

Abstract
Over 20 years ago, the synaptic protein alpha-synuclein was identified as the primary component of the Lewy bodies (LBs) that are a sine qua non of Parkinson’s disease (PD). Since that time, extensive research has demonstrated that alpha-synuclein pathology is not only a hallmark of PD, but can also cause neuronal dysfunction and death. Detailed staging of alpha-synuclein pathology in the brains of patients has revealed a progressive pattern of pathology that correlates with the symptoms of disease. Early in the disease course, PD patients exhibit motor dysfunction, and alpha-synuclein pathology at this stage is primarily found in regions controlling motor function. At later stages of disease as patients’ cognitive function deteriorates, alpha-synuclein pathology can be found in cortical structures responsible for higher cognitive processing. The stereotypical progression of alpha-synuclein pathology through the brain over time suggests that there may be a physical transmission of pathological alpha-synuclein from one area of the brain to another. The transmission hypothesis posits that an initial seed of pathological alpha-synuclein in one neuron may be released and taken up by another vulnerable neuron and thereby initiate pathological misfolding of alpha-synuclein in the recipient neuron. In recent years, convergent evidence from various studies has indicated that pathological protein transmission can occur in the human brain. Cell and animal models based on the transmission hypothesis have shown not only that pathological alpha-synuclein can be transmitted from cell-to-cell, but that this pathology can lead to neuronal dysfunction and degeneration. The alpha-synuclein transmission hypothesis has profound implications for treatment of what is currently an intractable neurodegenerative disease. In this review, we explore the evidence for cell-to-cell transmission of pathological alpha-synuclein, the current understanding of how pathological alpha-synuclein can move to a new cell and template misfolding, and the therapeutic implications of alpha-synuclein transmission.
Keywords: transmission, neurodegeneration, spread, genetic risk factor, therapeutic
1 |. INTRODUCTION
Parkinson’s disease (PD) is the second-most common neurodegenerative disease, affecting at least 0.3 percent of the worldwide population and over 3 percent of those over 80 years old [1]. Since James Parkinson’s “Essay on the Shaking Palsy” over 200 years ago [2], much has been discovered about the underlying etiology of PD, yet many of James Parkinson’s original observations remain cardinal features of PD. In its early stages, PD is primarily characterized as a movement disorder, with patients exhibiting a range of slowness of movement, rigidity, postural instability and tremors in addition to non-motor symptoms including REM sleep disorder, constipation and hyposmia, which may precede motor symptoms [3]. The motor symptoms are due in large part to the loss of dopaminergic neurons in the substantia nigra and commensurate dysregulation of basal ganglia activity [4]. However, with efficacious treatments of motor symptoms in PD, up to 80 percent of PD patients will also experience cognitive decline during the disease course, leading to Parkinson’s disease dementia (PDD) [5]. PDD is difficult to distinguish pathologically from dementia with Lewy bodies (DLB) [6, 7], and the diseases are widely regarded as two ends on the spectrum of a single disease [8, 9], distinguished by which symptoms develop initially—motor (PDD) or cognitive (DLB) [10].
Neuropathologically, PD, PDD and DLB are characterized by the presence of proteinaceous inclusions termed Lewy bodies (LBs) first described by Friedrich Lewy in 1912 [11]. Advances in histochemical and biochemistry techniques led to the identification of alpha-synuclein as the primary protein component of LBs [12–14] as well as the glial cytoplasmic inclusions (GCIs) of multiple system atrophy (MSA), a less common alpha-synucleinopathy distinct from PD, PDD and DLB [15]. The discovery that alpha-synuclein gene (SNCA) duplications [16, 17], triplications [18] and point mutations [19–25] cause familial forms of PD bolstered the hypothesis that alpha-synuclein is playing a direct role in disease pathogenesis. The pathological and genetic data implicating alpha-synuclein in PD, PDD and DLB led to the classification of these diseases as alpha-synucleinopathies.
Extensive research over the past 20 years has been aimed at understanding how alpha-synuclein aggregates and leads to neuronal dysfunction and death in vulnerable neurons. A preponderance of growing evidence suggests that stochastic formation of LBs in individual neurons does not alone explain the pattern of pathology and symptomatic progression observed in PD. Rather, recent research supports the cell-to-cell transmission of misfolded alpha-synuclein in a prion-like manner wherein misfolded alpha-synuclein released by one neuron can be taken up and act as a template for misfolding of alpha-synuclein in the recipient neuron. However, unlike prions, pathological alpha-synuclein shows no evidence of being infectious [26]. Here, we review: (1) data supporting the cell-to-cell transmission of alpha-synuclein in humans, (2) cell and animal models, (3) proposed mechanisms of spread, (4) the role of conformation in alpha-synuclein transmission, and (5) the therapeutic implications of spread. For many years the primary treatment for PD has been symptomatic dopamine-replacement therapy, which does not alter the progression of disease. Extracellular spread of misfolded alpha-synuclein provides many more therapeutic avenues to explore, especially for those synucleinopathies such as DLB for which dopamine replacement is not a viable therapy.
2 |. EVIDENCE FOR TRANSMISSION IN HUMANS
alpha-Synuclein is an intracellular protein predominantly localized in the presynaptic terminals [27] but under pathological conditions forms LBs in the soma of affected neurons [4]. LB formation can also be quite selective within a neuronal population, devastating some neurons, while nearby neurons show no sign of inclusion formation [28]. These observations, coupled with the etiology of PD as an age-related disease, suggest that alpha-synuclein inclusions may form in a set of vulnerable neurons over long periods and thereby lead to the death of those neurons. However, recent data from humans and model systems indicates that alpha-synuclein can be released from neurons [29], and that this released alpha-synuclein can be taken up by neurons in synaptically-connected regions, leading to pathology induction in those neurons and a pathological spread of disease through the brain that correlates with disease progression.
Brains from patients with PD have LB pathology not only in the substantia nigra but in other extranigral sites in the brainstem and throughout the brain [30, 31]. Studies carried out in the early 2000s by Braak and colleagues sought to understand the relationship between LB pathology and disease severity [30, 31]. These studies were based on the assumption that brain nuclei with LB pathology in asymptomatic or early PD would represent the earliest sites of disease initiation, while areas with LBs in severely symptomatic individuals would represent the late end of the disease spectrum. These studies examined over 168 brains from PD patients, patients with LBs but not diagnosed with PD and patients without LBs or any neurological diagnosis [30, 31]. In asymptomatic individuals, pathology was observed most consistently in the dorsal IX/X motor nucleus (DMX) and anterior olfactory nucleus. Moderate cases had pathology in the midbrain, and the most severe cases also had pathology in the neocortex. These data suggested to the authors that PD may begin in the dorsal IX/X motor nucleus of the medulla, or olfactory nucleus, and propagate up through the brainstem, then to cortical regions.
The spread hypothesis developed by Braak and colleagues provided a strong foundation for understanding the progression of PD, but 50 percent of alpha-synucleinopathy cases did not fall into the proposed stages of disease. A later study evaluated 417 subjects with PD, DLB, incidental PD, and Alzheimer’s disease with LBs [32]. The resulting staging system suggested that from an initial stage I where only the olfactory bulb was affected, cases diverged into two distinct stages—either stage IIa (brainstem predominant) or stage IIb (limbic predominant). Following stage II, the two pathways converge into a stage III in which both brainstem and limbic areas are affected, followed by stage IV in which neocortical areas are affected. Since most patients meeting clinical criteria for PD or DLB often have involvement of all the staged areas, it is possible they lie on a spectrum in which limbic-predominant pathology precipitates early cognitive impairment, while brainstem-predominant pathology precipitates the motor symptoms of PD. Importantly, pathological staging of subjects correlates with neuron loss and cognitive and motor symptoms [32, 33], suggesting that the spread of pathology in these diseases is predictive of clinical disease progression. Structural imaging of brains from patients with PD has also shown that brain atrophy follows a stereotyped pattern based on neuronal connectivity and that this atrophy correlates with clinical features of disease [34]. There is also recent evidence that oligodendroglial alpha-synuclein pathology may spread through white matter tracts in MSA [35], although studies staging MSA alpha-synuclein pathology are still in the early stages.
Further evidence that pathogenic alpha-synuclein can transmit between neurons has come from PD patients who had grafts of embryonic ventral mesencephalon neurons in their striatums in an experimental therapeutic effort to replace the neurons that were lost during disease. While patients with these grafts showed clinical improvement in motor symptoms, the grafted neurons formed LBs after the relatively short timescale of 11–22 years [36–39]. The young age of the implanted neurons suggests that the neurons did not form LB pathology on their own, but rather were induced to form alpha-synuclein pathology by the PD brain environment. This induction could be indirect or through direct uptake of misfolded alpha-synuclein from LB-containing neurons surrounding the graft [7, 39].
It is important to note that the cell-to-cell transmission of pathogenic alpha-synuclein as a modulator of disease progression is not divorced from the concept of intrinsic cell vulnerability. In no study has it been demonstrated or suggested that all neurons synaptically connected to regions with pathology will themselves develop pathology. Rather, evidence from human neuropathology supports a model in which alpha-synuclein pathology forms in vulnerable neurons and is followed by release of this alpha-synuclein from neurons and propagation along anatomically-connected pathways to other vulnerable neurons in the network.
3 |. CELL AND ANIMAL MODELS OF TRANSMISSION
If indeed extracellular alpha-synuclein can be taken up by other cells and initiate the recruitment and corruption of endogenous alpha-synuclein into misfolded alpha-synuclein pathology, then it should be possible to recapitulate this process in cell culture and animal models. alpha-synuclein can be overexpressed in Escherichia coli and purified by gel filtration and ion exchange [40]. Shaking monomeric alpha-synuclein at 37C and 1000 rpm will rapidly induce misfolding of alpha-synuclein into -sheet rich fibrils, which can be sonicated to produce smaller pre-formed fibrils (PFFs) (Fig. 1). These PFFs provide an experimental pool of misfolded alpha-synuclein. The first experiments to test whether exogenous alpha-synuclein PFFs can induce misfolding of intracellular alpha-synuclein used non-neuronal cells overexpressing alpha-synuclein and a lipophilic reagent to augment the transfer of PFFs to the cytosol [41, 42]. The exogenous PFFs induced misfolding of intracellular alpha-synuclein into LB-like structures that contained major hallmarks of LBs, including phosphorylation of S129 in alpha-synuclein and ubiquitin [41, 42].
