this local burning but only 2 in 100 will stop treatment because of this.
The one small study of the cannabis based mouth spray Setivax also showed reduced pain levels in patients, to a small extent. Pain was measured on a 0 to 5 point scale and there was an improvement in patients receiving Setivax of 0.74 points. About one in every three patients taking this medication developed a side effect, which was commonly dizziness (26%), dry mouth (13%) or light headedness (10%). Although this is only one study, weighing up these side effects and the minimal benefit on pain levels, until further trials are carried out we cannot recommend the use of this medication.
Summary of findings
Background
Description of the condition
Rheumatoid arthritis (RA) is a systemic, autoimmune, inflammatory arthritis characterised by progressive synovitis with resultant joint destruction, functional disability and significant pain. Even with significant advances in disease modifying therapy, many patients continue to experience pain and would like to see an improvement in their pain management (Heiburg 2002). Pain results in a significant burden of suffering and impacts on overall well being, sleep, mood, energy and quality of life (Cooper 2003; Skevington 1998).
In recent times there has been growing interest and substantial progress in our understanding of the underlying mechanisms of chronic pain. However, deciding on an optimal pain management strategy for an individual with RA is not easy and, when initial standard treatments for pain fail, physicians may consider a broader scope of pharmacologic agents. With the unravelling of the complex interplay between the neural and immune systems in the genesis of pain, neuromodulators have received increasing attention as analgesics. The evidence base for their use, however, is less clear.
Description of the intervention
Neuromodulators are broadly defined as substances which alter nerve impulse transmission. Those sometimes used in pain management include anticonvulsant agents, ketamine, nefopam, capsaicin and cannabinoids. More recently, botulinum toxin (BT) has also been trialled in a variety of pain states (Singh 2011; Waseem 2011). However, with widely varying mechanisms of action their role in pain management for patients with RA is not yet clear.
Anticonvulsants have several pharmacological actions that can interfere with processes involved in neuronal hyperexcitability, either by decreasing excitatory or increasing inhibitory neuronal transmission. The commonly used anticonvulsant agents include gabapentin, pregabalin, phenytoin, sodium valproate, lamotrigine, carbamazepine, levetiracetam, oxcarbazepine, tiagabine and topiramate. Their use is not without risk however, with serious adverse effects being reported including deaths from haematological reactions (Franceschi 1988).
Neuromodulation resulting in analgesia may also occur via inhibition of N‐methyl‐D‐aspartate (NMDA) receptors. Clinically available NMDA antagonists include dextromethorphan and ketamine, although they have a variety of neuropsychiatric side effects that limit their clinical usefulness (Finnerup 2007). Nefopam is another centrally acting agent, used mainly in Europe and New Zealand as a potent non‐opioid analgesic to improve postoperative pain (Kakkar 2009). It has also been used in rheumatic disease and other musculoskeletal disorders in the United Kingdom. Nefopam is generally considered to be safe and well tolerated with reported adverse effects most commonly including drowsiness, nausea and vomiting, and sweating. However, potentially more serious adverse effects can occur including confusion, anaphylaxis and tachycardia (Evans 2008).
Although controversial, cannabinoids are another neuromodulator that may be inhaled, taken orally or administered via the oromucosal route. In the majority of countries, the use of medicinal cannabis is illegal and hence standardised products are not available at all. In Canada, however, four cannabinoid agents are available and include naturally grown cannabis (available under the Medical Marihuana Access Regulations), a cannabis buccal spray (Sativex), nabilone (Cesamet), and dronabinol (Marinol). Any beneficial effects may however be offset by potentially serious harms including cognitive deficits, memory impairment, motor disturbances and psychotropic effects (Lynch 2005; Walker 2001).
Neuromodulation may also be achieved with drugs that act more peripherally. Capsaicin is an ingredient in chilies that inhibits the neuropeptide pain mediator named substance P. Topical application of either 0.025% or 0.075% preparations has been shown in placebo controlled studies to relieve joint pain and tenderness in patients with arthritis (Altman 1994; McCarthy 1992) as well as postherpetic neuralgia, diabetic neuropathy, trigeminal neuralgia and reflex sympathetic dystrophy (Morris 1997). Capsaicin containing sprays are also used as riot control and personal defence agents. Adverse events commonly include a minor cutaneous burning sensation at the site of local application that is usually well tolerated. Cough has also been reported in about 8% of patients using the stronger 0.075% cream. Eye exposure leads to intense tearing, pain, conjunctivitis and blepharospasm.
Botulinum toxin is a potent neurotoxin produced by Clostridium botulinum that can block acetylcholine release peripherally at the neuromuscular junction and interrupt neuromuscular transmission. Botulinum toxin has been shown to have an analgesic effect when administered via injection in patients with focal peripheral neuropathic pain and allodynia (Ranoux 2008), diabetic neuropathy (Yuan 2009) and rheumatoid arthritis (Singh 2008). Known complications include local pain at the site of injection, muscle weakness, fatigue, fever, dry mouth and ptosis, which can also occur one to two weeks after injection. All side effects are temporary and reversible.
How the intervention might work
Patients with RA are known to manifest a generalized increase in pain perception and in effect become 'pain sensitized'. In simple terms, a complex interaction of nerve signals is transmitted by neurochemicals which are stimulatory from the site of the pathology and inhibitory from the central nervous system (CNS). The cumulative result is an individual's perception of pain. A loss of synaptic inhibition in the spinal dorsal horn or excessive excitatory stimulation may occur for a variety of reasons and is believed to contribute significantly to pain pathology. In chronic joint inflammation, enhanced pain perception is the result of the impact of proinflammatory cytokines on both peripheral and central pain pathways (Edwards 2009). One of the reasons for this is that pain receptors become sensitized in the presence of ongoing inflammation thereby reducing their threshold for neuronal firing. Drugs that are capable of influencing central sensitization may, therefore, be beneficial in patients with RA. Many therapeutic neuromodulators are used with the aim of reducing the excitability of neurons in the peripheral nervous system or CNS by modulating the activity of ion channels (gabapentin, pregabalin, carbamazepine and capsaicin) or by mimicking and enhancing endogenous inhibitory mechanisms (dextromethorphan and ketamine).
Anticonvulsant drugs have been used in pain management for over 50 years although their precise mechanisms of action remain unclear. With wide variation in chemical structure, mechanism of action and therapeutic properties, they are known to depress abnormal neuronal discharges and raise the threshold for propagating neural impulses. In addition, they may also enhance gamma‐aminobutyric acid (GABA) inhibition, stabilize neuronal cell membranes and activate N‐methyl‐D‐aspartate (NMDA) receptor sites (Davis 2007; Woolf 1991). Gabapentin and pregabalin are two newer agents that have shown efficacy in fibromyalgia, painful diabetic neuropathy, postherpetic neuralgia (Gilron 2006), mixed neuropathic pain conditions, phantom limb pain and spinal cord injury (Dworkin 2007). Both agents act at the α2δ subunit of the voltage‐gated calcium channels resulting in a reduction in the release of several neurotransmitters including glutamate, norepinephrine, serotonin, dopamine and substance P. Carbamazepine has shown some efficacy in small studies in the treatment of trigeminal neuralgia and related syndromes (Wiffen 2005). It is thought to work by blocking voltage sensitive sodium channels thereby reducing the ability of the neurone to fire at high frequency (Chong 2000). Lamotrigine is another new anticonvulsant that blocks voltage dependent sodium channels and inhibits glutamate release (Tremont‐Lukats 2000). It has not been shown to be significantly effective in diabetic neuropathy, intractable neuropathic pain, spinal cord injury or trigeminal neuralgia (Wiffen 2007). Topiramate potentiates GABA responses, significantly increasing central nervous system GABA levels. It is an effective drug for migraine and has been variably effective in studies of neuropathic pain in cancer patients (Bendaly 2007).
Of the agents working via central sensitization, ketamine is an analogue of phencyclidine with a mechanism of action that is associated with antagonism of NMDA receptors. It currently has a role to play as an adjuvant to opioids in the treatment of refractory pain in cancer patients (Bell 2003), in the treatment of neuropathic pain (Fisher 2000) and in the treatment of acute postoperative pain (Bell 2006). The exact mechanism of action of nefopam's analgesic properties remains unknown. It is a centrally acting antinociceptive compound with supra‐spinal and spinal sites of action (Piercey 1981) that does not bind to opiate receptors (Heel 1980). It inhibits the reuptake of monoamines (Fuller 1993), modulates descending serotoninergic pathways (Hunskaar 1987) and may also interact with the dopaminergic pathway as well (Esposito 1986).
Cannabinoids modulate neural nociceptive mechanisms via two cannabinoid receptors (CB1 and CB2) in supra‐spinal, spinal or peripheral regions (Calignano 1998; Walker 2005). Activation of CB1 receptors is predominantly associated with a dampening down of neuronal excitability whereas activation of CB2 receptors is associated with decreases in immune cell function, including attenuated cytokine release. The presence of cannabinoid receptors in the pain processing areas of the brain, spinal cord and periphery and the ability of cannabinoids to modulate pain systems support the premise that cannabinoids exhibit significant potential as analgesic agents (Pertwee 2001).
Capsaicin is a naturally occurring component of many chilies and has been shown to be effective in neuropathic pain and chronic musculoskeletal pain via depletion of substance P (Mason 2004). Capsaicin binds to nociceptors in the skin causing an initial neuronal excitation and a period of enhanced sensitivity. After repeated applications this is followed by persistent desensitisation, possibly due to the depletion of substance P (Nolano 1999).
Finally, Clostridium botulinum neurotoxins are known to inhibit synaptic vesicle release of neurotransmitters at the nerve terminals and thus reversibly block neurotransmission. However, the precise mechanisms whereby botulinum toxin induces pain relief in patients with arthritis remains unknown. In vitro studies have demonstrated the ability of botulinum toxin to block substance P release, thereby disrupting nociceptor function and decreasing pain generation, transmission and neurogenic inflammation (Mahowald 2009). Botulinum toxin has also been demonstrated to reduce levels of inflammatory markers following injury in animal models (Singh 2009). More research is needed, however, to further explain the suppressant effect of this agent on pain receptors.
Why it is important to do this review
The management of pain remains an important yet often suboptimal aspect of care in patients with RA. With widespread repercussions that affect quality of life, even seemingly small changes in pain intensity may have a large impact on perceived health in these patients. A wide variety of adjuvant drugs including neuromodulators have shown variable efficacy in the treatment of other chronic, painful conditions. Their use is, however, not without risk. Despite more frequent prescription, there is a paucity of information to guide physicians in the safe use of neuromodulators for patients with RA.
Objectives
The objectives of this systematic review were to evaluate the analgesic effectiveness and safety of neuromodulators in patients with RA.
Methods
Criteria for considering studies for this review
Types of studies
All published randomised controlled trials (RCTs) or quasi‐randomised (that is where allocation was not truly random) controlled trials (CCTs) which compared neuromodulator therapy to another therapy (active or placebo, including non‐pharmacological therapies) for RA were considered for inclusion. Only trials that were published as full articles or were available as a full trial report were included.
Types of participants
Adult patients (aged 18 years or older) with a diagnosis of RA. Populations that included a mixed population with RA and other musculoskeletal pain were excluded unless results for the RA population could be separated out from the trial analysis.
Types of interventions
All formulations and doses of neuromodulators including anticonvulsant agents (gabapentin, pregabalin, phenytoin, sodium valproate, lamotrigine, carbamazepine, levetiracetam, oxcarbazepine, tiagabine and topiramate), ketamine, bupropion, methylphenidate, nefopam, capsaicin and cannabinoids were considered.
