

Highlights
► Viruses are the most abundant and most diverse biological entity of the biosphere. ► Rate of virus discoveries by metagenomics: higher than all other methods combined. ► Sequences with no homolog in public database are not all ‘junk’ — but treasure trove and blueprint of virus discovery. ► Metagenomics finds fields of application in clinical virology, public health and beyond. ► We propose the metagenomic Koch's postulates to identify unculturable etiological agents.
Abstract
Monitoring the emergence and re-emergence of viral diseases with the goal of containing the spread of viral agents requires both adequate preparedness and quick response. Identifying the causative agent of a new epidemic is one of the most important steps for effective response to disease outbreaks. Traditionally, virus discovery required propagation of the virus in cell culture, a proven technique responsible for the identification of the vast majority of viruses known to date. However, many viruses cannot be easily propagated in cell culture, thus limiting our knowledge of viruses. Viral metagenomic analyses of environmental samples suggest that the field of virology has explored less than 1% of the extant viral diversity. In the last decade, the culture-independent and sequence-independent metagenomic approach has permitted the discovery of many viruses in a wide range of samples. Phylogenetically, some of these viruses are distantly related to previously discovered viruses. In addition, 60–99% of the sequences generated in different viral metagenomic studies are not homologous to known viruses. In this review, we discuss the advances in the area of viral metagenomics during the last decade and their relevance to virus discovery, clinical microbiology and public health. We discuss the potential of metagenomics for characterization of the normal viral population in a healthy community and identification of viruses that could pose a threat to humans through zoonosis. In addition, we propose a new model of the Koch's postulates named the ‘Metagenomic Koch's Postulates’. Unlike the original Koch's postulates and the Molecular Koch's postulates as formulated by Falkow, the metagenomic Koch's postulates focus on the identification of metagenomic traits in disease cases. The metagenomic traits that can be traced after healthy individuals have been exposed to the source of the suspected pathogen.
Introduction
Direct-count epifluorescence and transmission electron microscopy have shown that viruses are highly abundant in most environments. Bergh et al. demonstrated that 1 l of seawater can contain as many as 1010 virus-like particles (VLPs) [1], approximately 10 times more than the number of prokaryotes. Terrestrial environments often have 109 VLPs per gram. By extrapolation from the estimated number of prokaryotes in different environments [2], viruses are the most abundant entities in the biosphere totaling an estimated number of 1.2 × 1030, 2.6 × 1030, 3.5 × 1031, and 0.25–2.5 × 1031 in the open ocean, in soil and in oceanic and terrestrial subsurfaces, respectively.
In the human holobiont, the 1013 human cells are outnumbered 10-fold by bacteria and 100-fold by viruses. Viral acquisition starts early in life in utero or perinatally during the first few weeks after birth as demonstrated by studies of the gut viral communities in infants. While no VLPs could be detected in the earliest infant stool samples, there were ∼108 virus particles per gram wet weight of feces by the end of the first week [2]. The majority of these VLPs appear to be bacteriophages, the bacteria-infecting viruses [2, 3, 4].
Culture techniques have been the gold standard for the detection of viruses for over a century. Despite the knowledge gained using the cultivation of viruses in cell culture, the consensus is that we have barely begun to chart the viral world, which is the ‘dark matter’ of the biological universe and a rich source of future discoveries [3]. Since the vast majority of viruses are not easily cultivatable, exploration of this dark matter requires culture-independent methods with larger detection coverage than culture.
While the sequencing of the 16S fragment of the small subunit of the ribosomal RNA (rRNA) gene has a proven track record for the detection of known and novel cellular organisms [4, 5, 6, 7, 8, 9, 10], this technique is not applicable to viruses because they lack the gene. Indeed, viruses do not share any common gene that could similarly qualify as a unified phylogenetic marker [11].
Metagenomics is an alternative culture-independent and sequence-independent approach that does not rely on the presence of any particular gene in all the subject entities. This approach was originally developed as a tool for ‘functional and sequence-based analysis of collective microbial genomes contained in environmental samples’ [12, 13]. Early metagenomic studies analyzing the genetic content of environmental samples yielded the identification of metabolic traits, the characterization of organisms and the discovery of new antibiotics and enzymes [12, 13, 14, 15, 16]. Metagenomic studies now encompass a wide scope of research fields including marine environmental research, plant and agricultural biotechnology, human genetics and diagnostics of human diseases. Accordingly, the number of metagenomics papers in peer-reviewed journals has increased greatly since 2002 (Figure 1a). The scope of applications for metagenomics will likely widen from environmental microbiome studies to routine clinical diagnostics for palliative care of patients, public health, industry and beyond.
Figure 1.
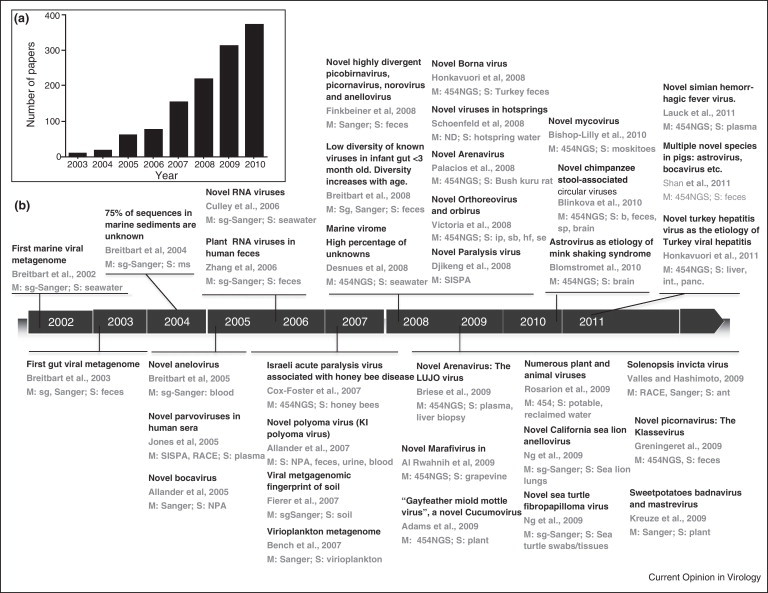
Overview of viral metagenomic studies between 2002 and 2011. (a) The number of published papers on metagenomics (2003–2010), as determined by Pubmed searches using the keywords ‘metagenome OR metagenomics’. (b) Timeline of important landmarks and virus discoveries achieved with the metagenomic approach between 2002 and 2011. The following studies were used to generate the figure: [17••, 18, 20•, 62, 66, 70, 71, 74, 84, 85, 86, 88, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123, 148, 161, 162]. M: Main characterization method used: 454NGS, 454 high-throughput sequencing using GS FLX or GS titanium platform; sg-Sanger, shotgun library with Sanger sequencing method. S: sample, Symbols used for sample type: ip, insect pool; sb, skunk brain; int: intestine; panc: pancreas; hf: human feces; se, sewer effluent; ms: marine sediment; nasopharyngeal aspirates (NPA).