FIGURE 1.
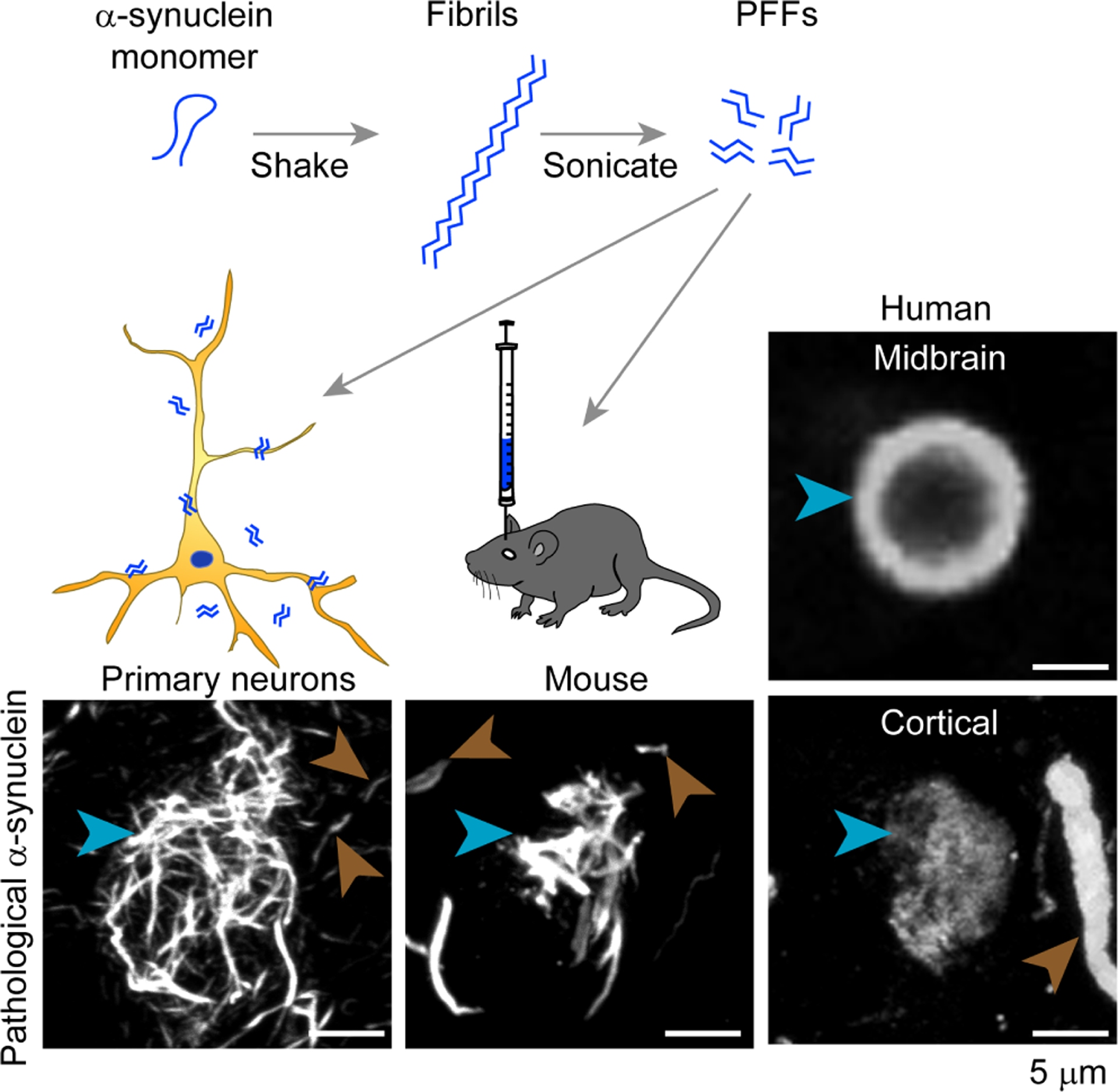
Cell and animal models of alpha-synuclein transmission recapitulate LB aggregates seen in PD. Purified recombinant alpha-synuclein monomer shaken at 37 degrees C and 1000 rotations per minute will spontaneously aggregate into long beta-sheet-rich fibrils. These fibrils can then be sonicated into smaller PFFs that can be added directly to wildtype primary neurons or inoculated into the brain of wildtype mice. This treatment leads to juxtanuclear alpha-synuclein inclusions in neurons and mice that resemble the LBs seen in human PD brains. Cell and mouse inclusions are stained for pS129 alpha-synuclein and the PD tissue is stained for Syn303, which recognizes misfolded alpha-synuclein and reveals LB or LB-like inclusions (blue arrowheads) and Lewy neurites (brown arrowheads). Scale bar = 5 micrometers.
Later, it was discovered that alpha-synuclein PFFs, in the absence of any other reagents, can be taken up into wildtype primary neurons and induce the misfolding of endogenous alpha-synuclein [43] (Fig. 1). The LB-like aggregates in these neurons induce time-dependent neuron dysfunction [43, 44] and death [43, 45]. Primary neuron inclusions contain many known components of LBs, including pS129 alpha-synuclein, ubiquitin and p62, and proteomics of these inclusions has been utilized to identify novel components of early LB formation [46]. While much of the work characterizing alpha-synuclein aggregates has been done in primary hippocampal neurons, alpha-synuclein PFFs induce similar LB-like structures in primary dopaminergic neurons [47, 48], inducing mitochondrial oxidant stress [48] that is thought to be part of PD pathogenesis. alpha-Synuclein extracted from PD and MSA patient brains can also induce pathology in primary neurons [47, 49], allowing the study of different strains/conformations of misfolded alpha-synuclein in culture. This experimental paradigm provides further evidence that the pathological inclusions seen in alpha-synucleinopathies have the ability to transfer potent, strain-specific conformations to normal alpha-synuclein [49].
alpha-synuclein PFFs are also able to template the misfolding of alpha-synuclein in living organisms, leading to the formation of LB-like inclusions in vulnerable neurons throughout the brain (Fig. 1). Initially, alpha-synuclein PFFs were injected into the brains of transgenic mice overexpressing human alpha-synuclein with the familial Ala53Thr mutation [50]. While these mice eventually develop alpha-synuclein inclusions due to expression of the transgene, the injection of alpha-synuclein PFFs dramatically accelerated the formation and spread of alpha-synuclein pathology. Later, injections of alpha-synuclein PFFs into wildtype mice were shown to induce LB-like inclusions which spread through the mouse brain in a defined spatiotemporal pattern [51]. This paradigm has been replicated by several laboratories using mice [52–55] and others using rats [56]. Mice and rats injected with alpha-synuclein PFFs undergo degeneration of vulnerable nigral neurons in an alpha-synuclein pathology-dependent manner, exhibiting motor deficits at later stages. Live imaging studies have also demonstrated that inclusions in cortical neurons lead to the death of these neurons, and that large, LB-like inclusions are not cleared from neurons without the concurrent death of those neurons [57].
The injection of alpha-synuclein PFFs into rodents recapitulates many of the main features of PD pathology and provides a valuable tool for assessing PD-relevant phenotypes and therapies. The alpha-synuclein pathology in rodents spreads in a time- and brain region-dependent manner that induces the death of inclusion-bearing neurons [51, 54, 57]. The induction of alpha-synuclein pathology in non-transgenic animals means that this paradigm can be used to assess the contribution of genetic risk factors to the development and toxicity of alpha-synuclein pathology. This paradigm is also ideal to test the efficacy of therapeutic interventions in sporadic PD since pathology can be induced without genetic or pharmacological manipulation of mice [58, 59]. The spread of pathology in injected rodents, as in PD patients, is complex. Future quantitative pathology experiments will be important to understand the network basis for when and how alpha-synuclein pathology spreads through the brain.
4 |. MECHANISMS OF TRANSMISSION
4.1 |. Uptake
There does not seem to be a significant barrier in cells to the uptake of misfolded alpha-synuclein. HEK and SH-SY5Y cell lines [60], primary neurons [43, 61], primary oligodendrocytes [62], primary astrocytes (unpublished results), and neurons in vivo [51] take up alpha-synuclein within minutes of exposure to the exogenous protein. Different methods of uptake have been proposed, including micropinocytosis [61, 63] and receptor-mediated endocytosis [64, 65]. Given the promiscuity of uptake in completely different cell types, it seems unlikely that a selectively expressed receptor would mediate the bulk of uptake. However, several mediators of alpha-synuclein uptake have been proposed and are reviewed in detail in section 6. Once endocytosed, misfolded alpha-synuclein is transported through the endo-lysosomal pathway where smaller units coalesce into mature lysosomes [61] (Fig. 2).
FIGURE 2.