Comparators included:
I. placebo;
II. other analgesics (e.g. paracetamol, NSAIDs, opioids, tramadol, antidepressants etc);
III. non‐pharmacological modalities (e.g. transcutaneous electrical nerve stimulation (TENS), acupuncture, etc);
IV. same drug at differing doses;
V. other neuromodulators.
Comparisons with placebo and with other controls were reported separately.
Drugs that have been withdrawn from the market due to safety concerns were excluded from the review.
Types of outcome measures
There is considerable variation in the outcome measures reported in clinical trials of interventions for pain. For the purpose of this systematic review, we have included outcome measures that are considered to be of the greatest importance to patients with persistent pain and the clinicians who care for them. The Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) has published consensus recommendations for determining clinically important changes in outcome measures in clinical trials of interventions for chronic pain. Reductions in pain intensity of ≥ 30% and ≥ 50% reflect moderate and substantial clinically important differences, respectively, and it has been recommended that the proportion of patients that respond with these degrees of pain relief be reported (Dworkin 2008).
Continuous outcome measures in pain trials (such as mean change on a 100 mm visual analogue scale) may not follow a Gaussian distribution. Often a bimodal distribution is seen instead, where patients tend to report either very good or very poor pain relief (Moore 2010). This creates difficulty in interpreting the meaning of average changes in continuous pain measures. For this reason, a dichotomous outcome measure (the proportion of participants reporting ≥ 30% pain relief) is likely to be more clinically relevant and is the primary efficacy measure in this review. It is recognised, however, that it has been the practice in most trials of interventions for chronic pain to report continuous measures and therefore the mean change in pain score was also included as a secondary efficacy measure.
The pain state at the end of a clinical trial of an analgesic intervention, in contrast to measures of pain improvement, has also been recommended as a clinically relevant dichotomous outcome measure and was included as a secondary efficacy measure in this review (Moore 2010). A global rating of treatment satisfaction, such as the Patient Global Impression of Change scale (PGIC) which provides an outcome measure that integrates pain relief, changes in function and side effects into a single, interpretable measure, is also recommended by IMMPACT and was included as a secondary outcome measure (Dworkin 2008).
Main outcomes
1. Efficacy: patient reported pain relief of 30% or greater.
2. Safety: number of withdrawals due to adverse events.
Secondary outcomes
3. Pain:
a. patient reported pain relief of 50% or greater,
b. patient reported global impression of clinical change (PGIC) much or very much improved,
c. proportion of patients achieving a pain score below 30/100 mm on a visual analogue scale (VAS),
d. mean change in pain score on a VAS or numerical rating scale (NRS).
4. Number and types of adverse events (AEs) and serious adverse events (SAEs) defined as AEs that were fatal, life‐threatening or required hospitalisation.
5. Function ‐ as measured by the Health Assessment Questionnaire (HAQ) or modified HAQ (Fries 1980; Pincus 1983).
6. Quality of life ‐ as measured by either generic instruments (such as the Short Form‐36 (SF‐36)) or disease‐specific tools (such as the Rheumatoid Arthritis Quality of Life instrument (RAQoL)).
7. Participant withdrawals due to inadequate analgesia.
8. Sleep as measured by any commonly used sleep scale (e.g. Insomnia Severity Index, Medical Outcomes Study (MOS) Sleep Scale, Pittsburgh Sleep Diary (PSD) and Pittsburgh Sleep Quality Index (PSQI)).
9. Depression, as measured by any commonly used depression scale (e.g. Hamilton Rating Scale for Depression (HRSD), Hospital Anxiety and Depression (HAD) score, Beck Depression Inventory (BDI), Zung self rating depression score).
The duration of trials of interventions for pain varies considerably. The efficacy of interventions, and the relative balance of benefits and harms, may vary according to the duration of the trial. Therefore the combination of results from trials of different durations may represent a source of bias in systematic reviews (Moore 2010). For the purpose of this review, trials were grouped into those of duration < 1 week, 1 to 6 weeks and > 6 weeks.
Outcomes at < 1 week, 1 to 6 weeks and > 6 weeks for the proportion reporting pain relief of 30% or greater, total number of withdrawals due to adverse effects, number of serious adverse events, function and quality of life were planned for inclusion in the summary of findings tables, when possible.
Search methods for identification of studies
Electronic searches
To identify relevant trials for this review, we used computer‐aided searches of the following databases for RCTs or CCTs using the search strategies detailed in the appendices.
1. Ovid MEDLINE (1950 to week 1 November 2010) (Appendix 1).
2. EMBASE Classic + EMBASE (Week 44, 2010) (Appendix 2).
3. The Cochrane Central Register of Controlled Trials (CENTRAL) (The Cochrane Library 2010, 4th quarter).
4. PsycINFO (1806 to week 2 November 2010).
No language restrictions were applied.
Searching other resources
The American College of Rheumatology (ACR) and European League Against Rheumatism (EULAR) conference abstracts, from 2008 and 2009, were also searched and a handsearch of references and relevant reviews was also performed to identify any additional trials not retrieved by the above methods.
Data collection and analysis
Selection of studies
All identified studies were assessed independently by two review authors (BR and SW) to identify trials that fulfilled the inclusion criteria. All possibly relevant articles were retrieved in full text and any disagreement in study selection was resolved by consensus or by discussion with a third review author (RB). Studies were translated into English where necessary.
Data extraction and management
Two independent review authors (BR and SW) extracted relevant information from the included trials including study design, characteristics of study population, treatment regimen and duration, outcomes and timing of outcome assessment using predetermined forms. The raw data (means and standard deviations for continuous outcomes and number of events or participants for dichotomous outcomes) were extracted for outcomes of interest.
Differences in data extraction were resolved by referring back to the original articles and establishing a consensus. A third review author (RB) was consulted to help resolve differences. when necessary.
Assessment of risk of bias in included studies
The potential for bias in the included studies was assessed using a risk of bias table (Higgins 2008). Two authors (BR, SW) independently assessed the risk of bias for all included studies for the following items: random sequence generation; allocation concealment; blinding of participants, care provider and outcome assessor for each outcome measure (see primary and secondary outcome measures); and incomplete outcome data and other biases. This conforms to the methods recommended by The Cochrane Collaboration (Higgins 2008). To determine the risk of bias of a study, for each criterion the provision of sufficient information and the likelihood of potential bias were evaluated. Each criterion was rated as 'Yes' (low risk of bias), 'No' (high risk of bias) or 'Unclear' (either lack of information or uncertainty over the potential for bias). In a consensus meeting, disagreements among the review authors were discussed and resolved. When a consensus was not reached, a third review author (RB) made the final decision.
Measures of treatment effect
For continuous data, results were analysed as mean differences between the intervention and comparator groups (MD) with 95% confidence intervals (CI). The mean difference between the treated group and control group was weighted by the inverse of the variance in the pooled treatment estimate. However, when different scales were used to measure the same conceptual outcome (for example functional status or pain) standardised mean differences (SMD) were calculated instead. SMDs were calculated by dividing the MD by the standard deviation, resulting in a unitless measure of treatment effect. For dichotomous data, a relative risk (RR) with corresponding 95% CI was calculated.
Unit of analysis issues
For studies containing more than two intervention groups, when making multiple pair‐wise comparisons between all possible pairs of intervention groups possible, we included the same group of participants only once in the meta‐analysis. When cross‐over trials were identified in which the reporting of continuous outcome data precluded paired analysis, these data were not to be included in a meta‐analysis in order to avoid unit‐of‐analysis error. Where carry‐over effects were thought to exist, and where sufficient data were present, data from the first period only was planned to be included in the analysis (Higgins 2009).
Dealing with missing data
In cases where individuals were missing from the reported results, we assumed that the missing value had a poor outcome. For dichotomous outcomes that measured adverse events (for example number of withdrawals due to adverse events), the withdrawal rate was calculated using the number of patients that received treatment as the denominator (worst case analysis). For dichotomous outcomes that measured benefits, the worst case analysis was calculated using the number of randomised participants as the denominator. For continuous outcomes (for example pain), we calculated the MD or SMD based on the number of patients analysed at the time point. If the numbers of patients analysed were not presented for each time point, the numbers of randomised patients in each group at baseline were used.
Where possible, missing standard deviations were computed from other statistics such as standard errors, CIs or P values according to the methods recommended in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2009). If standard deviations were not able to be calculated, we imputed them (for example from other studies in the meta‐analysis (Higgins 2009).
Assessment of heterogeneity
Prior to meta‐analysis, we assessed studies for clinical homogeneity with respect to type of therapy, control group and outcomes. For any studies judged as clinically homogeneous, statistical heterogeneity was estimated using the I2 statistic (Deeks 2008). We used the following as a rough guide for interpretation: 0% to 40%, might not be important; 30% to 60%, may represent moderate heterogeneity; 50% to 90%, may represent substantial heterogeneity; and 75% to 100%, considerable heterogeneity. In cases of considerable heterogeneity (defined as I2 ≥ 75%) we planned to explore the data further, including subgroup analyses, in an attempt to explain the heterogeneity; however statistical heterogeneity of this magnitude was not found in any of our meta‐analyses.
Assessment of reporting biases
In order to determine whether reporting bias was present, we planned to determine whether the protocol of the RCT was published before the recruitment of patients into the study was started. For studies published after 1st July 2005, we planned to screen the Clinical Trial Register at the International Clinical Trials Registry Platform of the World Health Organization (http://apps.who.int/trialssearch) (DeAngelis 2004). However, no studies were published after 2005. We assessed whether selective reporting of outcomes was present (outcome reporting bias).
We compared the fixed‐effect model estimate against the random‐effects model to assess the possible presence of small sample bias in the published literature (that is, where the intervention effect is more beneficial in smaller studies). In the presence of small sample bias, the random‐effects model estimate of the intervention is more beneficial than the fixed‐effect estimate (Sterne 2008). We planned to further explore the potential for reporting bias by funnel plots, however less than10 studies were included so this was not possible.
Data synthesis
Where studies were sufficiently homogeneous that it remained clinically meaningful for them to be pooled, a meta‐analysis was performed using a random‐effects model, regardless of the I2 statistic results. Analysis was performed using Review Manager 5 and forest plots were produced for all analyses.
Subgroup analysis and investigation of heterogeneity
Where sufficient data were available, the following subgroup analyses were planned:
1. patients' age (< 65 years versus ≥ 65 years);
2. gender (male versus female);
3. duration of RA (≤ 2 years versus > 2 years).
Sensitivity analysis
Where sufficient studies existed, sensitivity analyses were planned to assess the impact of any bias attributable to inclusion of trials with inadequate treatment allocation concealment (including studies with quasi‐randomised designs).
Presentation of key results
A summary of findings table was produced using GRADEpro software. This table provides key information concerning the quality of evidence, the magnitude of effect of the interventions examined, and the sum of available data on the outcomes (short and long term outcomes for pain, total number of withdrawals due to adverse effects, function and quality of life) as recommended by The Cochrane Collaboration. It includes an overall grading of the evidence related to each of the main outcomes using the GRADE approach (Schünemann 2008).
In addition to the absolute and relative magnitude of effect provided in the summary of findings table, for dichotomous outcomes the number needed to treat to benefit (NNTB) or the number needed to treat to harm (NNTH) were calculated from the control group event rate and the relative risk using the Visual Rx NNT calculator (Cates 2004). For continuous outcomes, the NNT will be calculated using the Wells calculator software available at the Cochrane Musculoskeletal Group (CMSG) editorial office (http://musculoskeletal.cochrane.org/). The minimal clinically important difference (MCID) for each outcome was determined for input into the calculator, using the formula where MCID was deemed to be 15% of the clinical outcome measure range being measured. If a study reported a mean difference, this was converted to a SMD for the purpose of this calculation.
Results
Description of studies
See: 'Characteristics of included studies' and 'Characteristics of excluded studies' tables.