The first application of metagenomics to the field of virology was in the analysis of the viral communities sampled at two near-shore marine locations in San Diego [17••]. Since then, it has been used to survey viruses in numerous environments including freshwater, marine sediment, soil and the human gut. Figure 1b shows an overview of diverse areas where the metagenomic approach has been applied for virus discovery since 2002. The success of these studies relied upon the advances observed in the past decade in the area of sequencing technology and in bioinformatics. Although the fundamental concept of metagenomics has not changed, several technical advances have proven valuable for the discovery of previously unidentified, uncultured viruses. While metagenomics originally depended upon cloning for the analysis of double-stranded DNA genomes [17••, 18, 19, 20•], high-throughput sequencing technologies can now be applied to all types of genomes, including single-stranded DNA and RNA [21].
Historically, diseases caused by viruses have been known before the discovery of their causative agents. The acquired immunodeficiency syndrome (AIDS), poliomyelitis, cervical cancers, and Burkitt's lymphoma were identified before their causative agents. Whereas poliomyelitis was documented in ancient Egyptian literature as early as approximately 3700 BC [22], poliomyelitis virus was not discovered by Landsteiner and Popper until 1909 [23]. Descriptions of clinical conditions likely to have smallpox have been found in ancient literature from Egypt (1100–1580 BC), China (1122 BC) and India (1500 BC)—long before both Jenner's discovery of smallpox vaccination and the later isolation of variola virus [24, 25, 26].
The future perspectives in virology appear that, the metagenomic approach will generate a plethora of genetic information from unknown and potentially infectious agents, some of which could be associated with human diseases. The discovery of viruses will start to precede the characterization of the diseases they cause, well before the pathogenicity of these agents is defined.
At this turning point in history, important questions need to be answered. For example, how far has this new viral metagenomics discipline evolved in its first decade? What has been learned so far that can be applied to viral discovery and the forecasting of future viral outbreaks? In this article, we review virus discovery techniques with a focus on metagenomic approaches that employ high-throughput sequencing technologies to characterize novel viruses.
Traditional techniques for virus discovery
Before the advent of molecular methods, many techniques including filtration, tissue culture, electron microscopy (EM), serology and vaccination have been used for the detection of viruses. In 1892, Ivanovski demonstrated the presence of infectious agents, coined ‘virus’ by Beijerinck in 1898, in filtrate of infected leaves passed through a Chamberland filter. This marks the discovery of the tobacco mosaic virus [27] and the birth of a new era in virology. Until then, the field of virology was not clearly defined. The instrumentation, from the discovery of tissue culture to modern molecular biology methods, has shaped the field and helped to discover many viruses. Since the invention of the technique of tissue culture in 1907 and the propagation of poliovirus in animal cells in 1909, cultivation of viruses has remained the gold standard for virus discovery for over a century [28, 29, 30]. Despite the achievements made by the culture technique, several limitations have hindered the discovery and detection of viruses in routine laboratory settings. Virus propagation requires the development of controlled conditions that mimic the natural ecosystem shared between viruses and their hosts [31•].
The invention of the electron microscope in 1933 provided the first visual proof of a virus. However, this technique is relatively expensive, tedious and lacks both sensitivity and specificity. Alternatively, serology can provide a hint of the acquisition of novel viruses — as was the case for hepatitis C virus [32, 33] — before the viral agents have been cultured or viewed by electron microscopy. The immune sera method has shown little value for virus discovery. The inoculation method, however, not only helped to identify novel viruses, but also was used as an immunization method to confer cross-protection against closely related viruses. Indeed, the cowpox-based inoculation developed by Jenner in 1796 was the first effective vaccine against an infectious disease. Nearly two centuries later, this strategy was used to eradicate smallpox. However, it is unlikely that Jenner's method would pass the scrutiny of modern ethical review boards for vaccine or virus discovery [34].
Molecular methods for virus discovery
The trends in clinical virology practices show gradual substitution of the traditional virus discovery methods with novel molecular biology technology. Nevertheless, traditional and the newer molecular biology techniques to isolate, identify, and characterize viruses play complementary roles in the viral discovery effort. For a comprehensive list and detailed description of molecular methods used for virus discovery, readers are referred to reviews by Delwart [31•] and Tang [35•]. Here, we focus on the viruses discovered using these methods and their future applications in clinical microbiology and public health settings.
Two types of molecular methods have been used for the virus discovery effort: sequence-dependent and sequence-independent methods.
Sequence-dependent methods, including PCR using consensus primers and hybridization methods such as microarrays, require the knowledge of the nucleic acid for the detection of novel viruses. Indeed, consensus sequences of previously known viruses have been used to identify novel viruses including highly divergent clades of human immunodeficiency virus [36], simian retroviruses [37, 38, 39, 40], and hepatitis E virus [41]. However, PCR using consensus primers based on previously characterized viruses have little or no value in detecting completely novel viruses. The microarray techniques were first introduced in 1995 to monitor the expression of multiple genes simultaneously [42]. For virus discovery, microarrays can be prepared with probes that hybridize known viral sequences and potentially novel viruses with sufficient sequence similarity. The method has been applied to detect a wide range of known viruses as well as novel highly divergent viral taxa [43]. Microarray screening has led to the identification and characterization of a novel gammaretrovirus, xenotropic murine leukemia virus-related virus (XMRV), in prostate tumors [43, 44]. Subsequent studies did not confirm these initial findings [45, 46], which points to potential limitations of the method. Another example of a well-known virus discovered with microarrays is SARS-CoV, a highly divergent coronavirus discovered amid a worldwide outbreak of the severe acute respiratory syndrome (SARS) in 2003 [43]. Reproducibility of results between microarray tests is frequently poor [47].
Unlike PCR and microarrays, the sequence-independent viral metagenomic approaches do not rely on prior knowledge of viruses in the samples. The suppression subtractive hybridization (SSH) and representational difference analysis (RDA) are examples of sequence-independent virus discovery methods. SSH was used first to study gene expression [48] and was later applied to investigate the etiology of diseases of unknown origin [49]. By hybridizing DNA obtained from patients and control subjects, nucleic acid from an unknown pathogen(s) can be detected [49, 50, 51]. Use of RDA led to the discovery of human herpes simplex virus type 8 (HHV8) [52], Torque Teno virus (TTV) [53], GBV-A, GBV-B viruses [54] and a novel highly divergent murine norovirus [55]. This method lacks sufficient sensitivity to detect viruses when the viral burden is low or when the DNA sequence of the suspected etiological agent is not clearly distinguishable from the control sample [31].
Sequence-independent single-primer amplification (SISPA) circumvents the viral load limitation of SSH. Although there are several variations to the original protocol published by Reyes et al. [56], the main strategy of SISPA is to exploit the sensitivity and the specificity of PCR amplification using primers that bind oligonucleotide fragments ligated to any putative viral DNA materials in the sample. SISPA has been modified to allow the detection of both DNA and RNA viruses after the removal of genomic and contaminating nucleic acids [57]. The SISPA method was used successfully for the discovery of Hepatitis E virus [58, 59], Norwalk virus [60], Human astrovirus [61, 62], and Parvoviruses 2 and 3 [63]. Another sequence-independent technique, the viral metagenomics (described in detail below), provides superior capability to detect known and unknown viruses than the traditional and molecular sequence-dependent and sequence-independent methods.
Viral metagenomics
Compared to virus discovery approaches outlined above, viral metagenomics is less biased. Potentially, any viruses in the samples, culturable or unculturable, known or novel can be readily detected with the viral metagenomic approach.