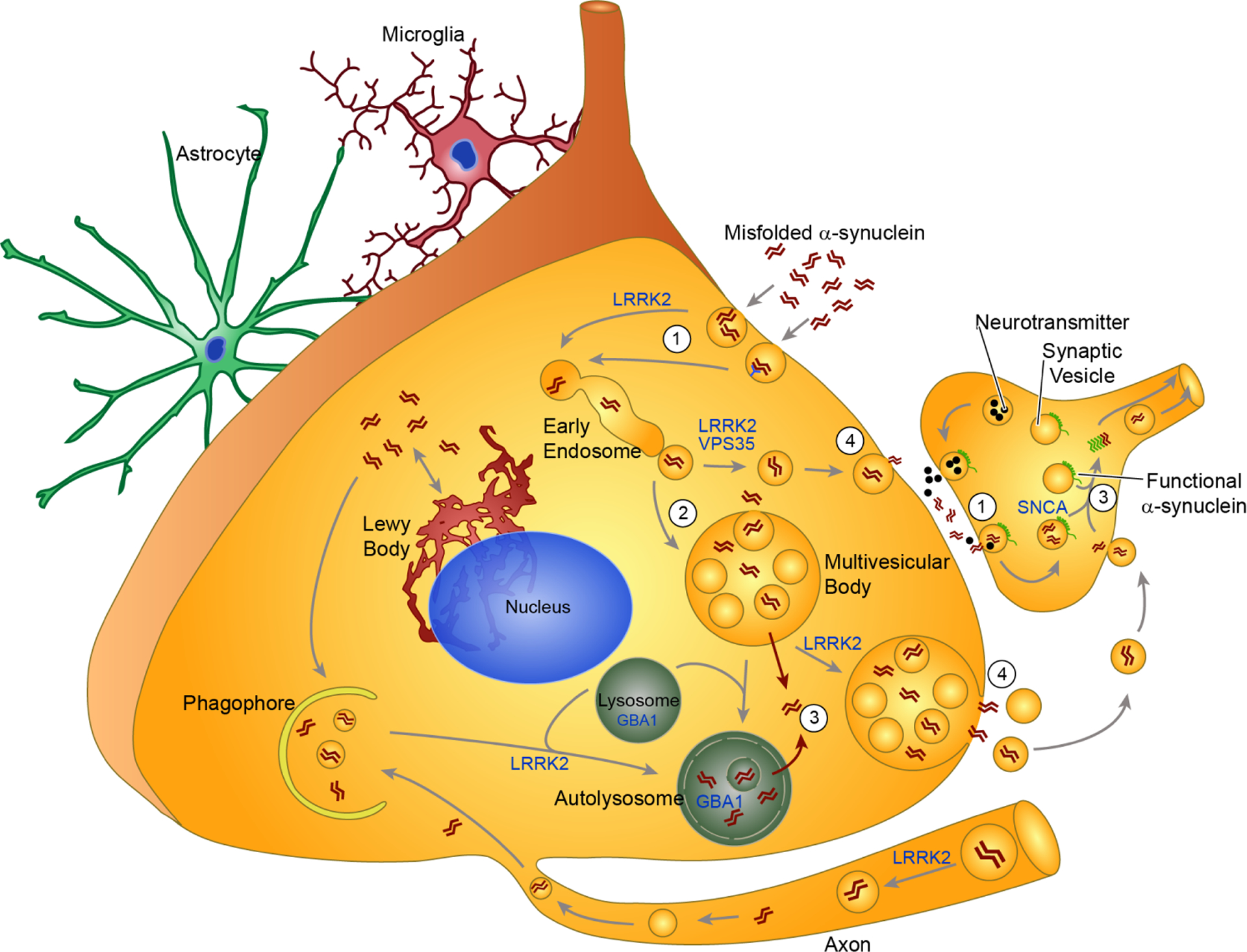
Mechanisms of pathogenic alpha-synuclein transmission. This schematic of alpha-synuclein transmission demonstrates current hypotheses about the uptake (1), processing (2), escape (3) and release (4) of misfolded alpha-synuclein in a neuron. Astrocytes and microglia are shown in the background because they may also play critical roles in this process. Additionally, several of the shown cellular processes may occur in astrocytes and microglia as well. Extracellular alpha-synuclein can be taken up into neurons either through non-selective endocytosis or receptor-mediated endocytosis (1). In presynaptic terminals, the synaptic vesicle cycle that releases neurotransmitter may also promote uptake of extracellular alpha-synuclein, explaining the retrograde spread of alpha-synuclein pathology in animal models. Following uptake, misfolded alpha-synuclein is processed through the endo-lysosomal pathway (2). At this point, alpha-synuclein may be directly targeted back to the plasma membrane through recycling vesicles, or into multivesicular bodies. At some point in processing, alpha-synuclein seeds can escape the endolysosomal pathway and become free in the cytosol (3). This allows recruitment of endogenous, functional alpha-synuclein into fibrillar structures, a process which would be most devastating at the presynaptic terminal where alpha-synuclein levels are highest (3). Autophagy has the ability to degrade both cytosolic and vesicular alpha-synuclein through engulfment by the phagophore and subsequent fusion with the lysosome. Vesicles or multivesicular bodies containing alpha-synuclein can be targeted for exocytosis, possible to unburden the lysosomal degradation pathway. This leads to release of pathogenic alpha-synuclein from the neuron (4). The LB may provide an additional method of protection for neurons that cannot degrade all misfolded alpha-synuclein. Instead, they deposit it in juxtanuclear inclusions that keep alpha-synuclein from moving around in a more damaging form. Several genetic risk factors for PD encode proteins that are involved in parts of this alpha-synuclein processing pathway. LRRK2 has been implicated in several of the vesicular trafficking pathways. VPS35 may also interact with LRRK2 in vesicle trafficking. GBA1 is a lysosomal enzyme, and mutations may shift the flux through this system.
4.2 |. Processing
Most of the alpha-synuclein taken up by neurons is retained in lysosomes for at least a week [61]. Lysosomal alpha-synuclein can be proteolytically processed and, possibly through lysosomal rupture [66, 67], a small amount of misfolded alpha-synuclein ends up in the cytoplasm (Fig. 2). It is not completely clear how alpha-synuclein escapes endosomal compartments, but lysosomal perturbation with chloroquine is sufficient to elevate the number of seeds in the cytoplasm and lead to increased recruitment of endogenous alpha-synuclein[61]. Lysosomal integrity is impaired with age, and elevated release from damaged lysosomes [66, 67] is one mechanism by which age could lead to pathogenic alpha-synuclein accumulation. Endogenous alpha-synuclein is recruited into thread-like neuritic inclusions that are trafficked to the soma of neurons where they form juxtanuclear LB-like inclusions that are modified by phosphorylation, ubiquitination in association with additional proteins such as p62 [43, 46]. Each new inclusion formed may act as a new pool of pathogenic alpha-synuclein. Large inclusions do not seem to be cleared over months in mice [57], but it is possible that small aggregates are released from neurons.
4.3 |. Release
Release of misfolded alpha-synuclein seems to be a much more regulated event, based on the sparse amount of alpha-synuclein that can be detected in the media and cerebrospinal fluid [29]. Cell culture studies have found that cellular stress on the proteostasis machinery can lead to unconventional alpha-synuclein release [68–72]. This action, while preventing the short-term consequences to an individual cell, may be the primary mechanism leading to the transmission of pathological alpha-synuclein. Aging neurons with impaired protein degradation machinery may rely on protein release to unburden lysosomal enzymes. The initial misfolding of alpha-synuclein may be stochastic, and only in aged neurons with impaired degradation machinery are seeds allowed to induce full-blown LBs. This suggests the concept of a “selfish” cell which, to prolong its own functionality, releases toxic alpha-synuclein into the interstitial fluid, and thereby puts at risk nearby vulnerable neurons. Synaptically-connected neurons may be at a particular risk. The presynaptic terminal undergoes rapid cycles of exo- and endocytosis to release neurotransmitter and retrieve membrane and excess neurotransmitter [73]. This rapid membrane cycling may make presynaptic terminals vulnerable to take up the pathogenic alpha-synuclein released by the post-synaptic neuron (Fig. 2). If a main location of uptake is the presynaptic bouton, propagation would proceed retrogradely from distal processes back to the cell body, which is consistent with what has been observed in animal models (unpublished results).
4.4 |. Genetic risk factors
Historically, many of the pathways thought to lead to PD have been described in single cells. However, in light of pathogenic alpha-synuclein transmission, PD pathways and genetic risk factors can be examined in a non-cell autonomous light. That is, genetic mutations that lead to PD may not always lead to cell death in the affected neurons. The primary dysfunctional pathway may actually lead to rescue of the affected neurons through redistribution of pathology to other neurons. Alternatively, pathological alpha-synuclein may affect non-neuronal cells such as astrocytes and microglia. Prominent genes with variants that predispose individuals to PD are expressed both in neurons and non-neuronal cells [74, 75]. It is currently unclear for many of these genes if pathogenesis is driven in a neuronal or non-neuronal cell type.
In addition to aggregated alpha-synuclein being the primary pathology present in PD and related alpha-synucleinopathies, alpha-synuclein gene mutations, duplications and triplications lead to familial PD [16–25]. Of all the potential PD risk factors, alpha-synuclein shows the most selective expression in neurons [76]. This does not, however, isolate the effects of alpha-synuclein to neurons. As reviewed here, there is extensive evidence that alpha-synuclein can be released by neurons and taken up by nearby neurons or other cell types. Remarkably, the primary pathology in MSA is alpha-synuclein inclusions in oligodendrocytes [15], despite the fact that oligodendrocytes express little or no alpha-synuclein under normal conditions [77]. It is not clear why pathology occurs in oligodendrocytes in MSA, but a prominent theory holds that alpha-synuclein released from neurons can be taken up by oligodendrocytes that surround the axons of neurons, and the cellular milieu of oligodendrocytes can lead to conversion of alpha-synuclein to a highly pathogenic conformational strain [78].
Leucine-rich repeat kinase 2 (LRRK2) is a large protein with scaffolding, GTPase and kinase domains [79]. LRRK2 mutations that lead to PD are largely thought to lead to elevated kinase function [80–83], and kinase inhibitors have been developed and are currently in clinical trials for PD. LRRK2 has been implicated in many cellular processes, but much recent evidence indicates that it is involved in vesicle trafficking, positioning it to regulate endocytosis, endo-lysosomal trafficking and exocytosis through Rab GTPase phosphorylation [82] (Fig. 2). LRRK2 is therefore in a critical position to be involved in pathogenic alpha-synuclein transmission through uptake of alpha-synuclein, lysosomal processing and release. LRRK2 is most highly expressed in astrocytes within the brain [84], and also within peripheral immune cells in the blood [85]. It is currently unclear if primary neurons expressing mutant LRRK2 can alter alpha-synuclein pathogenesis [47, 86] or if LRRK2 expression in non-neuronal cells is more critical for pathogenesis.