Results of the search
The computer‐assisted literature search yielded 927 articles (CENTRAL 113, MEDLINE 250, EMBASE 557 and PsycINFO 7). A review of the last two years of ACR and EULAR abstracts did not find any further relevant studies. Removal of 137 duplicates left 790 abstracts for review. Of these, 14 studies were assessed for detailed review, of which four studies fulfilled our selection criteria. No additional studies were identified through reference checking (Figure 1).
1.

Study flow diagram.
Included studies
Of the four studies deemed to meet our inclusion criteria (Blake 2006; Deal 1991; Emery 1986; Swinson 1988), two trials evaluated the centrally‐acting analgesic drug nefopam (Emery 1986; Swinson 1988) and one each assessed topical capsaicin (Deal 1991) and a cannabis based medication administered via the oro‐mucosal route (Blake 2006). All trials investigated pain as their primary outcome measure using a VAS (100 mm). A summary of the interventions in these four studies is listed below.
I. Neuromodulator versus placebo
a) Nefopam versus placebo (Emery 1986; Swinson 1988)
b) Topical capsaicin 0.25% versus placebo (Deal 1991)
c) Oro‐mucosal cannabis based medicine (Sativex) versus placebo (Blake 2006)
II. Neuromodulator versus other analgesics (e.g. paracetamol, NSAIDs, opioids, tramadol, antidepressants etc)
No studies
III. Neuromodulator versus non‐pharmacological modalities (e.g. TENS, acupuncture, etc)
No studies
IV. Neuromodulator versus same drug at differing doses
No studies
V. Neuromodulator versus other neuromodulators
No studies
Two cross‐over trials (52 participants) evaluated nefopam at the same dose (60 mg tds) against placebo, over either two weeks (Swinson 1988) (n = 27) or four weeks (Emery 1986) (n = 25). Only Emery 1986 included a one week washout period before each active treatment phase. Both studies were performed in the UK and were published in the late 1980s. Participants were outpatients who met the ACR criteria for RA and had persistent pain. The majority of patients were female (83%) and they were generally receiving maximal dose NSAID therapy with 51% of the Emery 1986 study participants also taking a variety of DMARDs (gold, penicillamine, chloroquine or prednisone). Co‐interventions were not reported in the Swinson 1988 trial. Neither study reported any specific information about the type of pain participants were suffering from. Pain was reported using a VAS 100 mm for both trials, with Swinson 1988 specifically measuring night pain and Emery 1986 measuring pain in general.
One trial evaluated 0.025% topical capsaicin (four times daily) versus a placebo vehicle cream on knee pain (Deal 1991). This study was conducted in the USA in patients with osteoarthritis (OA) (70 participants) and RA (31 participants), with the data presented separately. Participants were predominantly female (81%), mean age 54 years (20 to 79 years) with moderate to severe knee pain (mean VAS 56 mm). No patient had undergone an intra‐articular corticosteroid injection in the preceding three weeks and 84% were taking NSAIDs, 32% corticosteroids, 13% gold and 10% other immunosuppressive agents. The primary outcome measure was pain intensity on a 100 mm VAS.
One placebo controlled trial (58 participants) assessed the cannabis based medication Sativex®, an oro‐mucosal spray containing 2.7 mg tetrahydrocannabinol (THC) and 2.5 mg cannabidiol (CBD) per activation (Blake 2006). This was a five week, randomised, double‐blind, multi‐centre parallel group study conducted in the UK in 2005. Patients with active RA (mean disease activity score (DAS) 5.9) and pain were recruited and they were generally female (79%), mean age 62.8 years, and had not used cannabis before recreationally (97%) or medicinally (98%). As intoxication reactions (dizziness and fatigue) are known to occur frequently during the initial titration period, the trial restricted dosing to the evenings (30 minutes before going to bed). Patients were titrated up to a maximum dose of 6 sprays, which is less than the maximal dose recommended (up to 12 sprays/day). The primary outcome variable was pain on movement (0 to 10 numerical rating scale), while pain was also measured using the Short Form McGill Questionnaire (SF‐MPQ).
Excluded studies
Ten additional studies were assessed.The reasons for their exclusion are presented in the 'Characteristics of excluded studies' table. Three trials evaluated botulinum toxin however these studies were performed predominantly in patients with OA and any data regarding RA patients could not be extracted separately (Boon 2010; Jasvinder 2009; Singh 2009). Similarly two studies evaluated topical capsaicin in patients with OA or a mixed population and no separate data for the RA patients were available (Hersh 1994; McCarthy 1992). Two other studies did not have a control group (Dykstra 2007; Singh 2009a) and one paper was a review article (Mahowald 2009a).
Risk of bias in included studies
See: Figure 2.
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
All studies had high risk of bias (Blake 2006; Deal 1991; Emery 1986; Swinson 1988).
The most common methodological shortcomings in the studies involved (in order of frequency):
failing to apply intention‐to‐treat analysis (4 (100%) trials scored 'negative’ or 'unclear’);
failure to blind study personnel (4 (100%) trials scored 'unclear’);
inadequate concealment of the drug allocation procedures (4 (100%) trials scored 'negative’ or 'unclear’);
failing to evaluate compliance (3 (75%) scored 'negative’ or 'unclear’);
inadequate method of randomisation (3 (75%) trials scored 'negative’ or 'unclear’);
failure to blind participants (2 (50%) trials scored 'negative’ or 'unclear’);
non‐equivalent co‐interventions (2 (50%) trials scored 'negative’ or 'unclear’);
failure to address incomplete outcome data (2 (50%) trials scored 'negative’ or 'unclear’);
dissimilarity of the baseline characteristics (1 (25%) trial scored 'negative’ or 'unclear’);
inadequate dropouts (1 (25%) trial scored 'negative’ or 'unclear’);
only reporting selective outcomes (no trial scored 'negative’ or 'unclear’).
Allocation
None of the trials provided any information about whether allocation of treatment was adequately concealed and they were deemed to be 'unclear'.
Blinding
No study provided any specific information about study design in regards to blinding of the personnel involved. This is important and raises the possibility that positive results may have been an artefact of physician expectations rather than a true effect.
Both trials investigating the effects of nefopam reported using identical placebo tablets to keep their patients blind to treatment (Emery 1986; Swinson 1988). Blinding was however a problem in one trial (Deal 1991) and unclear in another (Blake 2006). Patients in the Deal 1991 trial received either topical capsaicin or placebo. A common and well known 'adverse event' associated with topical capsaicin is a local burning sensation at the site of application. This is thought to be due to enhanced substance P release. Although the placebo was a vehicle cream designed to appear indistinguishable from the active agent, blinding and placebo validity may have been compromised in this trial with 44% of patients treated with capsaicin and only one participant treated with placebo reporting local burning. To address this issue the authors performed a repeated‐measures analysis (two‐tailed) of the physician's global evaluation, comparing the response of capsaicin participants with burning versus those without burning. Although they reported finding no significant difference, the numbers in this analysis were small and may not have been adequately powered to detect a significant effect. In the Blake 2006 trial patients used an oro‐mucosal preparation which contained a mint flavouring. It was not clear whether the placebo preparation was also mint flavoured to maintain blinding.
Incomplete outcome data
Two of the trials (Emery 1986; Swinson 1988) accounted for all missing data and performed a 'completers only' analysis. The other two trials did not specify detailed information regarding missing data (Blake 2006; Deal 1991). In Deal 1991 two patients in the capsaicin group dropped out, however it was unclear whether this was due to an adverse event or because of protocol violation. These patients were subsequently excluded from the analysis. In Blake 2006 four patients withdrew from the study and were accounted for, however it was unclear at what time points they withdrew and how their data were treated.
Selective reporting
All trials reported all prespecified outcomes.
Other potential sources of bias
No study performed an intention‐to‐treat analysis. Three studies performed a completers only analysis (Deal 1991; Emery 1986; Swinson 1988) while Blake 2006 did not provide enough information on how they treated missing data. It is likely that they too performed a completers only analysis. Only one trial reported measuring patient compliance with their allocated treatment by asking patients if they had taken the treatment (Deal 1991). As participants tend to over report compliance this may have biased the results towards a more conservative estimate. Deal 1991 did not provide details of the volume of cream applied, which may have been variable in patients.
Only two trials reported measuring the use of co‐interventions (Deal 1991; Swinson 1988). The authors of the Deal 1991 study reported that they interviewed patients regarding the use of concomitant medications however there data were not published. In the Swinson 1988 trial, the 24/25 patients taking analgesics before the trial were asked to stop them. It is not specified if this was measured subsequently. Any use of corticosteroids or disease‐modifying antirheumatic drugs (DMARDs) was also not described. In the Blake 2006 trial, the active treatment group appeared to improve in their disease activity scores (DAS) scores over the course of the trial and it was unclear whether this was due to the active intervention or other medications not reported. There was no similar improvement in DAS scores in the placebo arm.
The Swinson 1988 trial did not report baseline characteristics and had a significantly high dropout rate with 12/25 (48%) patients withdrawing, predominantly due to adverse events. This is significantly higher than in the Emery 1986 trial, which used the same dose of nefopam (19%).
Effects of interventions
See: Table 1; Table 2; Table 3
for the main comparison.
Nefopam 60 mg tds compared to placebo for pain management in rheumatoid arthritis
| Nefopam 60 mg tds compared to placebo for pain management in rheumatoid arthritis | ||||||
| Patient or population: patients with pain management in rheumatoid arthritis Settings: outpatient Intervention: nefopam 60mg tds Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Nefopam 60mg tds | |||||
| Pain VAS 100mm Follow‐up: 2 weeks | The mean Pain in the intervention groups was 21.16 lower (35.61 to 6.71 lower) | 48 (2 studies) | ⊕⊕⊝⊝ low | Absolute risk difference 21% (7% to 36%), relative percent change 54% (17% to 91%). Number needed to treat (NNT) was 2 (95%CI 1.4‐9.5) | ||
| Withdrawal Due to adverse events | 0 per 1000 | 0 per 1000 (0 to 0) | RR 11 (0.64 to 189.65) | 54 (1 study) | ⊕⊕⊕⊝ moderate1 | Not statistically significant. Absolute risk difference 19% (3% to 34%), relative percent change 1000% (‐36% to 18865%) |
| Total adverse events | 77 per 1000 | 316 per 1000 (122 to 823) | RR 4.11 (1.58 to 10.69) | 104 (2 studies) | ⊕⊕⊝⊝ low1,2 | Absolute risk difference 27% (12% to 42%), relative percent change 311% (58% to 969%). Number needed to harm (NNTH) 9 (95% CI 2 to 367). |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Only small number of participants 2 Both trials had high risk of bias
2.
Capsaicin 0.025% compared to placebo for knee pain in patients with rheumatoid arthritis
| Capsaicin 0.025% compared to placebo for knee pain in patients with rheumatoid arthritis | ||||||
| Patient or population: patients with Knee pain in patients with rheumatoid arthritis Settings: outpatient Intervention: capsaicin 0.025% Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Capsaicin 0.025% | |||||
| Pain (% reduction from baseline) VAS 100mm Follow‐up: 2 weeks | The mean Pain (% reduction from baseline) in the intervention groups was 34.4 lower (54.66 to 14.14 lower) | 30 (1 study) | ⊕⊕⊝⊝ low1,2 | Absolute risk difference 34% (14% to 55%), relative percent change 63% (26% to 99%). Number needed to treat (NNT) 2 (95% CI 1.4 to 6). | ||
| Pain (% reduction from baseline) VAS 100mm Follow‐up: 4 weeks | The mean Pain (% reduction from baseline) in the intervention groups was 25 lower (51.76 lower to 1.76 higher) | 29 (1 study) | ⊕⊕⊝⊝ low1,2 | Not statistically significant. Absolute risk difference ‐25% (‐52% to 2%), relative percentage change ‐46% (‐94% to 3%). | ||
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Patients were not adequately blinded, short duration trial and inadequately powered . 2 Small number of participants
3.