Viral metagenomic methods have evolved significantly since they were first developed. In early studies [17••, 18, 19, 20•], preliminary sample preparation involved shearing of DNA and cloning. These steps were required in order to obtain sufficient DNA given the low amount of viral DNA in environmental samples (∼10 μg/100 l of sea water). Because viral DNA often contains modified nucleotides and because some viral genes (e.g. holins and lysozymes) are toxic to cells, the DNA was randomly sheared to produce small fragments before cloning [17••, 18, 19, 20•]. The process of sample preparation has since been streamlined and the sequencing speed increased with the advent of high-throughput sequencing technologies. The replacement of cloning with high-throughput methods has revolutionized metagenomics.
There are several high-throughput sequencing platforms commercially available that vary by the sequencing principle, the sequencing speed, the cost and read length. An overview of a typical viral metagenomic protocol that can be used in a virus discovery study is provided in Figure 2 . Essentially, a metagenomic analysis involves three main steps: (1) sample preparation, (2) high-throughput sequencing and (3) bioinformatic analysis. Below we provide an outline of each of these steps. More detailed descriptions have been previously published [64••].
Figure 2.
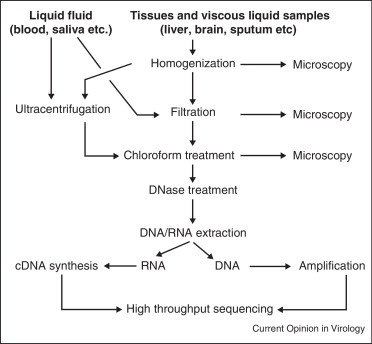
Flow chart for the generation of a viral metagenome using high-throughput sequencing.
Sample preparation. Theoretically, any type of sample can be analyzed using the metagenomic approach, including seawater [65], blood [66], horse feces [67], stool [20•, 68, 69, 70, 71], marine sediments [18], coral tissues [72, 73], and hot springs [74]. Because viral genomes are relatively short, bacterial or eukaryotic nucleic acids can severely interfere with the isolation and detection of viral DNA or RNA that typically represents only a small fraction. Thus, removal of non-viral nucleic acid is necessary [64••, 75]. Homogenization, filtration and ultracentrifugation are often necessary to concentrate the viral particles present in the sample (Figure 2). To ensure that viruses are not lost during the virus preparation, epifluorescence microscopy with SYBR-gold staining is used on aliquots of samples obtained after the homogenization, filtration, and chloroform treatments to monitor the presence of VLPs [64••].
Chloroform treatment followed by DNase digestion is used to remove contaminating DNA. The chloroform disrupts mitochondrial, bacterial and eukaryotic membranes, thereby exposing non-viral DNA to the subsequent nuclease treatment [76, 77]. Unfortunately, chloroform treatment may also cause enveloped viruses to lose their protective lipid membrane, thereby rendering their DNA subject to DNase digestion [66]. Moreover, DNase treatment does not always completely eliminate non-viral DNA in the sample [63, 64••]. After extraction, DNA may need to be amplified with random primers [78, 79]. The Whole Transcriptome Amplification (WTA) kit can be used for the synthesis of cDNA from viral RNA [80].
Single virus genomics (SVG) was introduced by Allen and collaborators to selectively isolate viruses before sequencing [81]. SVG uses flow cytometry to sort viruses based on a method originally described by Brussard et al. [82]. Following the sorting, DNA of different sizes is immobilized in agarose gel, and then amplified using the multiple displacement amplification (MDA) method. The SVG approach can also be applied to RNA viruses provided a reverse-transcription step is inserted between the flow cytometry and MDA.
High-throughput sequencing. Early metagenomic applications involved the generation of shotgun libraries and direct sequencing of the total DNA content using the Sanger enzymatic dideoxy-sequencing method. This approach permitted the discovery of novel phages in marine environments [61, 66]. The Sanger technique had been the standard method for sequencing since it was first described in 1977 [83]. Development of the ‘next-generation’ sequencing platforms offered the combined advantages of speed, automation and high-throughput, thereby increased sequencing capabilities by a factor of 100 to a million relative to the Sanger technology.
The Illumina/Solexa and Roche 454 next-generation sequencing platforms have been used most often in virus discovery (Figure 1). The Illumina/Solexa method is based on sequencing-by-synthesis chemistry using fragments of the sample DNA ligated to oligonucleotide adapters. The adapters on a solid support act as primers for DNA polymerase to incorporate reversible terminator nucleotides, each labeled with a different fluorescent dye. A typical sequencing run can generate up to 18 gigabases of data with an average read length of 75–100 nucleotides [21]. The Sweetpotato badnavirus and the Sweetpotato mastrevirus are examples of viruses discovered using the Illumina/Solexa sequencing platform [84].
The 454 FLX titanium pyrosequencer commercialized by Roche has been the most used for the discovery and characterization of novel viruses (http://www.454.com/publications-and-resources/publications.asp?postback=true). This platform was used for the identification of an uncharacterized mycovirus [85], Solenopsis invicta virus 3 [86], Merino Walk virus and a new arenavirus [87, 88], among others (Figure 1b). For sequencing, DNA is fragmented and ligated to biotinylated specific linkers. The complex DNA/linkers fragment is attached to streptavidin-coated beads that anchor the DNA inside a droplet of water and PCR reagents in oil emulsion. Each fragment is first amplified to produce the template for sequencing reaction. Sequencing is carried out by annealing primers to the linker portion of the template complex, followed by the incorporation of nucleotides by DNA polymerase, which facilitates the extension of the complementary DNA. The pyrophosphate released by this process is measurable by the production of light [89, 90]. The Roche 454 system measures the pyrophosphate released as the result of nucleotide incorporation during DNA synthesis mediated by DNA polymerase. The amount of light released is proportional to the intensity of the light signal captured by a charge-coupled device (CCD) camera, which then converts light signals into digital data [91, 92]. A typical optimum run using a 454 pyrosequencer yields about one million reads with an average length of 350–450 nucleotides, totaling about 0.4 gigabases.
Bioinformatic analyses. The analysis of the copious data generated by high-throughput sequencing is the most challenging aspect of metagenomics. An inherent difficulty in assigning taxonomic designations to viral sequences is that there is no universally homologous nucleic acid component present in all viruses that can be used to build phylogenetic trees — a factor that also fuels the debate over whether or not viruses belong in the tree of life [11, 93, 94, 95, 96]. In most metagenomic studies, sequences generated by high-throughput sequencing are queried by homology search tools to previously documented sequences stored either in a local database or in public databases such as the Genbank. Unfortunately, homology searches against known sequences in Genbank cannot characterize unknown viruses (Figure 3 ).
Figure 3.

The unknowns: sequences with no detectable homologs in Genbank. (a) Proportion of the unknowns reported in viral metagenomic studies of diverse environments. (b) Diagram illustrating the abundance of unknown and known sequences in the environment. The distinction between known and unknown depends on the thresholds used.