VPS35, one of three proteins that make up the retromer complex, has been implicated in diverse membrane trafficking events [87]. Similar to LRRK2, VPS35 is positioned as a potential effector of alpha-synuclein transmission (Fig. 2). In fact, LRRK2 and VPS35 may function in the same pathway, since it was recently shown that mutant VPS35 elevates Rab GTPase phosphorylation in a LRRK2-dependent manner [88].
Mutations in GBA1 are the most common genetic risk factor for PD and related synucleinopathies, with 2.3–9.4 percent of PD patients carrying a mutation in GBA1 [75]. GBA1 encodes the lysosomal enzyme glucocerebrosidase (GCase, Fig. 2), which metabolizes glucosylceramide and glucosylsphingosine [75]. GBA1 mutations in some cell and animal models elevate alpha-synuclein expression [75], and overexpressed alpha-synuclein can reduce GCase activity and protein levels [75], leading to the hypothesis that alpha-synuclein and GCase act in a pathogenic loop in which alpha-synuclein accumulation leads to decreased GCase activity, which leads to further alpha-synuclein accumulation [89]. The diminished lysosomal capacity induced by lipid accumulation [89] may result in enhanced escape of alpha-synuclein seeds from the lysosome, or the enhanced release of alpha-synuclein into the interstitial fluid. Either of these will modulate the vulnerability of neurons to LB formation and enhance pathological alpha-synuclein transmission. GCase is also highly expressed in non-neuronal cells [75], and gliosis is a primary feature in mouse models of GCase dysfunction [90, 91], suggesting potential non-cell autonomous features of PD pathogenesis.
5 |. THE ROLE OF CONFORMATION IN TRANSMISSION
Currently, the source of alpha-synuclein used for most experiments is produced in bacteria and fibrillized in de novo reactions into PFFs in vitro [40]. Primary neurons and mice treated with PFFs form aggregates that in many ways resemble LBs seen in synucleinopathy patients [43, 46, 51]. In addition, recombinant PFFs have high purity and can be chemically labelled for biochemistry and imaging experiments [61]. These advantages have allowed several groups to solve the structure of alpha-synuclein fibrils. The first of these studies solved the structure of isotopically-labeled alpha-synuclein PFFs using solid-state nuclear magnetic resonance (NMR) [92]. The -sheet folds of alpha-synuclein formed a Greek-key structure, although residues 55–62 forming the outer -sheet were not well resolved. Later studies using cryo-electron microscopy (EM), identified a similar Greek key structure, but found that two protofilaments are staggered and tightly bind to form a polar fibril with helical structure [93]. The fibrils form a left-handed helix with a width of 10 nm and a pitch of 239 nm [94]. Interestingly, many of the mutations that lead to early-onset familial PD are at the interface of the two protofibrils and may influence fibril structure. A recent report found that multiple polymorphs of fibrils could be formed from recombinant alpha-synuclein with a similar -sheet core, but with disparate protofibril interaction interfaces [95].
Despite the biological activity of recombinant alpha-synuclein PFFs, human-derived fibrillar alpha-synuclein may have critical deviations from the structure of recombinant fibrils. Also, different alpha-synucleinopathies may harbor different forms of pathological alpha-synuclein [49, 96]. A recent study using detergent-insoluble alpha-synuclein from LBs and MSA GCIs found that GCIs have a different proteolytic digestion pattern than LB alpha-synuclein [49]. Further, GCI alpha-synuclein had approximately 1,000-fold greater biological activity than LB or PFF alpha-synuclein. These and other findings from this study indicate that MSA alpha-synuclein has a different structure than LB or PFF alpha-synuclein. However, it has not yet been possible to solve the structure of human PD- or MSA-derived alpha-synuclein fibrils. The recent publication of tau fibril structures from AD [97] and Pick’s disease [98] brains using cryo-EM suggests that this technique has the ability to solve human biosample-derived amyloid structures. Knowing the structure of pathological alpha-synuclein fibrils will allow the in silico development of molecules that interfere with alpha-synuclein fibrilization and provide information for rational design of small molecule therapies or antibodies for immunotherapy of patients with synucleinopathies.
6 |. THERAPEUTIC IMPLICATIONS OF TRANSMISSION
No disease-modifying therapy currently exists for any neurodegenerative disease, including PD and related dementias. For many PD patients, motor symptoms can be managed for years with symptomatic dopamine replacement therapy [99, 100]. However, this symptomatic therapy does not alter the disease course, so the alpha-synuclein pathology spreads inexorably to higher cortical areas, leading to the cognitive deficits [6] that are more deleterious to quality of life than the original motor symptoms. Additionally, patients with DLB present with cognitive deficits which are not ameliorated by dopamine replacement, and other non-motor symptoms are also a primary complaint of PD patients [101]. Therefore, it is critical to develop therapies that target the underlying pathophysiology of disease rather than simply treat symptoms. The theory of cell-to-cell transmission of alpha-synuclein pathology has led to several novel strategies of therapy that target extracellular alpha-synuclein itself, alpha-synuclein internalization, non-cell autonomous effects of alpha-synuclein or alpha-synuclein transmission from outside of the central nervous system.
6.1 |. Glymphatic clearance
The presence of interstitial alpha-synuclein opens novel avenues of clearance by the brain and therapeutic intervention. Recent research has indicated that the brain has the ability to drain interstitial fluid through perivenous pathways [102]. While it has been previously recognized that solutes injected into parenchyma of the brain can diffuse towards blood vessels, a glial “lymphatic” (glymphatic) system was recently described in which CSF in periarterial spaces can be driven by a differential pressure gradient through the parenchyma, clearing out metabolic waste through the perivenous space (Fig. 3) [102]. While the brain had been thought until this point to be devoid of lymphatic vessels, the glymphatic system seems to serve this function for the brain. This was followed by the discovery of lymphatic vessels in the meninges [103, 104] which can clear macromolecules from interstitial fluids [105]. In addition, disruption of this clearance in a mouse model of AD lead to an exacerbation of amyloid- (A) accumulation and cognitive decline [105]. Disruption of lymphatic drainage has also been recently shown to exacerbate alpha-synuclein pathology and motor symptoms in a transgenic mouse model [106]. Therefore, treatments which enhance the glymphatic clearance of interstitial waste have the potential to remove extracellular alpha-synuclein and prevent its spread through the brain (Fig. 3).
FIGURE 3.
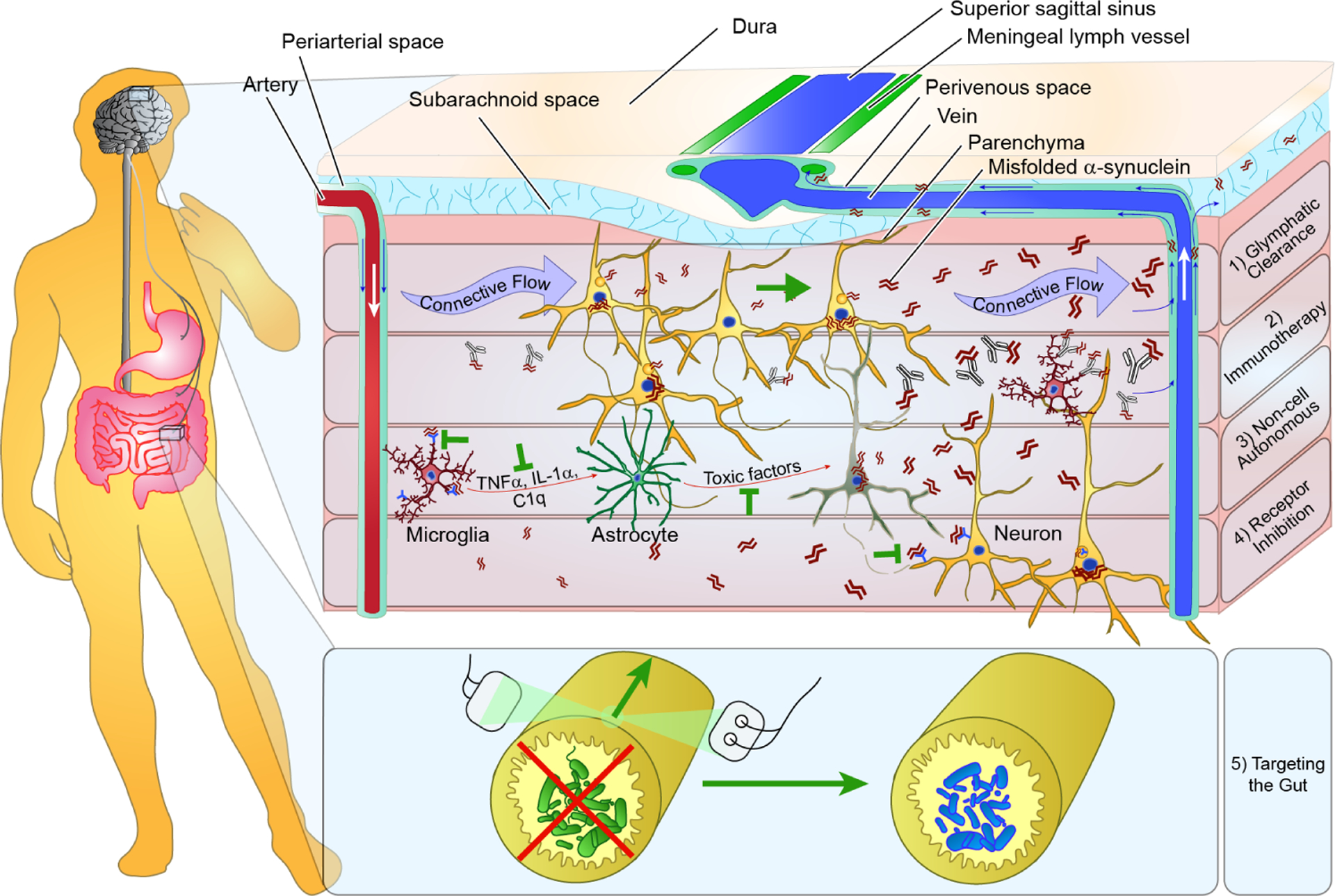
Therapeutic implications of alpha-synuclein transmission. The presence of extracellular pathogenic alpha-synuclein species has profound implications for how PD can be treated. This schematic drawing displays several of the therapeutic approaches that are possible to directly address alpha-synuclein transmission. A hypothetical patient is shown with both brain and gut treatments outlined. (1) The recent elucidation of a glymphatic pathway which clears metabolites through connective flow between periarterial and perivenous space presents the possibility of simply enhancing clearance of released alpha-synuclein. This alpha-synuclein would then enter the CSF and potentially meningeal lymph vessels. (2) Immunotherapy targeting alpha-synuclein would exclusively target extracellular species and thereby enhance their degradation through microglia or enhance their clearance into the CSF. (3) Previous work has shown that misfolded alpha-synuclein itself or some factor released by neurons can activate microglia which signal astrocytes and induce a neurotoxic phenotype. Blocking alpha-synuclein binding to microglia, microglial signaling to astrocytes or astrocyte release of neurotoxic factors are all potential therapeutic strategies. (4) Some studies have indicated that extracellular alpha-synuclein is taken up in a receptor-dependent manner and that blockade of binding to the receptor can reduce alpha-synuclein uptake and improve neuron health. (5) PD patients have elevated rates of constipation and deleterious microbiota population. Treatment of constipation via chemical or electrical stimulation or repopulation of intestines with healthy microbiota are treatments which may not only alleviate the GI symptoms experience by PD patients, but also neurological symptoms.