Oromucosal cannabis (Sativex) compared to placebo for pain management in patients with rheumatoid arthritis
| Oromucosal cannabis (Sativex) compared to placebo for pain management in patients with rheumatoid arthritis | ||||||
| Patient or population: patients with pain management in patients with rheumatoid arthritis Settings: outpatient Intervention: oromucosal cannabis (Sativex) Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Oromucosal Cannabis (Sativex) | |||||
| Pain at present McGill short form questionnaire ‐ Verbal rating scale (0‐5) Follow‐up: 5 weeks | The mean Pain at present in the intervention groups was 0.72 lower (1.31 to 0.13 lower) | 58 (1 study) | ⊕⊕⊝⊝ low1,2 | Number needed to treat (NNT) 3 (2‐18), absolute risk difference 14% (3%% to 26%), relative percentage change 23% (4% to 41%). | ||
| Quality of Sleep Numerical rating scale (0‐10). Scale from: 0 to 10. Follow‐up: 5 weeks | The mean Quality of Sleep in the intervention groups was 1.17 higher (0.13 to 2.21 higher) | 58 (1 study) | ⊕⊕⊝⊝ low1,2 | NNT 2 (1 to 18), absolute risk difference 12% (1% to 22%), relative percentage change 20% (2% to 38%). | ||
| Withdrawal due to adverse events Follow‐up: 5 weeks | 111 per 1000 | 14 per 1000 (1 to 265) | RR 0.13 (0.01 to 2.39) | 58 (1 study) | ⊕⊕⊝⊝ low1,2 | Not significant difference. Absolute risk difference 13% (1% to 239%), relative percentage change |
| Total adverse events | 407 per 1000 | 741 per 1000 (448 to 1000) | RR 1.82 (1.1 to 3) | 58 (1 study) | ⊕⊕⊝⊝ low1,2 | Number needed to harm (NNTH) of 3 (95% CI 3‐13), absolute risk difference 33% (9% to 58%), relative percentage change 82% (10% to 200%). |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Indaquate blinding and skewed data 2 Small number of participants
See: 'Summary of findings' table for the main comparisons.
Primary outcomes
Effectiveness of neuromodulators versus placebo: pain Intensity
Nefopam versus placebo
Two studies with a high risk of bias assessed pain outcomes in patients receiving oral nefopam 180 mg daily and reported conflicting outcomes (Emery 1986; Swinson 1988). Both studies used a cross‐over design, however only Emery 1986 had a washout period before each active treatment was given. Neither study reported the primary outcome measure of patient reported pain relief of 30% or greater. Emery 1986 reported a significant reduction in mean pain intensity (VAS 100 mm) compared to placebo after two (MD ‐21.16, 95% CI ‐35.61 to ‐6.71) (Analysis 1.1, Figure 3) and four weeks (MD ‐25, 95% CI ‐42.22 to ‐7.78) (Analysis 1.2). In contrast, Swinson 1988 reported no significant difference after two weeks of treatment in night pain on a 100 mm VAS (MD ‐12.00, 95% CI ‐38.59 to 14.59) (Analysis 1.1). This study was biased by the high dropout rate however making meaningful interpretation difficult. When the data were pooled using a random‐effects model there was still a significant reduction in pain favouring nefopam (WMD ‐21.16, 95% CI ‐35.61 to ‐6.71), with statistical homogeneity (I2 = 0, P = 0.42). Using the Emery 1986 study for the baseline control group SD and a minimum clinically important difference (MCID) of 15 mm, the NNT was 2 (95% CI 1.4 to 9.5).
1.1.

Comparison 1 Nefopam 60 mg tds versus placebo, Outcome 1 VAS Pain 2 weeks.
3.

Forest plot of comparison: 1 Nefopam 60 mg tds versus placebo, outcome: 1.1 VAS Pain 2 weeks.
1.2.

Comparison 1 Nefopam 60 mg tds versus placebo, Outcome 2 VAS Pain at 4 weeks.
Only the Emery 1986 study reported data withdrawals due to adverse events. Five patients in the nefopam group withdrew after developing nausea, with no withdrawals in the placebo group. This small trial showed a trend favouring placebo, however this did not reach statistical significance (RR 11.00, 95% CI 0.64 to 189.65) (Analysis 1.3). Swinson 1988 reported that 12 patients dropped out but did not describe which treatment they had received. Both trials reported adverse event data, with only Emery 1986 showing a statistically significant increase in adverse events in the nefopam group (RR 4.11, 95% CI 1.58 to 10.69) (Analysis 1.4, Figure 4) yielding a NNTH of 9 (95% CI 2 to 367). These were predominantly nausea (56%), sweating (44%), insomnia (11%), pruritis (11%) and malaise (11%).
1.3.

Comparison 1 Nefopam 60 mg tds versus placebo, Outcome 3 Withdrawal Due to adverse events.
1.4.

Comparison 1 Nefopam 60 mg tds versus placebo, Outcome 4 Total adverse events.
4.

Forest plot of comparison: 1 Nefopam 60 mg tds versus placebo, outcome: 1.4 Total adverse events.
No data were provided regarding functional status, quality of life, withdrawals due to inadequate analgesia, depression or sleep.
Topical capsaicin versus placebo
Deal 1991 used two separate measures to evaluate pain (percentage reduction of pain using a VAS 100 mm and categorical scale). Overall, both scales reported a statistically significant improvement in pain favouring topical use of 0.025% capsaicin at two weeks only. Using the VAS 100 mm, there was a significantly greater reduction in pain favouring capsaicin at one and two weeks, with a MD of ‐23.80 (95% CI ‐44.81 to ‐2.79) and ‐34.40 (95% CI ‐54.66 to ‐14.14) respectively. This corresponded to a NNT 3 (95% CI 2 to 47) at one week and NNT 2 (95% CI 1.4 to 6) at two weeks. There was only a trend towards improvement at four weeks (MD ‐25.00, 95% CI ‐51.76 to 1.76) (Analysis 2.1, Figure 5). With the categorical scale there was only a statistically significant difference at two weeks (MD ‐0.60, 95% CI ‐0.99 to ‐0.21), with a trend towards improvement at one (MD ‐0.37, 95% CI ‐0.77 to 0.03) and four weeks (MD ‐0.47, 95% CI ‐1.08 to 0.14) (Analysis 2.2). These discrepancies were likely to be related to type II error.
2.1.

Comparison 2 Capsaicin 0.025% versus placebo, Outcome 1 Pain VAS (% reduction from baseline).
5.

Forest plot of comparison: 2 Capsaicin 0.025% versus placebo, outcome: 2.1 Pain VAS (% reduction from baseline).
2.2.

Comparison 2 Capsaicin 0.025% versus placebo, Outcome 2 Pain Categorical pain score (change from baseline).
Change from baseline of the physician’s global evaluation (‐1 to 3, higher score indicating greater improvement) was not significantly different after one week (MD 0.48, 95% CI ‐0.06 to 1.02) however the findings favoured the capsaicin group, with a statistically significant improvement, after two (MD 0.60, 95% CI 0.10 to 1.10) and four weeks of treatment (MD 1.36, 95% CI 0.52 to 2.20) (Analysis 2.3).
2.3.

Comparison 2 Capsaicin 0.025% versus placebo, Outcome 3 Physician Global Evaluation *Global evaluation ( ‐1 to 3, worse to completely gone).
No data regarding withdrawal due to adverse events could be extracted for the RA population. Two patients withdrew after two weeks of capsaicin treatment due to burning at the site of application but it was unclear if these patients were from the RA or OA population. Likewise, no data were extracted regarding total adverse events. The only adverse reaction reported to be attributable to the intervention was the presence of burning at the site of application. This occurred in 44% of the trial patients receiving capsaicin (and one patient in the placebo group) but it was unclear what proportion of patients with RA were affected.
No data were presented regarding functional status, quality of life, withdrawals due to inadequate analgesia, depression or sleep.
Oro‐mucosal cannabis spray versus placebo
Blake 2006 did not report the primary outcome measure of patient reported pain relief of 30% or greater. Pain on movement and morning pain were measured using a numerical rating scale (NRS) (0 to 10). After five weeks of treatment there was a statistically significant improvement favouring cannabis over placebo (median difference ‐0.95, 95% CI ‐1.83 to ‐0.02, P = 0.044) (Table 7). The authors reported median values because of the skewed nature of their data. Significant improvements were seen in the degree of morning pain (median difference ‐1.04, 95% CI ‐1.90 to ‐0.18, P = 0.018). Using the SF‐MPQ, there was only a significant difference favouring cannabis in the verbal rating score (0 to 5) and 'pain at present' (MD ‐0.72, 95% CI ‐1.31 to ‐0.13) (Analysis 3.1). No significant difference was found in total intensity of pain or intensity of 'pain at present' measurements (Table 7) .
1.
Data from Blake 2006 trial
| Outcome | Median Difference | 95% Confidence Intervals | p value |
| Morning pain on movement | ‐0.95 | 1.83, 0.02 | 0.044 |
| Morning pain at rest | ‐1.04 | 1.90, 0.18 | 0.018 |
| SF‐MPQ, total intensity of pain | 3.00 | ‐3.00. 9.00 | 0.302 |
| SF‐MPQ, intensity of pain at present | ‐3.00 | 18.0, 9.00 | 0.572 |
These scores were not normally distributed and were analysed non‐parametrically (Wilcoxon rank‐sum test, Hodges–Lehmann median difference and 95% CI). SF‐MPQ, short form McGill Pain Questionnaire.
3.1.

Comparison 3 Cannabis (Setivax) versus placebo, Outcome 1 Short Form McGill Pain Questionnaire (SF‐MPQ).
There were no withdrawals due to adverse events in the cannabis group compared with three (11%) in the placebo arm (RR 0.13, 95% CI 0.01 to 2.39) (Analysis 3.3). There were no serious adverse events in the active treatment group compared with two (7%) in the placebo group. No further details about the nature of these events were available. Participants receiving cannabis were significantly more likely to suffer an adverse event (risk ratio (RR) 1.82, 95% CI 1.10 to 3.00) corresponding to a NNTH of 3 (95% CI 3 to 13). These were most commonly dizziness (26%), light headedness (10%), dry mouth (13%), nausea (6%) and falls (6%).
3.3.
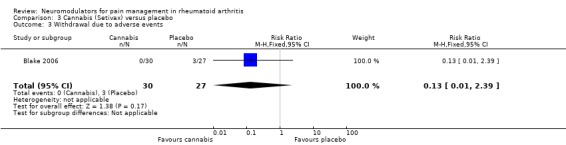
Comparison 3 Cannabis (Setivax) versus placebo, Outcome 3 Withdrawal due to adverse events.
Quality of sleep was also measured using an NRS (0 to 10). Patients receiving the cannabis based medication had a significant improvement in their quality of sleep when compared to placebo (MD 1.17, 95% CI 0.13 to 2.21) (Analysis 3.2). No functional outcomes, quality of life, depression or withdrawals due to inadequate analgesia were reported.
3.2.

Comparison 3 Cannabis (Setivax) versus placebo, Outcome 2 Sleep Numerical Rating Score (0‐10).
Other analyses
None of the planned subgroup analyses were performed for age, gender and duration of disease due to the insufficient data available.
Discussion
Summary of main results
This systematic review identified four randomised trials evaluating the use of neuromodulators for pain management in patients with RA. Two small cross‐over studies evaluated the centrally acting, non‐opioid analgesic nefopam (52 participants) (Emery 1986; Swinson 1988) while one parallel study each assessed topical capsaicin (31 participants) (Deal 1991) and the cannabis based medicine Sativex® (58 participants) (Blake 2006).