The analysis of metagenomic libraries requires fast computation and the right algorithms to characterize sequences as belonging to putative viruses. To ensure that bioinformatic analyses are performed only on high quality data, the reads are typically processed through a software pipeline to remove any background sequences including host and bacterial DNA that had not been removed by the filtration, chloroform, and DNase I treatments [97, 98, 99]. The resulting sequence reads are assembled with strict parameters to generate contigs, each made of sequences derived from the same organism quasi-species. Using a stringent assembly parameter is critical to avoid sequence chimerization. The contigs sequences are then compared to the Genbank non-redundant nucleotide database using BLAST [100] or USEARCH [101]. Note that using a database containing only viral sequences will not be able to identify bacterial, archaeal or eukaryotic sequences and lead to an overestimation of the fraction of unknowns (see below).
With the increasing number of data generated from different studies, there is a need for a cross-metagenome meta-analysis [102, 103]. This is particularly important because of the diversity of different viral metagenomic protocols and the lack of standard algorithm for downstream data analysis. The following items should be included in any report on viral metagenomic studies: firstly, the sequencing platform and its version number; secondly, raw sequence data accession numbers in a public database; thirdly, details about the bioinformatic analysis, including the homology search tool and the database being used to assign the taxonomy, and their versions; fourthly, a list of known and previously unknown viruses found, clearly showing if the ‘novel’ viruses are new strains of a previously described species or completely different viruses; and fifthly, causality evidence if any.
The challenge with the unknown sequences
The most intriguing aspect of viral metagenomics is the fact that a large number — usually the majority — of sequences has no significant similarity to anything known. In this review, we refer to these sequences as the ‘unknown’ (Figure 3a). A typical human or environmental viral metagenome can contain between 60% and 99% unknown sequences (Figure 3) [17••, 18, 20•, 62, 66, 70, 71, 74, 84, 85, 86, 88, 104, 105, 106, 107, 108, 109, 110, 111, 112, 113, 114, 115, 116, 117, 118, 119, 120, 121, 122, 123]. Factors contributing to this variation include the sample type, the length of the sequence reads, the homology search method (BLASTn, tBLASTx, etc.), the similarity threshold (E-value cutoff), the database and version of that database used for the homology searches (Figure 3b). Depending on how they are viewed, the unknowns can represent either a formidable challenge or a treasure trove for virus discovery. Although researchers often tend to consider the unknowns as ‘junk,’ these sequences could be a valuable blueprint for the discovery of novel viruses [112, 124, 125]. Thus far, there is a lack of suitable bioinformatic methods to characterize the unknown sequences.
A tentative solution is to compare the sequences between samples in order to at least gain some insight about the viral entities that are shared between them. A program such as PHACCS (PHAge Communities from Contig Spectra) can be used to assess the biodiversity of uncultured viral communities by mathematically modeling the community structure using the contig spectrum of metagenome assemblies [126]. This method was extended to assess cross-assemblies of reads from different samples [65], providing a homology-independent tool for the comparison of metagenomes with a high proportion of unknown sequences. Although PHACCS may provide a glimpse of the composition and difference between metagenomes, it has limited value for the characterization of novel viruses. Two tools can be used to predict whether unknown sequences are from bacteriophages undergoing lytic and lysogenic lifestyles. One such tool described by Deschavanne et al. [127] compares the genome signatures of query sequences against those of their host genome in order to identify host–phage relationship and information about the phage lifestyle. The second method, PHACTS, depends on residual homology between the putative unknown sequence and sets of randomly selected viral proteins from known viruses (K McNair et al., PHACTS: a computational approach to classifying the lifestyle of phages, unpublished data). Alternatively, viruses may be classified by basic sequence properties. For instance, the circularity of the contig, its oligonucleotide profile [128], and the open reading frame (ORF) structure (S Akhter et al., PhiSpy: A novel algorithm for finding prophages in bacterial genomes that combines similarity-based and composition-based strategies, under review) may all provide clues whether the unknown sequence could be from a potential novel virus. These properties can be combined into a prediction network used to classify viruses into lifestyle groups or taxonomic clades.
Although newly discovered viruses are often labeled ‘novel,’ the question remains whether these sequences represent truly novel viruses or ancient viruses that simply have never been observed before. The age of a sequence has traditionally been determined by multiple alignments of query sequences with their homologs and by calculating the divergence times from a common ancestral node on a phylogenetic tree. Dates can be estimated using either a molecular clock [129] or by assigning a calibration date to a specific node in the tree based on fossil or other evidence [130, 131, 132]. For viral metagenomic sequences, however, building a phylogenetic tree is itself problematic because often the sequenced reads may represent non-overlapping subregions of an unknown viral genome. Moreover, there is no fossil data available to calibrate the age of nodes in the tree. A promising approach might be to estimate divergence times from assembled viral contigs. De novo assembly allows non-overlapping regions to be combined into a single consensus sequence. For a given molecular clock, SNP analysis of the contributing reads could provide an estimation of how long ago the sequenced reads diverged. Such estimates may be critical when addressing the question of the origin of a newly identified infectious agent.
Koch's postulates, metagenomics and viral etiology of diseases
Until recently, virus discoveries were made in the context of disease etiology. Thus, virus discovery studies were biased mainly because of the use of convenient samples available from patients. Because of the difficulties involved, the investment of efforts and resources required to isolate viruses often could not be justified outside the disease context. It is likely that the context of the diseases has also led to the misconception that all viruses are pathogenic. This dogma was challenged by the discovery of viruses such as Torque Teno virus (TTV) and hepatitis G virus (GBV-C), originally associated with post-transfusion hepatitis [53, 133, 134, 135], and then were subsequently shown be classical examples of viral commensals [136, 137]. The widely accepted notion that viruses act as obligatory pathogens is beginning to give way to the concept that viruses can be part of the normal flora of the human body. Considering their high abundance in the gastrointestinal tract, on skin and even in blood and lungs [138] it is unlikely that viruses could only be pathogenic without any benefits for their hosts. The abundance of viruses, particularly phages, in the lung — an environment previously thought to be sterile — may reflect their beneficial role in keeping bacterial populations in check [138]. The pathogenicity of the GBV-C has shifted to a more radical designation as a ‘good’ virus in cases of co-infection with HIV. Indeed, GBV-C has been associated with a more favorable prognosis for patients with HIV infection by slowing the progression to AIDS [139, 140]. Similarly, dengue virus, a known pathogen, has been shown to limit HIV-1 replication and to reduce the viral load [141]. These examples need to be taken into account when metagenomic approach is applied to virus discovery. The characterization of a novel virus can be easily achieved in silico with limited bioinformatics tools but the determination of causation may not always be trivial.
The causality is not always conclusive even when the suspect virus is found in the scene of the crime. In other words, finding a virus in a sample from a patient with an illness of unknown etiology and even demonstrating the association does not always prove causation. For this reason, strict guidelines proposed by Robert Koch and later modified by Rivers [142] have been used to assign causality to infectious agents. One of Koch's postulates requires that the candidate etiological agent be isolated from a diseased organism and grown in pure culture. However, many viruses cannot be propagated by culture techniques [143].
New molecular biology techniques have been used for virus discovery bypassing the prerequisite of the Koch's postulates. For instance, the Merkel cell polyomavirus (MCV) was identified as the causative agent of Merkel's cell carcinoma without satisfying all of the requisites of Koch's postulates [144]. Similarly, the sea turtle tornovirus 1 was associated with fibropapillomatosis using a culture-independent metagenomic approach [118].