6.2 |. alpha-Synuclein immunotherapy
Immunotherapy has arisen as one of the most effective treatments of modern biology due to the ability of immune cells to generate highly selective antibodies against deleterious proteins. However, cells in the brain do not generate antibodies and of the antibodies circulating in the blood, only about 0.1
alpha-synuclein immunotherapy is a more recent approach to PD therapy, predicated on the hope that there is sufficient extracellular alpha-synuclein for immunotherapy to reduce the total alpha-synuclein internalized by vulnerable neurons or to specifically block transmission from affected areas of the brain to non-affected areas. Active immunization directly with alpha-synuclein provides long-term titers of anti-alpha-synuclein antibodies with a single inoculation of antigen [109]. This strategy has been shown to reduce aggregated alpha-synuclein in transgenic mouse [109] and rat [110] brains. The drawback of this approach is that individuals may mount different immune responses to the antigen, including a T-cell response which could lead to autoimmunity. This is of particular importance for alpha-synuclein immunization due to the high expression of alpha-synuclein in red blood cells [111]. The more recent development of short peptides that mimic the original antigen but are too short to induce a T cell response have been developed by AFFIRiS and show efficacy in clearing alpha-synuclein aggregates from the brains of transgenic mouse models [112]. More importantly, two of these vaccines, PD01A and PD03A, were found safe and tolerable in Phase Ib clinical trials (press release).
A second strategy for alpha-synuclein immunotherapy is passive immunization with monoclonal antibodies (Mabs) generated against alpha-synuclein. One of the benefits of this strategy is that the Mabs used can be highly optimized through screening to specifically identify pathogenic alpha-synuclein and/or to have exceptionally high affinity. In addition, after cloning, the Mabs can be modified to be smaller and hybridized with effector domains that enhance blood-brain barrier penetration. The drawback of this approach is that the injected antibodies have a relatively short half-life that will require patients to receive many injections throughout their lives. Passive immunization has proven to be highly effective in animal models of PD at reducing alpha-synuclein aggregates and rescuing neuron death [113–121]. Antibodies have been generated that are selective for alpha-synuclein oligomers [118–120], c-terminal truncated alpha-synuclein [114, 116] or fibrillar alpha-synuclein [113]. It is currently unclear whether antibodies directed against certain epitopes will be more efficacious than others, but multiple passive immunotherapies are proceeding to clinical trials. PRX002, developed by Prothena and Roche was recently found to be safe and tolerable in Phase Ib clinical trials and showed a dose-dependent reduction in free serum alpha-synuclein [122]. An alternate strategy for identifying antibodies for immunotherapy pans for autoantibodies from humans that recognize alpha-synuclein [123, 124]. BIIB054, which was developed in such a fashion by Neurimmune and Biogen, is currently in clinical trials for treatment of synucleinopathies [125].
alpha-Synuclein immunotherapy is one of the most developed clinical interventions to date. Multiple possible mechanisms exist for the efficacy of alpha-synuclein immunotherapy in PD (Fig. 3), including: (1) Antibody binding to alpha-synuclein aggregates may simply prevent further templating of monomeric alpha-synuclein. (2) alpha-synuclein antibodies may bind extracellular alpha-synuclein aggregates, prevent uptake by neurons, and facilitate clearance through glymphatic pathways. (3) Antibody-bound alpha-synuclein may be preferentially taken up by multiple cell types and promote the lysosomal degradation of alpha-synuclein. This degradation could be facilitated in microglia by recognition of the Fc (constant) domain of antibodies. While it is unclear whether the Fc domain is necessary for therapeutic efficacy of antibodies, microglia may play another role in PD pathogenesis that makes them a target of therapeutic intervention.
6.3 |. Non-cell autonomous targets
Inflammation and microglial activation have been observed in PD through the detection of inflammatory biomarkers in the cerebrospinal fluid of PD patients [126], PET imaging of activated microglia [127], and immunohistochemistry to label activated microglia [128–130]. While microglia function in healthy brains to maintain neurons, disease-state microglia can release pro-inflammatory cytokines. Recently, these cytokines were shown to act on astrocytes to initiate a state that is toxic to nearby neurons in experimental models [131] (Fig. 3). Misfolded alpha-synuclein can induce microglia to adopt an inflammatory state [132, 133]. Microglia can then induce the activation of astrocytes and subsequent neurotoxicity [133]. Blocking of the initial activation of microglia by misfolded alpha-synuclein is therefore an attractive strategy for treating synucleinopathies. One study found that oligomeric alpha-synuclein is an agonist for toll-like receptor 2 (TLR2), and that this binding leads to activation of microglia [132]. The same group subsequently found that genetic, pharmacological or immunotherapy antagonism of the TLR2 pathway led to reduced alpha-synuclein aggregation and neurotoxicity [134, 135]. Another study found that glucagon-like peptide-1 receptor (GLP1R) agonist NLY01 acting in microglia can rescue the cascade of inflammation initiation by misfolded alpha-synuclein, leading to rescue of neuron pathology and death [133]. GLP1R agonists have a long history in the treatment of various diseases. Exendin-4 was approved in 2005 to treat type 2 diabetes mellitus. In a recent clinical trial, PD patients treated with GLP1R agonist Exenatide showed marked improvement in motor scores over placebo group [136]. While both TLR2 and GLP1R are expressed on a multitude of cell types, recent studies suggest that targeting these receptors may reduce toxic inflammation in PD. Alternatively, therapies that target the activation of astrocytes by microglia-derived cytokines may provide alternative means of preventing neurodegeneration in PD [137].
6.4 |. alpha-Synuclein receptor inhibtion
It is clear from studies in wildtype primary neurons and mouse models that exogenous misfolded alpha-synuclein can be taken up into neurons and induce the misfolding of endogenous alpha-synuclein. It is not clear what process or processes mediate the uptake of exogenous alpha-synuclein, but once internalized alpha-synuclein is trafficked to lysosomes through endo-lysosomal intermediates [61]. The presence of alpha-synuclein in these membranous structures has suggested to some that they may be internalized through receptor-mediated endocytosis. Hence, blocking the interaction between alpha-synuclein and its receptor could prevent transmission between vulnerable neurons. Multiple groups have tested whether alpha-synuclein aggregates can be internalized via micropinocytosis, which is dependent on heparan sulfate proteoglycans (HSPGs). Both non-neuronal cells [63] and primary neurons [61] show reduction in alpha-synuclein uptake with heparin application, suggesting that uptake is at least partially dependent on HSPG binding.
Later, a screen for receptors binding misfolded alpha-synuclein identified lymphocyte-activation gene 3 (LAG3) as a high-affinity binding partner of pathological alpha-synuclein [64]. Despite the fact that LAG3 accounted for a minority of binding to neurons, LAG3 knockout reduced the amount of alpha-synuclein transmitted between neurons and rescued neuron death in vivo. Additionally, an antibody which interfered with the interaction between LAG3 and alpha-synuclein also reduced pathology.
A binding to the cellular prion protein (PrPC) has previously been shown to result in a synaptic toxicity and neuronal dysfunction via mGluR5 and Fyn [138, 139]. Two recent studies found that PrPc is also responsible for uptake of misfolded alpha-synuclein [65] and alpha-synuclein-induced neuronal dysfunction and cognitive impairment [140]. The first study suggested a novel function for PrPC, which is normally found on the extracellular membrane of neurons. Genetic knockout of PrPC resulted in reduction of misfolded alpha-synuclein uptake in primary neurons and reduced aggregated alpha-synuclein in vivo [65]. The second study found that misfolded alpha-synuclein can act in a manner similar to A. alpha-synuclein synaptotoxicity was dependent on PrPC expression, mGluR5 phosphorylation of Fyn kinase and activation of NMDA receptors [140]. These studies all indicate that blocking the interaction of misfolded alpha-synuclein with cellular receptors may prevent the transmission and/or toxicity of alpha-synuclein pathology (Fig. 3).