There is weak evidence that nefopam is an effective analgesic when compared with placebo in patients with RA, over four weeks. The magnitude of this benefit was on average 21.16 points on a 100 mm VAS scale. The NNT to achieve a positive result from these studies at two weeks was 2 (95% CI 1.4 to 9.5). Any benefits in pain reduction, however, may be limited by its side effect profile with both studies in this review having high dropout rates. Nausea is a well known side effect of nefopam and was the predominant reason for withdrawal in the included studies (19% in Emery 1986). Given the small number of participants, withdrawals due to adverse events did not reach statistical significance, however there was a strong trend for an increased risk with nefopam (RR 11.00, 95% CI 0.64 to 189.65). Total adverse events were significantly more common with nefopam and consisted of sweating, insomnia, pruritis and malaise (NNTH 9, 95% CI 2 to 367).
Topical capsaicin in a low dose also appears to provide some pain relief in patients with RA. The single trial that was identified assessed the efficacy of topical capsaicin 0.025% against placebo in a mixed population (RA and OA) and reported statistically significant improvements in pain favouring capsaicin (Deal 1991). After one week of treatment, patients receiving capsaicin scored on average 24 points less on a 0 to 100 mm pain scale than those in the placebo group. The NNT to achieve this benefit was 3 (95% CI 2 to 47). This improved to an average of 34 points less on the pain scale after two weeks of treatment (NNT 2, 95% CI 1.4 to 6). After four weeks there was a trend towards improvement of 25 points (on average) but this did not reach statistical significance. It should be noted, however, that this study was hampered by the inherent difficulties of truly blinding patients receiving this medication. The characteristic local burning sensation that alerts patients to their active treatment can be severe and limits the tolerability of this medication in some patients. Safety data regarding the RA population was not reported separately so our review is unable to assess the safety of this medication in patients with RA. Overall, 44% of patients (with RA and OA) were reported to suffer mild or moderate local burning at the site of application, which is consistent with rates reported in other trials. There were no serious adverse events. The limited amount of data means that these estimates for the number of participants achieving clinically useful levels of pain relief are not robust.
One small trial has also assessed the efficacy of the oro‐mucosal delivery of a cannabis based medicine (Sativex®) in pain reduction in patients with RA (Blake 2006). After five weeks of treatment there was a small but statistically significant improvement in pain (0.72 on a 0 to 5 point scale) and sleep (1.17 on a 0 to 10 point scale) with outcomes favouring cannabis over placebo. When compared with placebo, pain on movement and morning pain were on average improved by one extra point on a 10 point scale. There were no withdrawals due to adverse events or serious adverse events, however on average one in three patients (NNTH 3, 95% CI 3 to13) suffered a mild adverse event, most commonly dizziness (26%), light headedness (10%), dry mouth (13%) or nausea (6%).
Overall completeness and applicability of evidence
There have been few trials that have addressed the efficacy and safety of neuromodulators in patients with RA so confidence intervals for estimates of efficacy are wide. None of the commonly used anticonvulsants have been evaluated. In particular, no studies have evaluated gabapentin or pregabalin, drugs that are being increasingly prescribed for chronic pain. Overall, the trials were small, of short duration and lacking in safety data. No study provided adequate data to address the primary efficacy outcome variable (> 30% improvement in pain) nor the secondary outcomes of function and health‐related quality of life. Further research is required in these areas. There was also no information to judge the comparative efficacy and safety of neuromodulators in subpopulations defined by age, gender or duration of disease. The characteristics of the pain were not described in any of the included studies. The limited data also prevents any conclusions being drawn regarding optimal dosages of the medications.
Nefopam is only available in limited parts of the world, where it is often used as an intravenous preparation in the perioperative setting. The two nefopam trials were conducted in the 1980s in study populations that are not reflective of current day patients with RA. In general these patients had poorly controlled disease and were only receiving NSAIDs with occasional low dose corticosteroid or disease‐modifying antirheumatic drugs (DMARDs) only. No patients were receiving biological therapy.
Capsaicin was tested in one small trial in the lower (0.025%) of the two strengths currently available. Despite showing a benefit, in the early 1990s, there have not been any further trials in patients with RA. Cannabis based medications have restricted access around the world limiting research in this area. No trials have been performed using other cannabis preparations (oral, inhaled) or the stronger topical preparation in patients with RA.
Quality of the evidence
All four trials had a high risk of bias. A single, new large study assessing any of the included drugs could significantly alter the estimates of effect. The predominant methodological flaws of the included trials included failure to describe randomisation, allocation concealment, compliance and blinding of study personnel. There were also high dropout rates in many studies with no study performing an intention‐to‐treat analysis. The included studies also did not record the concomitant use of other analgesic agents, which is a surrogate measure and confounder of any improvement in pain outcome. Other limitations of the literature are relatively small numbers of placebo or head‐to‐head trials, short duration of follow‐up, and generally poor quality of adverse event assessment. Also, few studies have adequately evaluated depression, sleep, and functional or quality of life outcomes.
Potential biases in the review process
We believe that all relevant studies were identified using a thorough search strategy of all major databases. No language restrictions were applied. Two review authors assessed the trials for inclusion in the review and their risk of bias, with a third review author adjudicating if there was any discrepancy. All studies identified in recent reviews were also identified in our search. The biggest limitation of the review process was that many trials did not provide enough published data, or data in a form that could be extracted for meta‐analysis.
Agreements and disagreements with other studies or reviews
To our knowledge, this is the first systematic review to assess the efficacy and safety of neuromodulators in patients with RA.
Nefopam is used primarily for acute pain in the perioperative setting and systematic reviews have been performed in this patient population. It may be administered via oral, rectal, intravenous or intramuscular delivery. A recent systematic review with less stringent inclusion criteria included nine randomised trials (847 participants) and found there was some evidence that nefopam reduced postoperative pain scores (WMD ‐11.5 mm, 95% CI ‐15.1 to ‐7.85) but it increased tachycardia (NNH 7) and sweating (NNH 13) (Evans 2008).
A recent Cochrane review assessing herbal therapy for treating rheumatoid arthritis (Cameron 2011) included two capsaicin trials in the analysis (Deal 1991; McCarthy 1992). We excluded the McCarthy 1992 trial as it contained a mixed population (OA and RA) and data could not be separately extracted for the RA group. Despite this, our results support the observations made in the review in regards to both efficacy and relative safety. Looking more widely, topical capsaicin has been used for many years to treat a variety of other painful conditions. A comprehensive systematic review of capsaicin for the treatment of chronic pain conditions included neuropathic pain states and musculoskeletal pain disorders and found similar results to our review (Mason 2004). In three trials (368 participants) of patients with chronic musculoskeletal pain (OA or back or jaw pain) the relative benefit achieved with capsaicin was 1.5 compared to placebo and the NNT was 8.1 (95% CI 4.6 to 34). When all trials of musculoskeletal and neuropathic pain were combined the NNTH was 2.5 (95% CI 2.1 to 15) with 54% of patients suffering a local adverse event and 13% withdrawing due to adverse events.
No systematic reviews have assessed cannabis based medications in patients with RA. Our results are however similar to systematic reviews that have been done in patients with general pain. One earlier systematic review evaluated all trials that assessed 'human pain' (five trials cancer pain, two chronic nonmalignant pain, two acute postoperative pain) and reported that cannabinoids were more effective than placebo but no more effective than codeine. Doses of tetrahydrocannabinol (THC) above 5 mg were associated with depressant effects on the central nervous system limiting their use (Campbell 2001). The conclusions of this review were that treatment of chronic pain based on cannabinoid compounds would entail more risk than benefit. Similar conclusions were seen in a more recent systematic review that assessed the benefits and harms of cannabis in patients with chronic pain (Martín‐Sánchez 2009). The authors reported that cannabis treatment was moderately efficacious in the treatment of chronic pain (SMD ‐0.61, 95% CI ‐0.84 to ‐0.37) however any beneficial effects were offset by potentially serious harms. These included alterations to perception with an odds ratio (OR) of 4.51 (97% CI 3.05 to 6.66), NNH 7 (95% CI 6 to 9); impaired motor function, OR 3.93 (95% CI 2.83 to 5.47), NNH 5 (95% CI 4 to 6); and altered cognitive function, OR 4.46 (95% CI 2.37 to8.37), NNH 8 (95% CI 6 to12).
Authors' conclusions
Implications for practice.
Limited data suggest that nefopam, capsaicin and cannabis are all superior to placebo in reducing pain in patients with RA. However each agent is associated with a significant side effect profile that may offset the benefits.
Nefopam appears to have moderate analgesic efficacy in patients with active RA who are not taking other analgesic medications. Many patients cannot tolerate it though, with nausea and sweating often leading to patients stopping the medication. Although not seen in the included studies, potentially more serious adverse effects can occur such as confusion and tachycardia. With many other safer analgesics available on the market today and no head‐to‐head trials suggesting superior efficacy, our review does not support the use of nefopam in patients with RA.
Topical capsaicin may provide some relief in patients with RA who have persistent knee pain but at the expense of local skin irritation with associated burning and stinging. The confidence in our estimates are not strong however given the difficulties with blinding, the small numbers of participants evaluated and the lack of adverse event data. In some patients, however, even a small degree of pain relief may be considered worthwhile. Until further research is available, given the relatively mild nature of the adverse events, capsaicin could be considered as an add‐on therapy for patients with persistent local pain and inadequate response or intolerance to other treatments and who are able to tolerate the side effects.
Oro‐mucosal cannabis appears to improve pain and sleep to a modest degree but about one in three patients develop side effects involving the central nervous system, indicating that the potential harms outweigh the modest benefits. Our review does not support the use of cannabinoids in patients with RA.
Implications for research.
To better assess the efficacy and safety of neuromodulators in patients with RA, large double‐blind placebo controlled and head‐to‐head RCTs with homogenous RA populations are required. They should be methodologically sound, have a low risk of bias and involve longer term follow‐up (six months). Pain outcomes should be uniformly studied and we recommend that the proportion of patients with reductions in pain intensity of ≥ 30% and ≥ 50% (which reflect moderate and substantial clinically important differences) be reported. Descriptive measures of the type of pain should also be included to help readers decide if the data are applicable to their particular patient's pain.
The routine collection of functional and health‐related quality of life outcomes as well as sleep is also important. Safety data including deaths, withdrawals due to adverse events, serious adverse events and total numbers and types of adverse events should be evaluated. Compliance, which gives an indication of tolerability and acceptability, should also be routinely collected. Achieving double‐blind conditions in placebo controlled trials using capsaicin and cannabis will help to improve internal validity. Options may be to use a lower dose of capsaicin or another agent such as methyl nicotinate, which causes similar local burning effects without analgesic benefit, as a comparator. No study has assessed gabapentin or pregabalin to date. These are two neuromodulators now widely used for different chronic pain states.
What's new
| Date | Event | Description |
|---|---|---|
| 28 September 2010 | Amended | CMSG ID A057‐P |
Acknowledgements
Louise Falzon, Trials Search Co‐ordinator, Cochrane Musculoskeletal Group, for designing the search strategy.
Appendices
Appendix 1. MEDLINE search strategy
1. exp arthritis, rheumatoid/
2. ((rheumatoid or reumatoid or revmatoid or rheumatic or reumatic or revmatic or rheumat$ or reumat$ or revmarthrit$) adj3 (arthrit$ or artrit$ or diseas$ or condition$ or nodule$)).tw.
3. (felty$ adj2 syndrome).tw.
4. (caplan$ adj2 syndrome).tw.
5. (sjogren$ adj2 syndrome).tw.
6. (sicca adj2 syndrome).tw.
7. still$ disease.tw.
8. or/1‐7
9. exp Neurotransmitter Agents/
10. neuromodulator$.tw. or neurohumor$.tw.
11. exp Anticonvulsants/
12. Anticonvulsant$.tw.
13. gabapentin.tw.