The methodological shift, from culture to metagenomics, will likely create a paradigm shift in the demonstration of disease causation. In many instances Koch's postulates will no longer be satisfied if culture techniques are used to prove causality. Falkow [145••] proposed the modified Koch's postulates which uses molecular methods to monitor the role played by genes in distinct bacterial virulence. To satisfy the revised molecular Koch's postulates, a strong association must be established between the phenotype or property under investigation and the pathogenic members of a genus or pathogenic strains of a species. The gene of interest should be found in all pathogenic members of the genus or species but be absent in nonpathogenic strains. At best, the nonpathogenic strains could carry the gene with critical mutations that could render the strain non-virulent. However, new molecular methods do not always distinctively characterize virulence genes and make a clear association with a disease of unknown etiology. This could be because genes can be expressed at different time-points during infection. Genes can be turned on and off and may require intrinsic factors in order to trigger the disease process.
Alternatively, we propose the metagenomic Koch's postulates, which focus on the identification of metagenomic traits in disease subjects. The metagenomic traits are molecular markers such as sequence reads, assembled contigs, genes or full-genomes that can uniquely distinguish diseased metagenomes from those obtained from matched healthy control subjects (Figure 4 ). The metagenomic traits found in diseased patients can be monitored in healthy individuals exposed to the suspected infectious agent. Although this novel approach requires separation or isolation of remaining co-occurring disease candidates (Figure 4.3), it does not necessarily require the isolation of the pathogen in tissue culture or pure culture media unlike the original Koch's postulates. Therefore, the genetic make-up of the agent responsible for a disease can provide early clues before its isolation by tissue culture.
Figure 4.
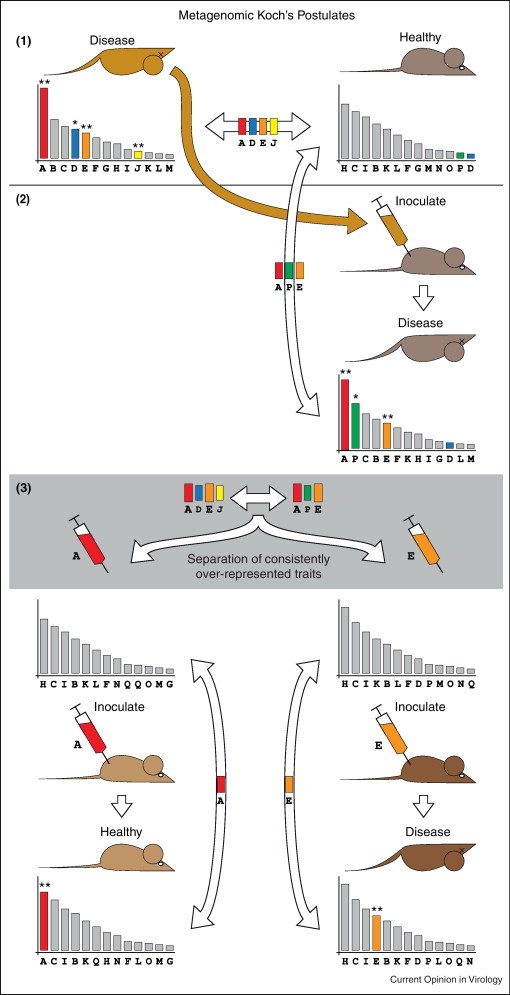
Metagenomic Koch's postulates. Comparison between a diseased and healthy control animal shows a significant difference between the metagenomic libraries (depicted by the histograms of relative abundance reads). In order to fulfill the metagenomic Koch's postulates: (1) The metagenomic traits in diseased subject must be significantly different from healthy subject. For example traits A, D, E and J found in the disease animal that are not present in the healthy control; (2) Inoculation of samples from the disease animal into the healthy control must lead to the induction of the disease state. Comparison of the metagenomes before and after inoculation should suggest the acquisition or increase of new metagenomic traits (A, E and P). New traits can be purified by methods such as serial dilution or time-point sampling of specimens from a disease animal. (3) Inoculation of the suspected purified traits into a healthy animal will induce disease if the traits form the etiology of the disease.
The modified metagenomic Koch's postulates proposed in this paper require that: Firstly, the diseased metagenome be significantly different from the metagenome constructed with the same sample type obtained from a healthy matched control subject. The suspected metagenomic traits must be present and more abundant in the diseased subject compared to matched control (Figure 4.1). Secondly, inoculating a healthy individual with a sample from a diseased subject must result in disease state (Figure 4.2). Differential metagenomic traits in step (1) recovered in the newly induced diseased subject may be the biomarker of the candidate etiological agent; and finally, selective inoculation of samples from the disease subject (in step 2) must induce disease in another healthy control subject if the metagenomic contains the trait associated with the etiological agent of the disease, or phenotype under investigation (Figure 4.3). Assuming that the metagenomic trait ‘E’ (Figure 4.3) is a contig sequence from a previously unknown and unculturable virus, its early identification using the metagenomic approach could spearhead the effort to generate diagnostic assays such as ELISA and PCR, well before the isolation and the characterization of the viruses by culture techniques.
Fulfilling this metagenomic model of the Koch's postulates is possible when one or multiple viral agents are involved in disease causation. With the original Koch's postulates or the modified molecular Koch's postulates, it is difficult enough to prove causality with one suspected agent using the culturing prerequisite. The complexity is even greater when multiple viruses are involved in the causation of a disease.
A similar approach, the siRNA-ome used previously by Kreuze et al. [84] led to the detection of etiological viruses causing diseases in plants despite the low copy number of the suspected traits [84]. The modified metagenomic Koch's postulates could also be tested in human diseases such as the murine mink cell leukemia caused by a C-type retrovirus, named the mink cell focus-inducing virus (MCFIV) [146]. MCFIV requires the cooperative interaction with other viruses to increase its propensity to cause leukemia [146]. The Burkitt's lymphoma caused by others Epstein-Barr virus (EBV) in regions holoendemic for Plasmodium falciparum, the etiology of malaria [147]. Metagenomics could become the future method of choice enabling the simultaneous analysis of multiple agents in a sample and assessment of the association and disease causality without the limitations imposed by culture techniques [138, 148, 149].
Future application of metagenomics to public health
The etiology of many diseases remains unknown. These ailments are collectively defined as diseases of unknown etiology when all conventional testing laboratory techniques are unsuccessful. Yet, the diseases with unknown origin have high rates of morbidity and mortality. For example, as many as 40% of cases of the infantile diarrhea, which alone claims ∼1.8 million fatalities annually, have no known specific causative agent [112]. Infantile diarrhea, the pyrexia of unknown origin, influenza-like illnesses, chronic fatigue syndrome, Alzheimer's disease, various forms of tumors such as diffuse large B-cell lymphoma and many other diseases of unknown origin can benefit directly from the metagenomic technology.