6.5 |. Targeting the gut
The remarkable appearance of alpha-synuclein inclusions in the olfactory bulb and DMX nucleus prior to the onset of clinical symptoms in PD [30, 31] suggests that PD pathology may initially occur at peripheral sites, then travel to the central nervous system via neurons and induce the central nervous system disorder that is recognized as PD. Substantial research effort has been directed to the examination of peripheral nerves for alpha-synuclein pathology, especially gastric neurons innervated by DMX neurons. alpha-synuclein inclusions have been identified, initially in the gastric submucosa of the fundus [141], and later in other areas of the gastrointestinal (GI) tract [142]. However, the specificity of these inclusions as a marker of PD pathogenesis is still a matter of debate [143]. Studies in rodents have demonstrated that alpha-synuclein is able to spread from the gut through the vagus nerve into the brain [144–147], supporting the notion that alpha-synuclein pathology could begin in the periphery.
There is a long history of an association between PD and GI dysfunction [2, 148–150]. One of the most highly reported non-motor symptoms of PD is constipation [151–154], which may lead to a buildup of abnormal gut microbiota. In fact, gut microbiota are different between PD and control subjects [155]. In a remarkable recent study in transgenic mice overexpressing human alpha-synuclein, ablation of the gut microbiome led to reduced alpha-synuclein pathology, decreased microglial activation and improved motor function [156]. These germ-free mice were then given gut microbiota from healthy controls or PD patients. Mice populated with PD microbiota had diminished motor performance compared to mice with healthy control microbiota.
Together, these results suggest that GI dysfunction may initiate or exacerbate underlying pathophysiology of PD patients. The connection between GI dysfunction and PD is consistent with the hypothesis by Braak and colleagues that PD could be caused by a “neurotropic pathogen” in the gut. It is currently unclear what this pathogen may be, but treatments are available for GI dysfunction, which may not only alleviate the painful constipation experienced by 80–90 percent of PD patients [154], but also provide corresponding benefits to neurological sequelae. Potential treatments to alleviate GI function include laxatives, interferential current therapy to stimulate gastric motility [157], antibiotic treatment and microbiota replacement [154, 158] (Fig. 3). If GI function is a cause of initial alpha-synuclein misfolding, then treatment may need to commence before the onset of PD symptoms in order to be effective, necessitating the identification of early biomarkers. However, if gut-brain signaling can directly modulate PD pathogenesis, as was seen in one PD mouse model [156], then treatment of current PD patients for their GI dysfunction well into the course of their illness could slow progression of neurological symptoms as well.
7 |. CONCLUSIONS
For 20 years, alpha-synuclein pathology been a sine qua non of PD and related alpha-synucleinopathies. However, it has been unclear what cellular factors lead to aggregation of alpha-synuclein and why specific neurons are affected at different stages of disease. alpha-Synuclein pathology staging suggested that the appearance of LBs is not random, but rather occurs progressively in synaptically-connected networks. Studies of grafted neurons as an experimental treatment of PD further suggested that pathological alpha-synuclein could be transmitted from diseased endogenous neurons to non-diseased grafted neurons. There has now been extensive work to show that the aggregated alpha-synuclein in PD brains and recombinant alpha-synuclein PFFs have the ability to be taken up by neurons and initiate the seeding of intracellular alpha-synuclein into fibrillar LB-like structures. Further, this misfolded alpha-synuclein can be released by neurons and can travel through the brain of animals in a conserved spatiotemporal pattern. While alpha-synuclein inclusions lead to neuronal dysfunction and death in affected neurons, extracellular alpha-synuclein may also lead to neurotoxic responses by microglia and astrocytes. The transmission of pathogenic alpha-synuclein is a new window for therapeutic treatment of PD, and several novel treatments targeting this pathway have shown promise in animal models and are currently in clinical trials for treatment of alpha-synucleinopathies.
8 | REFERENCES
- 1.Pringsheim T, et al. , The prevalence of Parkinson’s disease: a systematic review and meta-analysis. Mov Disord, 2014. 29(13): p. 1583–90. [DOI] [PubMed] [Google Scholar]
- 2.Parkinson J, An essay on the shaking palsy. 1817. J Neuropsychiatry Clin Neurosci, 2002. 14(2): p. 223–36; discussion 222. [DOI] [PubMed] [Google Scholar]
- 3.Poewe W, et al. , Parkinson disease. Nat Rev Dis Primers, 2017. 3: p. 17013. [DOI] [PubMed] [Google Scholar]
- 4.Dauer W and Przedborski S, Parkinson’s disease: mechanisms and models. Neuron, 2003. 39(6): p. 889–909. [DOI] [PubMed] [Google Scholar]
- 5.Irwin DJ, Lee VM, and Trojanowski JQ, Parkinson’s disease dementia: convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat Rev Neurosci, 2013. 14(9): p. 626–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Irwin DJ, et al. , Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol, 2017. 16(1): p. 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsuboi Y, Uchikado H, and Dickson DW, Neuropathology of Parkinson’s disease dementia and dementia with Lewy bodies with reference to striatal pathology. Parkinsonism Relat Disord, 2007. 13 Suppl 3:p. S221–4. [DOI] [PubMed] [Google Scholar]
- 8.Postuma RB, et al. , Abolishing the 1-year rule: How much evidence will be enough? Mov Disord, 2016. 31(11): p. 1623–1627. [DOI] [PubMed] [Google Scholar]
- 9.Jellinger KA, Neurobiology of cognitive impairment in Parkinson’s disease. Expert Rev Neurother, 2012. 12(12): p. 1451–66. [DOI] [PubMed] [Google Scholar]
- 10.McKeith IG, et al. , Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology, 2017. 89(1): p. 88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodrigues e Silva AM, et al. , Who was the man who discovered the “Lewy bodies”? Mov Disord, 2010. 25(12): p. 1765–73. [DOI] [PubMed] [Google Scholar]
- 12.Spillantini MG, et al. , Alpha-synuclein in Lewy bodies. Nature, 1997. 388(6645): p. 839–40. [DOI] [PubMed] [Google Scholar]
- 13.Spillantini MG, et al. , Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett, 1998. 251(3): p. 205–8. [DOI] [PubMed] [Google Scholar]
- 14.Baba M, et al. , Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol, 1998. 152(4): p. 879–84. [PMC free article] [PubMed] [Google Scholar]
- 15.Tu PH, et al. , Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble alpha-synuclein. Ann Neurol, 1998. 44(3): p. 415–22. [DOI] [PubMed] [Google Scholar]
- 16.Chartier-Harlin MC, et al. , Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet, 2004. 364(9440): p. 1167–9. [DOI] [PubMed] [Google Scholar]
- 17.Ibanez P, et al. , Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet, 2004. 364(9440):p. 1169–71. [DOI] [PubMed] [Google Scholar]
- 18.Singleton AB, et al. , alpha-Synuclein locus triplication causes Parkinson’s disease. Science, 2003. 302(5646): p. 841. [DOI] [PubMed] [Google Scholar]
- 19.Appel-Cresswell S, et al. , Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord, 2013. 28(6): p. 811–3. [DOI] [PubMed] [Google Scholar]
- 20.Kiely AP, et al. , alpha-Synucleinopathy associated with G51D SNCA mutation: a link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol, 2013. 125(5):p. 753–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kruger R, et al. , Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet, 1998. 18(2): p. 106–8. [DOI] [PubMed] [Google Scholar]
- 22.Pasanen P, et al. , Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol Aging, 2014. 35(9): p. 2180 e1–5. [DOI] [PubMed] [Google Scholar]
- 23.Polymeropoulos MH, et al. , Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science, 1997. 276(5321): p. 2045–7. [DOI] [PubMed] [Google Scholar]
- 24.Proukakis C, et al. , A novel alpha-synuclein missense mutation in Parkinson disease. Neurology, 2013. 80(11): p. 1062–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zarranz JJ, et al. , The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol, 2004. 55(2): p. 164–73. [DOI] [PubMed] [Google Scholar]
- 26.Irwin DJ, et al. , Evaluation of potential infectivity of Alzheimer and Parkinson disease proteins in recipients of cadaver-derived human growth hormone. JAMA Neurol, 2013. 70(4): p. 462–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maroteaux L, Campanelli JT, and Scheller RH, Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci, 1988. 8(8): p. 2804–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Surmeier DJ, Obeso JA, and Halliday GM, Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci, 2017. 18(2): p. 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.El-Agnaf OM, et al. , Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J, 2003. 17(13): p. 1945–7. [DOI] [PubMed] [Google Scholar]
- 30.Braak H, et al. , Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson’s disease (preclinical and clinical stages). J Neurol, 2002. 249 Suppl 3: p. III/1–5. [DOI] [PubMed] [Google Scholar]
- 31.Braak H, et al. , Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging, 2003. 24(2): p. 197–211. [DOI] [PubMed] [Google Scholar]
- 32.Beach TG, et al. , Unified staging system for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol, 2009. 117(6): p. 613–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Braak H, Rub U, and Del Tredici K, Cognitive decline correlates with neuropathological stage in Parkinson’s disease. J Neurol Sci, 2006. 248(1–2): p. 255–8. [DOI] [PubMed] [Google Scholar]
- 34.Dagher A and Zeighami Y, Testing the Protein Propagation Hypothesis of Parkinson Disease. J Exp Neurosci, 2018. 12: p. 1179069518786715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brettschneider J, et al. , Converging Patterns of alpha-Synuclein Pathology in Multiple System Atrophy. J Neuropathol Exp Neurol, 2018. 77(11): p. 1005–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kordower JH, et al. , Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med, 2008. 14(5): p. 504–6. [DOI] [PubMed] [Google Scholar]
- 37.Li JY, et al. , Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med, 2008. 14(5): p. 501–3. [DOI] [PubMed] [Google Scholar]
- 38.Li JY, et al. , Characterization of Lewy body pathology in 12- and 16-year-old intrastriatal mesencephalic grafts surviving in a patient with Parkinson’s disease. Mov Disord, 2010. 25(8): p. 1091–6. [DOI] [PubMed] [Google Scholar]
- 39.Kurowska Z, et al. , Signs of degeneration in 12–22-year old grafts of mesencephalic dopamine neurons in patients with Parkinson’s disease. J Parkinsons Dis, 2011. 1(1): p. 83–92. [DOI] [PubMed] [Google Scholar]