14. neurontin.tw.
15. (Carbamazepine or Tegretol).tw.
16. (Clonazepam or Klonopin or Rivotril or Rivatril).tw.
17. (Lamotrigine or Lamictal).tw.
18. (Oxcarbazepine or Trileptal or Oxaleptal).tw.
19. (Phenytoin or Phenytek or Dilantin or Eptoin or Epanutin or Diphenin or Dipheninum or Phydum).tw.
20. (Pregabalin or Lyrica).tw.
21. (Topiramate or Topamax).tw.
22. (Valproic Acid or Valproate or Epilim or Depakote).tw.
23. (Ketamine or Ketalar or nefopam or acupan).tw.
24. (Capsaicin or Zostrix or Capsin or Capzasin‐P).tw.
25. exp Capsaicin/
26. exp Ketamine/
27. exp Bupropion/
28. (Bupropion or Wellbutrin or Zyban).tw.
29. exp Methylphenidate/
30. (Dexmethylphenidate or Focalin).tw.
31. (Methylphenidate or Ritalin or Concerta or bromocriptine).tw.
32. or/9‐31
33. randomized controlled trial.pt.
34. controlled clinical trial.pt.
35. randomized.ab.
36. placebo.ab.
37. drug therapy.fs.
38. randomly.ab.
39. trial.ab.
40. groups.ab.
41. or/33‐40
42. exp animals/ not humans.sh.
43. 41 not 42
44. 8 and 32 and 43
Appendix 2. EMBASE Classic + EMBASE search strategy
1. exp arthritis, rheumatoid/
2. ((rheumatoid or reumatoid or revmatoid or rheumatic or reumatic or revmatic or rheumat$ or reumat$ or revmarthrit$) adj3 (arthrit$ or artrit$ or diseas$ or condition$ or nodule$)).tw.
3. (felty$ adj2 syndrome).tw.
4. (caplan$ adj2 syndrome).tw.
5. (sjogren$ adj2 syndrome).tw.
6. (sicca adj2 syndrome).tw.
7. still$ disease.tw.
8. or/1‐7
9. exp Neurotransmitter Agents/
10. neuromodulator$.tw.
11. neurohumor$.tw.
12. exp Anticonvulsants/
13. Anticonvulsant$.tw.
14. gabapentin.tw.
15. neurontin.tw.
16. (Carbamazepine or Tegretol).tw.
17. (Clonazepam or Klonopin or Rivotril or Rivatril).tw.
18. (Lamotrigine or Lamictal).tw.
19. (Oxcarbazepine or Trileptal or Oxaleptal).tw.
20. (Phenytoin or Phenytek or Dilantin or Eptoin or Epanutin or Diphenin or Dipheninum or Phydum).tw.
21. (Pregabalin or Lyrica).tw.
22. (Topiramate or Topamax).tw.
23. (Valproic Acid or Valproate or Epilim or Depakote).tw.
24. (Ketamine or Ketalar or nefopam or Acupan).tw.
25. (Capsaicin or Zostrix or Capsin or Capzasin‐P).tw.
26. exp Cannabinoids/
27. endocannabinoid$.tw.
28. (cannabinoids$ or Rimonabant or SR141716 or Acomplia or Bethin or Monaslim or Remonabent or Riobant or Slimona or Rimoslim or Zimulti or Riomont).mp.
29. exp Capsaicin/
30. (Zostrix or Capsin or Capzasin‐P).tw.
31. exp Ketamine/
32. exp Bupropion/
33. (Bupropion or Wellbutrin or Zyban).tw.
34. exp Methylphenidate/
35. (Dexmethylphenidate or Focalin).tw.
36. (Methylphenidate or Ritalin or Concerta).tw.
37. exp botulinum toxin/
38. botulinum toxin E/ or botulinum toxin B/ or botulinum toxin A/ or botulinum toxin F/
39. botulin$.tw.
40. botox.tw.
41. OnabotulinumtoxinA.tw.
42. RimabotulinumtoxinB.tw.
43. AbobotulinumtoxinA.tw.
44. BTXA.tw.
45. (dyslor or dysport or lanzox or myobloc or neurobloc or oculinum or prosigne or vistabel or vistabex).tw.
46. or/9‐45
47. (random$ or placebo$).ti,ab.
48. ((single$ or double$ or triple$ or treble$) and (blind$ or mask$)).ti,ab.
49. controlled clinical trial$.ti,ab.
50. RETRACTED ARTICLE/
51. or/47‐50
52. (animal$ not human$).sh,hw.
53. 51 not 52
54. 8 and 46 and 53
Data and analyses
1.
Nefopam 60 mg tds versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 VAS Pain 2 weeks | 2 | 48 | Mean Difference (IV, Random, 95% CI) | ‐21.16 [‐35.61, ‐6.71] |
| 2 VAS Pain at 4 weeks | 1 | 22 | Mean Difference (IV, Fixed, 95% CI) | ‐25.0 [‐42.22, ‐7.78] |
| 3 Withdrawal Due to adverse events | 1 | 54 | Risk Ratio (M‐H, Fixed, 95% CI) | 11.0 [0.64, 189.65] |
| 4 Total adverse events | 2 | 104 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.11 [1.58, 10.69] |
2.
Capsaicin 0.025% versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Pain VAS (% reduction from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.1 Week 1 | 1 | 31 | Mean Difference (IV, Fixed, 95% CI) | ‐23.8 [‐44.81, ‐2.79] |
| 1.2 Week 2 | 1 | 30 | Mean Difference (IV, Fixed, 95% CI) | ‐34.4 [‐54.66, ‐14.14] |
| 1.3 Week 4 | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | ‐25.0 [‐51.76, 1.76] |
| 2 Pain Categorical pain score (change from baseline) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.1 Week 1 | 1 | 31 | Mean Difference (IV, Fixed, 95% CI) | ‐0.37 [‐0.77, 0.03] |
| 2.2 Week 2 | 1 | 30 | Mean Difference (IV, Fixed, 95% CI) | ‐0.6 [‐0.99, ‐0.21] |
| 2.3 Week 4 | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | ‐0.47 [‐1.08, 0.14] |
| 3 Physician Global Evaluation *Global evaluation ( ‐1 to 3, worse to completely gone) | 1 | Std. Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3.1 Week 1 | 1 | 31 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.61 [‐0.11, 1.33] |
| 3.2 Week 2 | 1 | 30 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.83 [0.08, 1.58] |
| 3.3 Week 4 | 1 | 29 | Std. Mean Difference (IV, Fixed, 95% CI) | 1.16 [0.36, 1.96] |
3.
Cannabis (Setivax) versus placebo
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Short Form McGill Pain Questionnaire (SF‐MPQ) | 1 | Mean Difference (Fixed, 95% CI) | Subtotals only | |
| 1.1 Pain at present | 1 | Mean Difference (Fixed, 95% CI) | ‐0.72 [‐1.31, ‐0.13] | |
| 2 Sleep Numerical Rating Score (0‐10) | 1 | Mean Difference (Fixed, 95% CI) | 1.17 [0.13, 2.21] | |
| 3 Withdrawal due to adverse events | 1 | 57 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.13 [0.01, 2.39] |
| 4 Total adverse events | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.82 [1.10, 3.00] |
3.4.

Comparison 3 Cannabis (Setivax) versus placebo, Outcome 4 Total adverse events.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
| Methods | Randomised, double‐blind, multi‐centre parallel group study | |
| Participants | 58 patients with RA 12 male, 46 female Mean age 62.8yrs Inclusion: active RA (ACR criteria), stable NSAIDs or steroids for one month and stable DMARDs for 3 months Exclusion: history of psychiatric disorders or substance misuse, severe cardiovascular, renal or hepatic disorder, or a history of epilepsy Sample size calculation: not reported |
|
| Interventions | Sativex oromucosal spray (2.7 mg Tetrahydrocannabinol (THC) and 2.5 mg cannabidiol (CBD) with each activation (dose)) vs Placebo Single dose nights 1 and 2, increased by one dose every 2 days to a maximum of six doses (according to individual response). Stable dosing was then maintained for a further 3 weeks Dosing was restricted to the evening to minimize possible intoxication‐type reactions |
|
| Outcomes | Baseline, day 14, 28, 49 and 57‐59 Primary: pain on movement (0–10 numerical rating scale (NRS)) Secondary: 1) Pain at rest (0‐10 NRS), Short Form McGill Pain Questionnaire (SF‐MPQ) 2) Sleep quality (0‐10 NRS) 3) Morning stiffness 4) 28‐joint disease activity score (DAS28) |
|
| Notes | Conclusion: statistically significant improvements in pain on movement, pain at rest, quality of sleep, DAS28 and the SF‐MPQ pain at present component were seen following CBM in comparison with placebo. Data was skewed as median differences were reported for most measures. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomised treatment allocation using permuted blocks of four" Comment: adequate |
| Allocation concealment (selection bias) | Unclear risk | No information specified |
| Blinding (performance bias and detection bias) Participant | Unclear risk | Quote: "versus placebo" Comment: no further information specified. Sativex has a mint flavour and it is likely the placebo did not replicate this |
| Blinding (performance bias and detection bias) Personnel | Unclear risk | No information specified |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 4 patients withdrew from the study and were accounted for, however it was unclear at what time point they withdrew and how their data were treated Comment: it is likely they were excluded from the analysis |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Compliance? | Unclear risk | No information specified |
| Co‐interventions? | Unclear risk | No information specified |
| Baseline characteristics? | Low risk | Quote: "There were no significant differences in demographics between groups" Comment: baseline characteristics were similar |
| intention to treat analysis? | Unclear risk | 4 patients withdrew from the study. It is not clear how their data were dealt with Comment: no information specified |
| Drop Outs? | Low risk | 4/58 (8.3%) patients dropped out, 3 from the placebo group and 1 active treatment |
| Summary Assessment of Bias? | High risk | High risk of bias |
| Methods | Double‐blind randomised controlled trial in patients wit RA and OA (excluded) | |
| Participants | 31 patients with ACR criteria for RA 6 male, 25 female Mean age 20‐79, mean pain 55‐57 (VAS 100mm) 84% taking NSAIDs, 32% corticosteroids, 13% gold, 10% other immunosuppressive agent Inclusion: >18yrs, moderate to severe unilateral or bilateral knee pain Exclusion: intrarticular corticosteroid injection ≤3 weeks or application of topical corticosteroids ≤7 days prior to study onset, skin disorder in affected knee Sample size calculation: not reported |
|
| Interventions | Topical 0.025% capsaicin versus placebo (vehicle cream) applied to the front, back, and both sides of the selected knee four times per day | |
| Outcomes | Baseline, 1, 2 and 4 weeks Primary: 1) Pain Intensity (VAS 100mm; no pain to worst imaginable pain) 2) Pain Relief (VAS 100mm; no pain relied to complete pain relief) 3) Physician Global Response:
|
|
| Notes | Capsaicin was significantly superior to placebo after one week of treatment in RA patients and after two weeks in OA patients. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were then randomly assigned..." Comment: no information specified |
| Allocation concealment (selection bias) | Unclear risk | No information specified |
| Blinding (performance bias and detection bias) Participant | High risk | A placebo vehicle cream was used, however topical capsaicin gives a characteristic burning sensation which was not blinded Comment: patients likely to have known they were receiving the active treatment |
| Blinding (performance bias and detection bias) Personnel | Unclear risk | No information specified Comment: likely to know that patients reporting burning were receiving the active treatment, however authors performed a repeated‐measures analysis (two‐tailed) of the physician's global evaluation, comparing the response of capsaicin patients with burning versus those without burning and found no significant difference. The numbers in this analysis were small however and may not have been able to detect a significant effect |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 2 patients in the capsaicin group dropped out. It was unclear whether this was due to an adverse event or because of protocol violation. These patients were excluded from the analysis |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Compliance? | Low risk | Quote: "Patients were interviewed at each visit to assess side effects, compliance..." Comment: no further information specified. It is likely that participants will have over reported compliance which would lead to a more conservative estimate. |
| Co‐interventions? | Unclear risk | Quote: "Patients were allowed to take standard oral arthritis medications during the study provided that the doses were stabilized before study start and the medications continued without interruption during the study." Quote: "Patients were interviewed at each visit to assess.....use of concomitant medications" Comment: co‐analgesics not specifically reported, but is likely they remained stable |