The success of metagenomics in identifying novel viruses in a wide variety of samples opens doors to new application areas particularly in public health and the prevention of infectious diseases. Although the metagenomic technology is not yet part of the routine diagnostics, results from clinical virology research provides valuable proof of concepts for a new era in clinical virology practices. For example, Finkbeiner et al. analyzed samples from 12 children using metagenomics and identified a large number of known eukaryotic viruses as well as sequences from putatively novel viruses [112]. Another study identified a corona-like virus, the Human Cosavirus E1 (HcoSV-E1), in a child with acute diarrhea [150]. These initial studies identified promising viral candidates to establish the etiology in these cases of diarrhea. The 2009 pandemic of influenza A (2009 H1N1) provided proof of concept in that metagenomics was effective to rapidly characterize the full genome of the flu virus [151]. Using the metagenomic approach, Palacios et al. discovered an arenavirus in samples which had tested negative by culture, PCR, serology and a microarray assay using oligonucleotide probes from a wide range of infectious agents [87], suggesting a potential causative agent for unexplained cases of post-transplantation death. In another study, Towner et al. described a new Ebola virus responsible for an outbreak of a hemorrhagic fever in the District of Bindibugyo, Uganda [152]. Rapid identification of these agents would provide the blueprint for the development of therapeutic regimen or preventive vaccine.
Prevention is better than cure. Potentially, a single or multiple jump of an animal virus to humans can have serious consequences. One way to prevent infectious diseases is through vaccine development. But the development of a vaccine takes time and demands a huge amount of resources. Preventing the introduction of an unknown virus to human populations is rather a far-reaching goal unless the methods of virus identification and characterization are put in place. A simple and practical strategy would be to assess the danger posed by viruses that thrive in animals and could cross to human through zoonosis.
Zoonosis is a source of up to 75% of emerging infectious diseases in humans [153]. As such, cross-species transfer from animals to humans has serious repercussions not only in public health but also in the socio-economical and political stability [68, 154, 155, 156, 157, 158]. The detection and characterization of novel viruses are of paramount importance in the forecasting of future outbreaks of viral diseases in humans. Surveying natural reservoirs for potential zoonotic infection [69] and human populations such as bush meat hunters who are exposed to animals could help prevent major outbreaks before the wide spread of viruses to human population. Data obtained in early identification of viruses are valuable for forecasting new emerging and re-emerging viral epidemics.
The experience gained from studying marine environments and hostile mine environments can be applied in public health programs that seek to determine the normal viral population and monitor changes in different geographical settings. We have termed such an approach as Public Health Viral Metagenomics Surveillance (PHVMS). Viral metagenomics surveillance is defined as the survey of the functional and taxonomic signatures representing the viruses normally circulating within that population in the absence of noticeable epidemics. In the event of a zoonotic outbreak, these functional and taxonomic signatures of the virome will likely show detectable shifts. Figure 5 shows a hypothetical rank abundance curve for six viruses (a–f). The introduction of a highly pathogenic species (g) can be expected to result in a disruption of the normal virome, including the appearance of opportunistic viral infections (h).
Figure 5.

Monitoring of emerging infectious diseases using a metagenomic approach. A hypothetical example of the potential use of the Public Health Viral Metagenomics Surveillance (PHVMS) approach for virus discovery based on comparison of viromes sampled before (I) and during (II) an epidemic. Depicted here are the rank abundance curves for viral species (a–h), where g represents a newly introduced, highly pathogenic species and h a less virulent virus.
Using PHACCS analysis [126], several parameters can be compared between the normal and disturbed viromes including the total number of viral species (richness) and their relative abundance (evenness). Another approach would be to determine the normal virome, a background viral metagenome to refer to in case of an outbreak. Lessons learned from studies of bacterial microbial metagenomes suggest that different environments often have different microbial signatures [159], including the functional metabolic information, the nucleotide usage, proportion of different species. Disrupting key metabolic processes of an environment can lead to disruption of the balance in that ecosystem. Similarly, the viromes in different human populations in different locations may display functional profiles characteristic of their respective environment, lifestyle and viruses circulating in each region. The magnitude of disturbance of the virome profile will depend on the fitness and virulence of the newly introduced pathogens and the immune fitness of the host. The viral communities in two different metagenomes can be compared using XIPE [160]. This statistical approach was developed for comparing metagenomic sequences derived from samples collected from the Sargasso Sea and from acid mine drainage and was able to accurately predict the physiology, metabolic potential and ecology of each ecosystem [160].
Conclusion
During the last decade, we have witnessed the emergence of metagenomics as a powerful novel tool with endless areas of applications in virology. Epidemiological data suggest that novel viruses are likely to be introduced into the human population through zoonosis [153, 158]. Also, the danger of intentional introduction of viruses through bioterrorism cannot be ignored. Viral metagenomics is a powerful, fast and sensitive technique available for identifying viruses including those that cannot be detected by conventional culture and sequence-dependent methods.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
Acknowledgements
We are grateful to Merry Youle for helpful suggestions and editing of the manuscript. JM was supported by a grant from the UCSD Center for AIDS Research (NIAID 5 P30 AI36214) and Moores UCSD Cancer Center (NCI 5P30 CA23100). BED was supported by the Dutch Science Foundation (NWO) Veni grant (016.111.075).
References
- 1.Bergh O., Borsheim K.Y., Bratbak G., Heldal M. High abundance of viruses found in aquatic environments. Nature. 1989;340:467–468. doi: 10.1038/340467a0. [DOI] [PubMed] [Google Scholar]
- 2.Whitman W.B., Coleman D.C., Wiebe W.J. Prokaryotes: the unseen majority. Proc Natl Acad Sci U S A. 1998;95:6578–6583. doi: 10.1073/pnas.95.12.6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rohwer F., Youle M. Consider something viral in your research. Nat Rev Microbiol. 2011;9:308–309. doi: 10.1038/nrmicro2563. [DOI] [PubMed] [Google Scholar]