- 40.Volpicelli-Daley LA, Luk KC, and Lee VM, Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nat Protoc, 2014. 9(9): p. 2135–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luk KC, et al. , Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A, 2009. 106(47): p. 20051–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nonaka T, et al. , Seeded aggregation and toxicity of alpha-synuclein and tau: cellular models of neurodegenerative diseases. J Biol Chem, 2010. 285(45): p. 34885–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Volpicelli-Daley LA, et al. , Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron, 2011. 72(1): p. 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Froula JM, et al. , alpha-Synuclein fibril-induced paradoxical structural and functional defects in hippocampal neurons. Acta Neuropathol Commun, 2018. 6(1): p. 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luna E, et al. , Differential alpha-synuclein expression contributes to selective vulnerability of hippocampal neuron subpopulations to fibril-induced toxicity. Acta Neuropathol, 2018. 135(6): p. 855–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Henderson MX, et al. , Unbiased proteomics of early Lewy body formation model implicates active microtubule affinity-regulating kinases (MARKs) in synucleinopathies. J Neurosci, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Henderson MX, et al. , LRRK2 activity does not dramatically alter alpha-synuclein pathology in primary neurons. Acta Neuropathol Commun, 2018. 6(1): p. 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dryanovski DI, et al. , Calcium entry and alpha-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J Neurosci, 2013. 33(24): p. 10154–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peng C, et al. , Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luk KC, et al. , Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med, 2012. 209(5): p. 975–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luk KC, et al. , Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science, 2012. 338(6109): p. 949–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Masuda-Suzukake M, et al. , Prion-like spreading of pathological alpha-synuclein in brain. Brain, 2013. 136(Pt 4): p. 1128–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Steiner JA, Quansah E, and Brundin P, The concept of alpha-synuclein as a prion-like protein: ten years after. Cell Tissue Res, 2018. 373(1): p. 161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rey NL, et al. , Spread of aggregates after olfactory bulb injection of alpha-synuclein fibrils is associated with early neuronal loss and is reduced long term. Acta Neuropathol, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Koller EJ, et al. , Inflammatory pre-conditioning restricts the seeded induction of alpha-synuclein pathology in wild type mice. Mol Neurodegener, 2017. 12(1): p. 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paumier KL, et al. , Intrastriatal injection of pre-formed mouse alpha-synuclein fibrils into rats triggers alpha-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol Dis, 2015. 82:p. 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Osterberg VR, et al. , Progressive aggregation of alpha-synuclein and selective degeneration of lewy inclusion-bearing neurons in a mouse model of parkinsonism. Cell Rep, 2015. 10(8): p. 1252–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Henderson MX, et al. , LRRK2 inhibition does not impart protection from alpha-synuclein pathology and neuron death in non-transgenic mice. Acta Neuropathol Commun, 2019. 7(1): p. 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhao HT, et al. , LRRK2 Antisense Oligonucleotides Ameliorate alpha-Synuclein Inclusion Formation in a Parkinson’s Disease Mouse Model. Mol Ther Nucleic Acids, 2017. 8: p. 508–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hansen C, et al. , alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest, 2011. 121(2): p. 715–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Karpowicz RJ Jr., et al. , Selective imaging of internalized proteopathic alpha-synuclein seeds in primary neurons reveals mechanistic insight into transmission of synucleinopathies. J Biol Chem, 2017. 292(32): p. 13482–13497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kisos H, et al. , Increased neuronal alpha-synuclein pathology associates with its accumulation in oligodendrocytes in mice modeling alpha-synucleinopathies. PLoS One, 2012. 7(10): p. e46817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Holmes BB, et al. , Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A, 2013. 110(33): p. E3138–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mao X, et al. , Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science, 2016. 353(6307). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Aulic S, et al. , alpha-Synuclein Amyloids Hijack Prion Protein to Gain Cell Entry, Facilitate Cell-to-Cell Spreading and Block Prion Replication. Sci Rep, 2017. 7(1): p. 10050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jiang P, et al. , Impaired endo-lysosomal membrane integrity accelerates the seeding progression of alpha-synuclein aggregates. Sci Rep, 2017. 7(1): p. 7690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Flavin WP, et al. , Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol, 2017. 134(4): p. 629–653. [DOI] [PubMed] [Google Scholar]
- 68.Jang A, et al. , Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J Neurochem, 2010. 113(5): p. 1263–74. [DOI] [PubMed] [Google Scholar]
- 69.Bae EJ, et al. , Lipid peroxidation product 4-hydroxy-2-nonenal promotes seeding-capable oligomer formation and cell-to-cell transfer of alpha-synuclein. Antioxid Redox Signal, 2013. 18(7): p. 770–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee HJ, et al. , Dopamine promotes formation and secretion of non-fibrillar alpha-synuclein oligomers. Exp Mol Med, 2011. 43(4): p. 216–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee HJ, et al. , Autophagic failure promotes the exocytosis and intercellular transfer of alpha-synuclein. Exp Mol Med, 2013. 45: p. e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fussi N, et al. , Exosomal secretion of alpha-synuclein as protective mechanism after upstream blockage of macroautophagy. Cell Death Dis, 2018. 9(7): p. 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kononenko NL and Haucke V, Molecular mechanisms of presynaptic membrane retrieval and synaptic vesicle reformation. Neuron, 2015. 85(3): p. 484–96. [DOI] [PubMed] [Google Scholar]
- 74.Schapansky J, Nardozzi JD, and LaVoie MJ, The complex relationships between microglia, alpha-synuclein, and LRRK2 in Parkinson’s disease. Neuroscience, 2015. 302: p. 74–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aflaki E, Westbroek W, and Sidransky E, The Complicated Relationship between Gaucher Disease and Parkinsonism: Insights from a Rare Disease. Neuron, 2017. 93(4): p. 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Taguchi K, et al. , Expression of alpha-synuclein is regulated in a neuronal cell type-dependent manner. Anat Sci Int, 2019. 94(1): p. 11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Richter-Landsberg C, et al. , alpha-synuclein is developmentally expressed in cultured rat brain oligodendrocytes. J Neurosci Res, 2000. 62(1): p. 9–14. [DOI] [PubMed] [Google Scholar]
- 78.Peng C, et al. , Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature, 2018. 557(7706): p. 558–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Price A, et al. , The LRRK2 signalling system. Cell Tissue Res, 2018. 373(1): p. 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Greggio E, et al. , Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis, 2006. 23(2): p. 329–41. [DOI] [PubMed] [Google Scholar]
- 81.Sheng Z, et al. , Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci Transl Med, 2012. 4(164): p. 164ra161.. [DOI] [PubMed] [Google Scholar]
- 82.Steger M, et al. , Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife, 2016. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.West AB, et al. , Parkinson’s disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A, 2005. 102(46): p. 16842–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Henry AG, et al. , Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression. Hum Mol Genet, 2015. 24(21): p. 6013–28. [DOI] [PubMed] [Google Scholar]
- 85.Hakimi M, et al. , Parkinson’s disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J Neural Transm (Vienna), 2011. 118(5): p. 795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Volpicelli-Daley LA, et al. , G2019S-LRRK2 Expression Augments alpha-Synuclein Sequestration into Inclusions in Neurons. J Neurosci, 2016. 36(28): p. 7415–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Rahman AA and Morrison BE, Contributions of VPS35 Mutations to Parkinson’s Disease. Neuroscience, 2019. 401: p. 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mir R, et al. , The Parkinson’s disease VPS35[D620N] mutation enhances LRRK2-mediated Rab protein phosphorylation in mouse and human. Biochem J, 2018. 475(11): p. 1861–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mazzulli JR, et al. , Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell, 2011. 146(1): p. 37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rocha EM, et al. , Sustained Systemic Glucocerebrosidase Inhibition Induces Brain alpha-Synuclein Aggregation, Microglia and Complement C1q Activation in Mice. Antioxid Redox Signal, 2015. 23(6): p. 550–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Soria FN, et al. , Glucocerebrosidase deficiency in dopaminergic neurons induces microglial activation without neurodegeneration. Hum Mol Genet, 2017. 26(14): p. 2603–2615. [DOI] [PubMed] [Google Scholar]
- 92.Tuttle MD, et al. , Solid-state NMR structure of a pathogenic fibril of full-length human alpha-synuclein. Nat Struct Mol Biol, 2016. 23(5): p. 409–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Guerrero-Ferreira R, et al. , Cryo-EM structure of alpha-synuclein fibrils. Elife, 2018. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li Y, et al. , Amyloid fibril structure of alpha-synuclein determined by cryo-electron microscopy. Cell Res, 2018. 28(9): p. 897–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li B, et al. , Cryo-EM of full-length alpha-synuclein reveals fibril polymorphs with a common structural kernel. Nat Commun, 2018. 9(1): p. 3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Prusiner SB, et al. , Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A, 2015. 112(38): p. E5308–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fitzpatrick AWP, et al. , Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature, 2017. 547(7662): p. 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Falcon B, et al. , Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yahr MD, et al. , Treatment of parkinsonism with levodopa. Arch Neurol, 1969. 21(4): p. 343–54. [DOI] [PubMed] [Google Scholar]
- 100.Cotzias GC, Papavasiliou PS, and Gellene R, Modification of Parkinsonism–chronic treatment with L-dopa. N Engl J Med, 1969. 280(7): p. 337–45. [DOI] [PubMed] [Google Scholar]