| Baseline characteristics? | Low risk | Baseline characteristics were similar |
| intention to treat analysis? | High risk | Completers only analysis |
| Drop Outs? | Low risk | 2/31 patients dropped out and were excluded |
| Summary Assessment of Bias? | High risk | High risk of bias |
| Methods | Double‐blind cross‐over trial | |
| Participants | 27 patients with RA 2 male, 25 female Mean age 59 years, disease 4‐30 years, 51% receiving gold, penicillamine, chloroquine or prednisone, "All were receiving maximal doses of one or more nonsteroidal anti‐inflammatory drugs, but had persistent pain" Inclusion: not specified Exclusion: not specified Sample size calculation: not reported |
|
| Interventions | Nefopam 60mg tds or placebo for 4 weeks, followed by one week washout then further 4 weeks on the alternative preparation | |
| Outcomes | Baseline, 2 weeks and 4 weeks for each cross‐over period Primary 1) Pain (VAS 100mm) 2) EMS (VAS 100mm) 3) Joint tenderness 4) Grip strength 5) Proximal interphalangeal joint (PIP) size |
|
| Notes | Conclusion: nefopam was a more effective analgesic than placebo when given as a supplement to anti‐inflammatory drugs | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information specified |
| Allocation concealment (selection bias) | Unclear risk | No information specified |
| Blinding (performance bias and detection bias) Participant | Low risk | Quote: "Placebo tablets were identical in appearance" Comment: it is likely the patients remained blinded |
| Blinding (performance bias and detection bias) Personnel | Unclear risk | Quote: "Neither doctor nor patient was aware of the treatment order" Comment: no information specified |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 5 patients dropped out and were accounted for. They were excluded from the analysis |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Compliance? | Unclear risk | Information not specified |
| Co‐interventions? | Unclear risk | Quote: "Patients were asked to discontinue pure analgesics one week prior to the study" Comment: no further information on co‐interventions during the trial were supplied |
| Baseline characteristics? | Low risk | Cross‐over trial. Quote: "Baseline measurements in the first and second periods were not significantly different for any variable", "Evidence for an order of treatment or carry‐over drug effect was sought by applying the approach described by Hill and Armitage" Comment: patients underwent a one week washout period and there was no evidence of a carry over effect |
| intention to treat analysis? | High risk | Completers only analysis |
| Drop Outs? | Low risk | 5/27 (18.5%) patients withdrew due to adverse events |
| Summary Assessment of Bias? | High risk | High risk of bias |
| Methods | Double‐blind randomised cross‐over trial | |
| Participants | 25 patients with RA 7 male, 18 female Mean age 58yrs (35‐71), mean duration RA12.6 years (2‐30), most patients taking NSAIDs which were continued Inclusion: classical or definite RA Exclusion: information not specified Sample size calculation: not reported |
|
| Interventions | Nefopam was 60 mg tds versus placebo for 2 weeks then cross‐over | |
| Outcomes | Baseline weeks 2 and 4 1) Pain: resting, standing, exercise and night pain (VAS100mm) 2) EMS (minutes) 3) Grip strength 4) Ring size |
|
| Notes | Conclusion: overall there was no statistically significant improvement in pain in patients receiving nefopam compared to placebo. Recruitment into the study was stopped because it was felt unethical to continue given the high dropout rate. High risk of bias as a consequence of dropouts. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information specified |
| Allocation concealment (selection bias) | Unclear risk | No information specified |
| Blinding (performance bias and detection bias) Participant | Low risk | Quote: "placebo tablets were identical" Comment: it is likely that patients remained blind |
| Blinding (performance bias and detection bias) Personnel | Unclear risk | No further information specified |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 12 patients dropped out and were accounted for |
| Selective reporting (reporting bias) | Low risk | All prespecified outcomes were reported |
| Compliance? | Unclear risk | No information specified |
| Co‐interventions? | Low risk | 24/25 patient were taking analgesics before the trial and these were ceased for the trial period. Any use of corticosteroids or DMARDs was not described |
| Baseline characteristics? | Unclear risk | Baseline characteristics not described. No washout period before or during the trial |
| intention to treat analysis? | High risk | Completers only analysis |
| Drop Outs? | High risk | 12/25 (48%) patients dropped out and were excluded |
| Summary Assessment of Bias? | High risk | High risk of bias |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Boon 2010 | Wrong patient population (OA) |
| Dykstra 2007 | Case series |
| Hersh 1994 | Mixed population, unable to extract RA data topical capsaicin in temporomandibular joint (TMJ) pain |
| Jasvinder 2009 | Mixed population, unable to extract RA data |
| Mahowald 2009a | Review article |
| McCarthy 1992 | Unable to extract RA data from mixed patient population (OA) |
| Rao 1995 | Comparator was DMARD |
| Richards 1987 | Comparator was DMARD |
| Singh 2009 | Case series |
| Singh 2009a | Mixed population, unable to extract RA data |
Contributions of authors
BR wrote the protocol and review.
SW contributed to the search strategy, study selection, data extraction, risk of bias analysis and provided comments and suggestions on draft versions of the protocol and review.
RB contributed to the search strategy, adjudicated on study selection, risk of bias assessments and data extraction and provided comments and suggestions on draft versions of the protocol and review.
All authors approved the current version.
Sources of support
Internal sources
-
Royal Prince Alfred Hospital, Australia.
In kind support
-
The Queen Elizabeth Hospital, Australia.
In kind support
-
Cabrini Hospital, Australia.
In kind support
-
School of Public Health & Preventive Medicine, Monash University, Australia.
In kind support
External sources
No sources of support supplied
Declarations of interest
None known
Edited (no change to conclusions)
References
References to studies included in this review
Blake 2006 {published data only}
- Blake DR, Robson P, Ho M, Jubb RW, McCabe CS. Preliminary assessment of the efficacy, tolerability and safety of a cannabis‐based medicine (Sativex) in the treatment of pain caused by rheumatoid arthritis. Rheumatology 2006;45 (1):50‐2. [DOI] [PubMed] [Google Scholar]
Deal 1991 {published data only}
- Deal CL, Schnitzer TJ, Lipstein E, Seibold JR, Stevens RM, Levy MD, et al. Treatment of arthritis with topical capsaicin: a double‐blind trial. Clinical Therapeutics 1991;13:383‐95. [PubMed] [Google Scholar]
Emery 1986 {published data only}
- Emery P, Gibson T. A double‐blind study of the simple analgesic nefopam in rheumatoid arthritis. British Journal of Rheumatology 1986;25(1):72‐6. [DOI] [PubMed] [Google Scholar]
Swinson 1988 {published data only}
- Swinson DR, Booth J, Baker RD. Nefopam in rheumatoid arthritis. Results of a double‐blind placebo controlled study. Clinical Rheumatology 1988;7(3):411‐2. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Boon 2010 {published data only}
- Boon AJ, Smith J, Dahm DL, et al. Efficacy of intra‐articular botulinum toxin type A in painful knee osteoarthritis: a pilot study. PM and R 2010;2:268‐76. [DOI] [PubMed] [Google Scholar]
Dykstra 2007 {published data only}
- Dykstra D, Stuckey M, Schimpff S, et al. The effects of intra‐articular botulinum toxin on sacroiliac, cervical/lumbar facet and sterno‐clavicular joint pain and C‐2 root and lumbar disc pain: a case series of 11 patients. The Pain Clinic 2007;19:27–32. [Google Scholar]
Hersh 1994 {published data only}
- Hersh EV, Pertes RA, Ochs HA. Topical capsaicin‐pharmacology and potential role in the treatment of temporomandibular pain. The Journal of Clinical Dentistry 1994;5(2):54‐9. [PubMed] [Google Scholar]
Jasvinder 2009 {published data only}
- Singh JA, Mahowald ML, Noorbaloochi S. Intra‐articular botulinum toxin A for refractory shoulder pain: a randomized, double‐blinded, placebo‐controlled trial. Translational Research 2009;153:205‐16. [DOI] [PubMed] [Google Scholar]
Mahowald 2009a {published data only}
- Mahowald ML, Krug HE, Singh JA, Dykstra D. Intra‐articular botulinum toxin type A: A new approach to treat arthritis joint pain. Toxicon 2009;54:658‐67. [DOI] [PubMed] [Google Scholar]
McCarthy 1992 {published data only}
- McCarthy GM, McCarthy DJ. Effect of topical capsaicin in the therapy of painful osteoarthritis of the hands. Journal of Rheumatology 1992;19:604–7. [PubMed] [Google Scholar]
Rao 1995 {published data only}
- Rao UR, Naidu MU, Kumar TR, Shobha U, Askar MA, Ahmed N, et al. Comparison of phenytoin with auranofin and chloroquine in rheumatoid arthritis‐‐a double blind study. Journal of Rheumatology 1995;22:1235‐40. [PubMed] [Google Scholar]
Richards 1987 {published data only}
- Richards IM, Fraser SM, Hunter JA, Capell HA. Comparison of phenytoin and gold as second line drugs in rheumatoid arthritis. Annals of the Rheumatic Diseases 1987;46:667‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Singh 2009 {published data only}
- Singh JA, Mahowald ML. Intra‐articular botulinum toxin A as an adjunctive therapy for refractory joint pain in patients with rheumatoid arthritis receiving biologics: A report of two cases. Joint, Bone, Spine 2009;76:190‐4. [DOI] [PubMed] [Google Scholar]
Singh 2009a {published data only}
- Mahowald ML, Krug HE, Singh JA, Dykstra D. Intra‐articular botulinum toxin A for refractory shoulder pain: a randomised, double‐blinded, placebo‐controlled trial. Toxicon 2009;153:205‐16. [DOI] [PubMed] [Google Scholar]
Additional references
Altman 1994
- Altman RD, Aven A, Holmburg CE, et al. Capsaicin cream, 0.025% as monotherapy for osteoarthritis: A double‐blind study. Seminars in Arthritis and Rheumatism 1994;23:25‐33. [Google Scholar]
Bell 2003
- Bell RF, Eccleston C, Kalso E. Ketamine as an adjuvant to opioids for cancer pain. Cochrane Database of Systematic Reviews 2003, Issue 1. [DOI: 10.1002/14651858.CD003351; PUBMED: 12243612 ] [DOI] [PubMed] [Google Scholar]
Bell 2006
- Bell RF, Dahl JB, Moore RA, Kalso EA. Perioperative ketamine for acute postoperative pain. Cochrane Database of Systematic Reviews 2006, Issue 1. [DOI: 10.1002/14651858.CD004603.pub2; PUBMED: 16437490 ] [DOI] [PubMed] [Google Scholar]
Bendaly 2007
- Bendaly EA, Jordan CA, Staehler SS, Rushing DA. Topiramate in the treatment of neuropathic pain in patients with cancer. Supportive Cancer Therapy 2007;4:241‐6. [DOI] [PubMed] [Google Scholar]
Calignano 1998
- Calignano A, Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature 1998;394:277–81. [DOI] [PubMed] [Google Scholar]
Cameron 2011
- Cameron M, Gagnier JJ, Chrubasik S. Herbal therapy for treating rheumatoid arthritis. Cochrane database of systematic reviews (Online) 2011;2:CD002948. [PUBMED: 21328257] [DOI] [PubMed] [Google Scholar]
Campbell 2001
- Campbell F, Tramèr M, Carroll D, et al. Are cannabinoids an effective and safe treatment option in the management of pain? A qualitative systematic review. BMJ 2001;323:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cates 2004
- Cates C. Dr Chris Cates' EBM Web Site. Visual Rx Version 3. Available from: http://www.nntonline.net/visualrx/ 2004.