- 4.Schmidt T.M., DeLong E.F., Pace N.R. Analysis of a marine picoplankton community by 16S rRNA gene cloning and sequencing. J Bacteriol. 1991;173:4371–4378. doi: 10.1128/jb.173.14.4371-4378.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shah N., Tang H., Doak T.G., Ye Y. Comparing bacterial communities inferred from 16S rRNA gene sequencing and shotgun metagenomics. Pac Symp Biocomput. 2011:165–176. doi: 10.1142/9789814335058_0018. [DOI] [PubMed] [Google Scholar]
- 6.Gross E.L., Leys E.J., Gasparovich S.R., Firestone N.D., Schwartzbaum J.A., Janies D.A., Asnani K., Griffen A.L. Bacterial 16S sequence analysis of severe caries in young permanent teeth. J Clin Microbiol. 2010;48:4121–4128. doi: 10.1128/JCM.01232-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tringe S.G., Hugenholtz P. A renaissance for the pioneering 16S rRNA gene. Curr Opin Microbiol. 2008;11:442–446. doi: 10.1016/j.mib.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 8.Manichanh C., Chapple C.E., Frangeul L., Gloux K., Guigo R., Dore J. A comparison of random sequence reads versus 16S rDNA sequences for estimating the biodiversity of a metagenomic library. Nucleic Acids Res. 2008;36:5180–5188. doi: 10.1093/nar/gkn496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Streit W.R., Schmitz R.A. Metagenomics — the key to the uncultured microbes. Curr Opin Microbiol. 2004;7:492–498. doi: 10.1016/j.mib.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 10.Liles M.R., Manske B.F., Bintrim S.B., Handelsman J., Goodman R.M. A census of rRNA genes and linked genomic sequences within a soil metagenomic library. Appl Environ Microbiol. 2003;69:2684–2691. doi: 10.1128/AEM.69.5.2684-2691.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rohwer F., Edwards R. The Phage Proteomic Tree: a genome-based taxonomy for phage. J Bacteriol. 2002;184:4529–4535. doi: 10.1128/JB.184.16.4529-4535.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Riesenfeld C.S., Schloss P.D., Handelsman J. Metagenomics: genomic analysis of microbial communities. Annu Rev Genet. 2004;38:525–552. doi: 10.1146/annurev.genet.38.072902.091216. [DOI] [PubMed] [Google Scholar]
- 13.Schloss P.D., Handelsman J. Biotechnological prospects from metagenomics. Curr Opin Biotechnol. 2003;14:303–310. doi: 10.1016/s0958-1669(03)00067-3. [DOI] [PubMed] [Google Scholar]
- 14.Krause D.O., Denman S.E., Mackie R.I., Morrison M., Rae A.L., Attwood G.T., McSweeney C.S. Opportunities to improve fiber degradation in the rumen: microbiology, ecology, and genomics. FEMS Microbiol Rev. 2003;27:663–693. doi: 10.1016/S0168-6445(03)00072-X. [DOI] [PubMed] [Google Scholar]
- 15.Rondon M.R., August P.R., Bettermann A.D., Brady S.F., Grossman T.H., Liles M.R., Loiacono K.A., Lynch B.A., MacNeil I.A., Minor C. Cloning the soil metagenome: a strategy for accessing the genetic and functional diversity of uncultured microorganisms. Appl Environ Microbiol. 2000;66:2541–2547. doi: 10.1128/aem.66.6.2541-2547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brady S.F., Clardy J. Palmitoylputrescine, an antibiotic isolated from the heterologous expression of DNA extracted from bromeliad tank water. J Nat Prod. 2004;67:1283–1286. doi: 10.1021/np0499766. [DOI] [PubMed] [Google Scholar]
- 17••.Breitbart M., Salamon P., Andresen B., Mahaffy J.M., Segall A.M., Mead D., Azam F., Rohwer F. Genomic analysis of uncultured marine viral communities. Proc Natl Acad Sci U S A. 2002;99:14250–14255. doi: 10.1073/pnas.202488399. [DOI] [PMC free article] [PubMed] [Google Scholar]; First proof-of-concept paper applying the metagenomic on marine viruses. This is regarded as the first viral metagenomic study that described the high abundance and high diversity of marine viruses and documentation of a high number of sequences with no homologs in existing databases including Genbank.
- 18.Breitbart M., Felts B., Kelley S., Mahaffy J.M., Nulton J., Salamon P., Rohwer F. Diversity and population structure of a near-shore marine-sediment viral community. Proc Biol Sci. 2004;271:565–574. doi: 10.1098/rspb.2003.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Breitbart M., Wegley L., Leeds S., Schoenfeld T., Rohwer F. Phage community dynamics in hot springs. Appl Environ Microbiol. 2004;70:1633–1640. doi: 10.1128/AEM.70.3.1633-1640.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20•.Breitbart M., Hewson I., Felts B., Mahaffy J.M., Nulton J., Salamon P., Rohwer F. Metagenomic analyses of an uncultured viral community from human feces. J Bacteriol. 2003;185:6220–6223. doi: 10.1128/JB.185.20.6220-6223.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; First application of the metagenomic approach to study viral communities in human feces, spearheading the virus discovery effort on clinical samples. Most sequences were unrelated to previously documented taxa in Genbank.
- 21.Ansorge W.J. Next-generation DNA sequencing techniques. N Biotechnol. 2009;25:195–203. doi: 10.1016/j.nbt.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 22.Paul J.R. Yale Studies in the History of Science and Medicine. Yale University Press; 1971. A history of poliomyelitis. [Google Scholar]
- 23.Kristensson K. The discovery of the poliovirus. Brain Res Bull. 1999;50:461. doi: 10.1016/s0361-9230(99)00134-3. [DOI] [PubMed] [Google Scholar]
- 24.Dixon C.W. Churchill; London: 1962. Smallpox. [Google Scholar]
- 25.Fenner F., Henderson D.A., Arita I., Jezek Z., Ladnyi I.D. In: Smallpox and Its Eradication. Fenner F., editor. WHO; 1988. [Google Scholar]
- 26.Hopkins D.R. Chicago University of Chicago Press; 1983. The Greatest Killer — Smallpox in History. [Google Scholar]
- 27.Lecoq H. Discovery of the first virus, the tobacco mosaic virus: 1892 or 1898? C R Acad Sci III. 2001;324:929–933. doi: 10.1016/s0764-4469(01)01368-3. [DOI] [PubMed] [Google Scholar]
- 28.Hamza I.A., Jurzik L., Uberla K., Wilhelm M. Methods to detect infectious human enteric viruses in environmental water samples. Int J Hyg Environ Health. 2011;214:424–436. doi: 10.1016/j.ijheh.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leland D.S., Ginocchio C.C. Role of cell culture for virus detection in the age of technology. Clin Microbiol Rev. 2007;20:49–78. doi: 10.1128/CMR.00002-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spector S.A., Dankner W.M. Rapid viral diagnostic techniques. Adv Pediatr Infect Dis. 1986;1:37–59. [PubMed] [Google Scholar]
- 31•.Delwart E.L. Viral metagenomics. Rev Med Virol. 2007;17:115–131. doi: 10.1002/rmv.532. [DOI] [PMC free article] [PubMed] [Google Scholar]; A very comprehensive description of metagenomic methods and important benchmarks achieved in the virus discovery effort.
- 32.Choo Q.L., Kuo G., Weiner A.J., Overby L.R., Bradley D.W., Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 33.Kuo G., Choo Q.L., Alter H.J., Gitnick G.L., Redeker A.G., Purcell R.H., Miyamura T., Dienstag J.L., Alter M.J., Stevens C.E. An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science. 1989;244:362–364. doi: 10.1126/science.2496467. [DOI] [PubMed] [Google Scholar]
- 34.Davies H. Ethical reflections on Edward Jenner's experimental treatment. J Med Ethics. 2007;33:174–176. doi: 10.1136/jme.2005.015339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35•.Tang P., Chiu C. Metagenomics for the discovery of novel human viruses. Future Microbiol. 2010;5:177–189. doi: 10.2217/fmb.09.120. [DOI] [PubMed] [Google Scholar]; A comprehensive review describing metagenomic methods and important benchmarks achieved.