- 101.Poewe W, Non-motor symptoms in Parkinson’s disease. Eur J Neurol, 2008. 15 Suppl 1: p. 14–20. [DOI] [PubMed] [Google Scholar]
- 102.Plog BA and Nedergaard M, The Glymphatic System in Central Nervous System Health and Disease: Past, Present, and Future. Annu Rev Pathol, 2018. 13: p. 379–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Louveau A, et al. , Structural and functional features of central nervous system lymphatic vessels. Nature, 2015. 523(7560): p. 337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Aspelund A, et al. , A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J Exp Med, 2015. 212(7): p. 991–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Da Mesquita S, et al. , Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature, 2018. 560(7717): p. 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zou W, et al. , Blocking meningeal lymphatic drainage aggravates Parkinson’s disease-like pathology in mice overexpressing mutated alpha-synuclein. Transl Neurodegener, 2019. 8: p. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Banks WA, et al. , Passage of amyloid beta protein antibody across the blood-brain barrier in a mouse model of Alzheimer’s disease. Peptides, 2002. 23(12): p. 2223–6. [DOI] [PubMed] [Google Scholar]
- 108.Schenk D, et al. , Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature, 1999. 400(6740): p. 173–7. [DOI] [PubMed] [Google Scholar]
- 109.Masliah E, et al. , Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron, 2005. 46(6): p. 857–68. [DOI] [PubMed] [Google Scholar]
- 110.Sanchez-Guajardo V, et al. , alpha-Synuclein vaccination prevents the accumulation of parkinson disease-like pathologic inclusions in striatum in association with regulatory T cell recruitment in a rat model. J Neuropathol Exp Neurol, 2013. 72(7): p. 624–45. [DOI] [PubMed] [Google Scholar]
- 111.Scherzer CR, et al. , GATA transcription factors directly regulate the Parkinson’s disease-linked gene alpha-synuclein. Proc Natl Acad Sci U S A, 2008. 105(31): p. 10907–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mandler M, et al. , Next-generation active immunization approach for synucleinopathies: implications for Parkinson’s disease clinical trials. Acta Neuropathol, 2014. 127(6): p. 861–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tran HT, et al. , Alpha-synuclein immunotherapy blocks uptake and templated propagation of misfolded alpha-synuclein and neurodegeneration. Cell Rep, 2014. 7(6): p. 2054–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Masliah E, et al. , Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS One, 2011. 6(4): p. e19338. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 115.Bae EJ, et al. , Antibody-aided clearance of extracellular alpha-synuclein prevents cell-to-cell aggregate transmission. J Neurosci, 2012. 32(39): p. 13454–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Games D, et al. , Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J Neurosci, 2014. 34(28): p. 9441–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Shahaduzzaman M, et al. , Anti-human alpha-synuclein N-terminal peptide antibody protects against dopaminergic cell death and ameliorates behavioral deficits in an AAV-alpha-synuclein rat model of Parkinson’s disease. PLoS One, 2015. 10(2): p. e0116841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Spencer B, et al. , ESCRT-mediated uptake and degradation of brain-targeted alpha-synuclein single chain antibody attenuates neuronal degeneration in vivo. Mol Ther, 2014. 22(10): p. 1753–67. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 119.Spencer B, et al. , alpha-synuclein conformational antibodies fused to penetratin are effective in models of Lewy body disease. Ann Clin Transl Neurol, 2016. 3(8): p. 588–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.El-Agnaf O, et al. , Differential effects of immunotherapy with antibodies targeting alpha-synuclein oligomers and fibrils in a transgenic model of synucleinopathy. Neurobiol Dis, 2017. 104: p. 85–96. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 121.Spencer B, et al. , Anti-alpha-synuclein immunotherapy reduces alpha-synuclein propagation in the axon and degeneration in a combined viral vector and transgenic model of synucleinopathy. Acta Neuropathol Commun, 2017. 5(1): p. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Schenk DB, et al. , First-in-human assessment of PRX002, an anti-alpha-synuclein monoclonal antibody, in healthy volunteers. Mov Disord, 2017. 32(2): p. 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Papachroni KK, et al. , Autoantibodies to alpha-synuclein in inherited Parkinson’s disease. J Neurochem, 2007. 101(3): p. 749–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Neff F, et al. , Immunotherapy and naturally occurring autoantibodies in neurodegenerative disorders. Autoimmun Rev, 2008. 7(6): p. 501–7. [DOI] [PubMed] [Google Scholar]
- 125.Weihofen A, et al. , Development of an aggregate-selective, human-derived alpha-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson’s disease models. Neurobiol Dis, 2019. 124: p. 276–288. [DOI] [PubMed] [Google Scholar]
- 126.Lindqvist D, et al. , Cerebrospinal fluid inflammatory markers in Parkinson’s disease–associations with depression, fatigue, and cognitive impairment. Brain Behav Immun, 2013. 33: p. 183–9. [DOI] [PubMed] [Google Scholar]
- 127.Gerhard A, et al. , In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis, 2006. 21(2): p. 404–12. [DOI] [PubMed] [Google Scholar]
- 128.McGeer PL, et al. , Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology, 1988. 38(8): p. 1285–91. [DOI] [PubMed] [Google Scholar]
- 129.Imamura K, et al. , Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol, 2003. 106(6): p. 518–26. [DOI] [PubMed] [Google Scholar]
- 130.Croisier E, et al. , Microglial inflammation in the parkinsonian substantia nigra: relationship to alpha-synuclein deposition. J Neuroinflammation, 2005. 2: p. 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Liddelow SA, et al. , Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 2017. 541(7638): p. 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kim C, et al. , Neuron-released oligomeric alpha-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun, 2013. 4: p. 1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Yun SP, et al. , Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med, 2018. 24(7): p. 931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kim C, et al. , Antagonizing Neuronal Toll-like Receptor 2 Prevents Synucleinopathy by Activating Autophagy. Cell Rep, 2015. 13(4): p. 771–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kim C, et al. , Immunotherapy targeting toll-like receptor 2 alleviates neurodegeneration in models of synucleinopathy by modulating alpha-synuclein transmission and neuroinflammation. Mol Neurodegener, 2018. 13(1): p. 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Athauda D, et al. , Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet, 2017. 390(10103): p. 1664–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Liddelow SA and Barres BA, Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity, 2017. 46(6): p. 957–967. [DOI] [PubMed] [Google Scholar]
- 138.Lauren J, et al. , Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature, 2009. 457(7233): p. 1128–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Brody AH and Strittmatter SM, Synaptotoxic Signaling by Amyloid Beta Oligomers in Alzheimer’s Disease Through Prion Protein and mGluR5. Adv Pharmacol, 2018. 82: p. 293–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Ferreira DG, et al. , alpha-synuclein interacts with PrP(C) to induce cognitive impairment through mGluR5 and NMDAR2B. Nat Neurosci, 2017. 20(11): p. 1569–1579. [DOI] [PubMed] [Google Scholar]
- 141.Braak H, et al. , Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett, 2006. 396(1): p. 67–72. [DOI] [PubMed] [Google Scholar]
- 142.Breen DP, Halliday GM, and Lang AE, Gut-brain axis and the spread of alpha-synuclein pathology: Vagal highway or dead end? Mov Disord, 2019. 34(3): p. 307–316. [DOI] [PubMed] [Google Scholar]
- 143.Ruffmann C and Parkkinen L, Gut Feelings About alpha-Synuclein in Gastrointestinal Biopsies: Biomarker in the Making? Mov Disord, 2016. 31(2): p. 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pan-Montojo F, et al. , Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS One, 2010. 5(1): p. e8762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pan-Montojo F, et al. , Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci Rep, 2012. 2: p. 898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Holmqvist S, et al. , Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol, 2014. 128(6): p. 805–20. [DOI] [PubMed] [Google Scholar]
- 147.Manfredsson FP, et al. , Induction of alpha-synuclein pathology in the enteric nervous system of the rat and non-human primate results in gastrointestinal dysmotility and transient CNS pathology. Neurobiol Dis, 2018. 112: p. 106–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lee HJ, et al. , Relation of Enteric alpha-Synuclein to Gastrointestinal Dysfunction in Patients With Parkinson’s Disease and in Neurologically Intact Subjects. J Neurogastroenterol Motil, 2018. 24(3): p. 469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Villumsen M, et al. , Inflammatory bowel disease increases the risk of Parkinson’s disease: a Danish nationwide cohort study 1977–2014. Gut, 2018. [DOI] [PubMed] [Google Scholar]
- 150.Weimers P, et al. , Inflammatory Bowel Disease and Parkinson’s Disease: A Nationwide Swedish Cohort Study. Inflamm Bowel Dis, 2018. [DOI] [PubMed] [Google Scholar]
- 151.Edwards LL, et al. , Gastrointestinal symptoms in Parkinson’s disease. Mov Disord, 1991. 6(2): p. 151–6. [DOI] [PubMed] [Google Scholar]
- 152.Chaudhuri KR, Healy DG, and Schapira AH, Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol, 2006. 5(3): p. 235–45. [DOI] [PubMed] [Google Scholar]
- 153.Martinez-Martin P, et al. , Prevalence of nonmotor symptoms in Parkinson’s disease in an international setting; study using nonmotor symptoms questionnaire in 545 patients. Mov Disord, 2007. 22(11): p. 1623–9. [DOI] [PubMed] [Google Scholar]
- 154.Fasano A, et al. , Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol, 2015. 14(6): p. 625–39. [DOI] [PubMed] [Google Scholar]
- 155.Scheperjans F, et al. , Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov Disord, 2015. 30(3): p. 350–8. [DOI] [PubMed] [Google Scholar]
- 156.Sampson TR, et al. , Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell, 2016. 167(6): p. 1469–1480 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Moore JS, Gibson PR, and Burgell RE, Neuromodulation via Interferential Electrical Stimulation as a Novel Therapy in Gastrointestinal Motility Disorders. J Neurogastroenterol Motil, 2018. 24(1): p. 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Felice VD, et al. , Microbiota-gut-brain signalling in Parkinson’s disease: Implications for non-motor symptoms. Parkinsonism Relat Disord, 2016. 27: p. 1–8. [DOI] [PubMed] [Google Scholar]