Chong 2000
- Chong MS, Smith TE. Anticonvulsants for the management of pain. Pain Reviews 2000;7:129–49. [Google Scholar]
Cooper 2003
- Cooper RG, Booker CK, Spanswick CC. What is pain management, and what is its relevance to the rheumatologist?. Rheumatology 2003;42:1133‐7. [DOI] [PubMed] [Google Scholar]
Davis 2007
- Davis MP. What is new in neuropathic pain?. Support Care Cancer 2007;15 (4):363‐72. [DOI] [PubMed] [Google Scholar]
DeAngelis 2004
- DeAngelis CD, Drazen JM, Frizelle FA, Haug C, Hoey J, Horton R, et al. Clinical trial registration: a statement from the International Committee of Medical Journal Editors. JAMA 2004;292:1363‐4. [DOI] [PubMed] [Google Scholar]
Deeks 2008
- Deeks JJ, Higgins JPT, Altman DG (editors). Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JPT, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.1 (updated September 2008). The Cochrane Collaboration, 2008. Available from www.cochrane‐handbook.org.
Dworkin 2007
- Dworkin RH, O’Connor AB, Backonja M, et al. Pharmacologic management of neuropathic pain: evidence‐based recommendations. Pain 2007;132:237–51. [DOI] [PubMed] [Google Scholar]
Dworkin 2008
- Dworkin RH, Turk DC, Wyrwich KW, et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. Journal of Pain 2008;9(2):105‐21. [DOI] [PubMed] [Google Scholar]
Edwards 2009
- Edwards RR, Wasan AD, Bingham CO 3rd, et al. Enhanced reactivity to pain in patients with rheumatoid arthritis. Arthritis Research & Therapy 2009;11:R61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Esposito 1986
- Esposito E, Romandini S, Merlo‐Pich E, et al. Evidence of the involvement of dopamine in the analgesic effect of nefopam. European Journal of Pharmacology 1986;128:57‐64. [DOI] [PubMed] [Google Scholar]
Evans 2008
- Evans M, Lysakowski C, Tramèr MR. Nefopam for the prevention of postoperative pain: quantitative systematic review. British Journal of Anaesthesia 2008;101(5):610–7. [DOI] [PubMed] [Google Scholar]
Finnerup 2007
- Finnerup NB, Otto M, McQuay HJ, et al. Algorithm for neuropathic pain treatment: an evidence based proposal. Pain 2005;118:289–305. [DOI] [PubMed] [Google Scholar]
Fisher 2000
- Fisher K, Coderre TJ, Hagen NA. Targeting the N‐methyl‐D‐aspartate receptor for chronic pain management: preclinical animal studies,recent clinical experience and future research directions. Journal of Pain and Symptom Management 2000;20(5):358–73. [DOI] [PubMed] [Google Scholar]
Franceschi 1988
- Franceschi M, Ciboddo G, Truci G, Borri A, Canal N. Fatal aplastic anemia in a patient treated with carbamazepine. Epilepsia 1988;29(5):582‐3. [PUBMED: 3409843] [DOI] [PubMed] [Google Scholar]
Fries 1980
- Fries JF, Spitz P, Kraines RG, Holman HR. Measurement of patient outcome in arthritis. Arthritis and Rheumatism 1980;23(2):137‐45. [PUBMED: 7362664] [DOI] [PubMed] [Google Scholar]
Fuller 1993
- Fuller RW, Snoddy HD. Evaluation of nefopam as a monoamine uptake inhibitor in vivo in mice. Neuropharmacology 1993;32:995‐9. [DOI] [PubMed] [Google Scholar]
Gilron 2006
- Gilron I, Flatters SJ. Gabapentin and pregabalin for the treatment of neuropathic pain: a review of laboratory and clinical evidence. Pain Research and Management 2006;11:16‐29A. [Google Scholar]
Heel 1980
- Heel RC, Brogden RN, Pakes GE, et al. Nefopam: a review of its pharmacological properties and therapeutic efficacy. Drugs 1980;19:249‐67. [DOI] [PubMed] [Google Scholar]
Heiburg 2002
- Heiberg T, Kvien TK. Preferences for improved health examined in 1,024 patients with rheumatoid arthritis: pain has highest priority. Arthritis and Rheumatism 2002;47(4):391‐7. [DOI] [PubMed] [Google Scholar]
Higgins 2008
- Higgins JPT, Altman DG (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.1 (updated September 2008). The Cochrane Collaboration, 2008. Available from www.cochrane‐handbook.org.
Higgins 2009
- Higgins JPT, Deeks JJ, Altman DG (editors). Chapter 16: Special topics in statistics. In: Higgins JPT, Green S (editors), Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2 (updated September 2009). The Cochrane Collaboration, 2009. Available from www.cochrane‐handbook.org.
Hunskaar 1987
- Hunskaar S, Fasmer OB, Broch OJ, et al. Involvement of central serotonergic pathways in nefopam‐induced anti‐nociception. European Journal of Pharmacology 1987;138:77‐82. [DOI] [PubMed] [Google Scholar]
Kakkar 2009
- Kakkar M, Derry S, Moore RA, et al. Single dose oral nefopam for acute postoperative pain in adults. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD007442.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lynch 2005
- Lynch ME. Preclinical science regarding cannabinoids as analgesics: an overview. Pain Research and Management 2005;10:7A–14A. [DOI] [PubMed] [Google Scholar]
Mahowald 2009
- Mahowald M, Singh JA, Kushnaryov A, et al. Repeat injections of intra‐articular botulinum toxin A for the treatment of chronic arthritis joint pain – a case series review. Journal of Clinical Rheumatology 2009;15(1):35–8. [DOI] [PubMed] [Google Scholar]
Martín‐Sánchez 2009
- Martín‐Sánchez E, Furukawa T, Taylor J, et al. Systematic review and meta‐analysis of cannabis treatment for chronic pain. Pain Medicine 2009;10(8):1353‐68. [DOI] [PubMed] [Google Scholar]
Mason 2004
- Mason L, Moore A, Derry S. Systematic review of topical capsaicin for the treatment of chronic pain. BMJ 2004;328:991. [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2010
- Moore RA, Eccleston C, Derry S, Wiffen P, Bell RF, Straube S, et al. "Evidence" in chronic pain‐establishing best practice in the reporting of systematic reviews. Pain 2010;150(3):386‐9. [DOI] [PubMed] [Google Scholar]
Morris 1997
- Morris V, Cruwys S, Kidd B. Characterisation of capsaicin‐induced mechanical hyperalgesia as a marker for altered nociceptive processing in patients with rheumatoid arthritis. Pain 1997;71:179‐86. [DOI] [PubMed] [Google Scholar]
Nolano 1999
- Nolano M, Simone DA, Wendelschafer‐Crabb G, et al. Topical capsaicin in humans: parallel loss of epidermal nerve fibres and pain sensation. Pain 1999;81:135‐45. [DOI] [PubMed] [Google Scholar]
Pertwee 2001
- Pertwee RG. Cannabinoid receptors and pain. Progress in Neurobiology 2001;63:569–609. [DOI] [PubMed] [Google Scholar]
Piercey 1981
- Piercey MF, Schroeder LA. Spinal and supraspinal sites for morphine and nefopam analgesia in the mouse. European Journal of Pharmacology 1981;74(2‐3):135‐40. [DOI] [PubMed] [Google Scholar]
Pincus 1983
- Pincus T, Summey JA, Soraci SA Jr, Wallston KA, Hummon NP. Assessment of patient satisfaction in activities of daily living using a modified Stanford Health Assessment Questionnaire. Arthritis and Rheumatism 1983;26(11):1346‐53. [DOI] [PubMed] [Google Scholar]
Ranoux 2008
- Ranoux D, Attal N, Morain F, Bouhassira D. Botulinum toxin type A induces direct analgesic effects in chronic neuropathic pain. Annals of Neurology 2008;64:274–83. [DOI] [PubMed] [Google Scholar]
Schünemann 2008
- Schünemann HJ, Oxman AD, Higgins JPT, Vist GE, Glasziou P, Guyatt GH. Chapter 11: Presenting results and ‘Summary of findings' tables. In: Higgins JPT, Green S, editors, Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.1 (updated September 2008). The Cochrane Collaboration, 2008. Available from www.cochrane‐handbook.org.
Singh 2008
- Singh J, Mahowald ML. Intra‐articular botulinum toxin A as an adjunctive therapy for refractory joint pain in patients with rheumatoid arthritis receiving biologics: A report of two cases. Joint, Bone, Spine 2009;76:190‐4. [DOI] [PubMed] [Google Scholar]
Singh 2011
- Singh JA, Fitzgerald PM. Botulinum toxin for shoulder pain: a Cochrane systematic review. The Journal of Rheumatology 2011;38(3):409‐18. [PUBMED: 21285169] [DOI] [PubMed] [Google Scholar]
Skevington 1998
- Skevington SM. Investigating the relationship between pain and discomfort and quality of life, using the WHOQOL. Pain 1998;76:395‐406. [DOI] [PubMed] [Google Scholar]
Sterne 2008
- Sterne JAC, Egger M, Moher D (editors). Chapter 10: Addressing reporting biases. In: Higgins JPT, Green S, editors. Cochrane Handbook for Systematic Reviews of Intervention. Version 5.0.1 (updated September 2008). The Cochrane Collaboration, 2008. Available from www.cochrane‐handbook.org.
Tremont‐Lukats 2000
- Tremont‐Lukats IW, Megeff C, Backonja MM. Anticonvulsants for neuropathic pain syndromes: mechanisms of action and place in therapy. Drugs 2000;60:1029‐52. [DOI] [PubMed] [Google Scholar]
Walker 2001
- Walker JM, Strangman NM, Huang SM. Cannabinoids and pain. Pain Research and Management 2001;6:74–9. [DOI] [PubMed] [Google Scholar]
Walker 2005
- Walker JM, Hohmann AG. Cannabinoid mechanisms of pain suppression. Handbook of Experimental Pharmacology 2005;168:509–54. [DOI] [PubMed] [Google Scholar]
Waseem 2011
- Waseem Z, Boulias C, Gordon A, Ismail F, Sheean G, Furlan AD. Botulinum toxin injections for low‐back pain and sciatica. Cochrane Database of Systematic Reviews (Online) 2011;1:CD008257. [PUBMED: 21249702] [DOI] [PubMed] [Google Scholar]
Wiffen 2005
- Wiffen PJ, McQuay HJ, Edwards JE, Moore RA. Gabapentin for acute and chronic pain. Cochrane Database of Systematic Reviews 2005, Issue 3. [DOI: 10.1002/14651858.CD005452] [DOI] [PubMed] [Google Scholar]
Wiffen 2007
- Wiffen PJ, Rees J. Lamotrigine for acute and chronic pain. Cochrane Database of Systematic Reviews 2007, Issue 2. [DOI: 10.1002/14651858.CD006044.pub2] [DOI] [PubMed] [Google Scholar]
Woolf 1991
- Woolf CJ, Mannion R. Neuropathic pain:aetiology, symptoms, mechanisms, and management. Lancet 1999;353:1959–64. [DOI] [PubMed] [Google Scholar]
Yuan 2009
- Yuan RY, Sheu JJ, Yu JM, et al. Botulinum toxin for diabetic neuropathic pain: a randomised double‐blind crossover trial. Neurology 2009;72:1473–8. [DOI] [PubMed] [Google Scholar]