- 36.Mokili J.L., Rogers M., Carr J.K., Simmonds P., Bopopi J.M., Foley B.T., Korber B.T., Birx D.L., McCutchan F.E. Identification of a novel clade of human immunodeficiency virus type 1 in Democratic Republic of Congo. AIDS Res Hum Retroviruses. 2002;18:817–823. doi: 10.1089/08892220260139567. [DOI] [PubMed] [Google Scholar]
- 37.Takemura T., Ekwalanga M., Bikandou B., Ido E., Yamaguchi-Kabata Y., Ohkura S., Harada H., Takehisa J., Ichimura H., Parra H.J. A novel simian immunodeficiency virus from black mangabey (Lophocebus aterrimus) in the Democratic Republic of Congo. J Gen Virol. 2005;86:1967–1971. doi: 10.1099/vir.0.80697-0. [DOI] [PubMed] [Google Scholar]
- 38.Barlow K.L., Ajao A.O., Clewley J.P. Characterization of a novel simian immunodeficiency virus (SIVmonNG1) genome sequence from a mona monkey (Cercopithecus mona) J Virol. 2003;77:6879–6888. doi: 10.1128/JVI.77.12.6879-6888.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Osterhaus A.D., Pedersen N., van Amerongen G., Frankenhuis M.T., Marthas M., Reay E., Rose T.M., Pamungkas J., Bosch M.L. Isolation and partial characterization of a lentivirus from talapoin monkeys (Myopithecus talapoin) Virology. 1999;260:116–124. doi: 10.1006/viro.1999.9794. [DOI] [PubMed] [Google Scholar]
- 40.Clewley J.P., Lewis J.C., Brown D.W., Gadsby E.L. A novel simian immunodeficiency virus (SIVdrl) pol sequence from the drill monkey, Mandrillus leucophaeus. J Virol. 1998;72:10305–10309. doi: 10.1128/jvi.72.12.10305-10309.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reyes G.R., Purdy M.A., Kim J.P., Luk K.C., Young L.M., Fry K.E., Bradley D.W. Isolation of a cDNA from the virus responsible for enterically transmitted non-A, non-B hepatitis. Science. 1990;247:1335–1339. doi: 10.1126/science.2107574. [DOI] [PubMed] [Google Scholar]
- 42.Schena M., Shalon D., Davis R.W., Brown P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 43.Wang D., Urisman A., Liu Y.T., Springer M., Ksiazek T.G., Erdman D.D., Mardis E.R., Hickenbotham M., Magrini V., Eldred J. Viral discovery and sequence recovery using DNA microarrays. PLoS Biol. 2003;1:E2. doi: 10.1371/journal.pbio.0000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Urisman A., Molinaro R.J., Fischer N., Plummer S.J., Casey G., Klein E.A., Malathi K., Magi-Galluzzi C., Tubbs R.R., Ganem D. Identification of a novel Gammaretrovirus in prostate tumors of patients homozygous for R462Q RNASEL variant. PLoS Pathog. 2006;2:e25. doi: 10.1371/journal.ppat.0020025. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Farley S.J. Prostate cancer: XMRV — contaminant, not cause? Nat Rev Urol. 2011;8:409. doi: 10.1038/nrurol.2011.101. [DOI] [PubMed] [Google Scholar]
- 46.Switzer W.M., Jia H., Zheng H., Tang S., Heneine W. No association of xenotropic murine leukemia virus-related viruses with prostate cancer. PLoS ONE. 2011;6:e19065. doi: 10.1371/journal.pone.0019065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Draghici S., Khatri P., Eklund A.C., Szallasi Z. Reliability and reproducibility issues in DNA microarray measurements. Trends Genet. 2006;22:101–109. doi: 10.1016/j.tig.2005.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morales P., Thurston C.F. Efficient isolation of genes differentially expressed on cellulose by suppression subtractive hybridization in Agaricus bisporus. Mycol Res. 2003;107:401–407. doi: 10.1017/s0953756203007366. [DOI] [PubMed] [Google Scholar]
- 49.Ambrose H.E., Clewley J.P. Virus discovery by sequence-independent genome amplification. Rev Med Virol. 2006;16:365–383. doi: 10.1002/rmv.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dios S., Poisa-Beiro L., Figueras A., Novoa B. Suppression subtraction hybridization (SSH) and macroarray techniques reveal differential gene expression profiles in brain of sea bream infected with nodavirus. Mol Immunol. 2007;44:2195–2204. doi: 10.1016/j.molimm.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 51.Diatchenko L., Lukyanov S., Lau Y.F., Siebert P.D. Suppression subtractive hybridization: a versatile method for identifying differentially expressed genes. Methods Enzymol. 1999;303:349–380. doi: 10.1016/s0076-6879(99)03022-0. [DOI] [PubMed] [Google Scholar]
- 52.Chang Y., Cesarman E., Pessin M.S., Lee F., Culpepper J., Knowles D.M., Moore P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science. 1994;266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 53.Nishizawa T., Okamoto H., Konishi K., Yoshizawa H., Miyakawa Y., Mayumi M. A novel DNA virus (TTV) associated with elevated transaminase levels in posttransfusion hepatitis of unknown etiology. Biochem Biophys Res Commun. 1997;241:92–97. doi: 10.1006/bbrc.1997.7765. [DOI] [PubMed] [Google Scholar]
- 54.Simons J.N., Pilot-Matias T.J., Leary T.P., Dawson G.J., Desai S.M., Schlauder G.G., Muerhoff A.S., Erker J.C., Buijk S.L., Chalmers M.L. Identification of two flavivirus-like genomes in the GB hepatitis agent. Proc Natl Acad Sci U S A. 1995;92:3401–3405. doi: 10.1073/pnas.92.8.3401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karst S.M., Wobus C.E., Lay M., Davidson J., Virgin HWt. STAT1-dependent innate immunity to a Norwalk-like virus. Science. 2003;299:1575–1578. doi: 10.1126/science.1077905. [DOI] [PubMed] [Google Scholar]
- 56.Reyes G.R., Kim J.P. Sequence-independent, single-primer amplification (SISPA) of complex DNA populations. Mol Cell Probes. 1991;5:473–481. doi: 10.1016/s0890-8508(05)80020-9. [DOI] [PubMed] [Google Scholar]
- 57.Bexfield N., Kellam P. Metagenomics and the molecular identification of novel viruses. Vet J. 2010;190:191–198. doi: 10.1016/j.tvjl.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reyes A., Haynes M., Hanson N., Angly F.E., Heath A.C., Rohwer F., Gordon J.I. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature. 2010;466:334–338. doi: 10.1038/nature09199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reyes G.R., Yarbough P.O., Tam A.W., Purdy M.A., Huang C.C., Kim J.S., Bradley D.W., Fry K.E. Hepatitis E virus (HEV): the novel agent responsible for enterically transmitted non-A, non-B hepatitis. Gastroenterol Jpn. 1991;26(Suppl 3):142–147. doi: 10.1007/BF02779285. [DOI] [PubMed] [Google Scholar]
- 60.Matsui S.M., Kim J.P., Greenberg H.B., Su W., Sun Q., Johnson P.C., DuPont H.L., Oshiro L.S., Reyes G.R. The isolation and characterization of a Norwalk virus-specific cDNA. J Clin Invest. 1991;87:1456–1461. doi: 10.1172/JCI115152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Finkbeiner S.R., Li Y., Ruone S., Conrardy C., Gregoricus N., Toney D., Virgin H.W., Anderson L.J., Vinje J., Wang D. Identification of a novel astrovirus (astrovirus VA1) associated with an outbreak of acute gastroenteritis. J Virol. 2009;83:10836–10839. doi: 10.1128/JVI.00998-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Blomstrom A.L., Widen F., Hammer A.S., Belak S., Berg M. Detection of a novel astrovirus in brain tissue of mink suffering from shaking mink syndrome by use of viral metagenomics. J Clin Microbiol. 2010;48:4392–4396. doi: 10.1128/JCM.01040-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Allander T., Emerson S.U., Engle R.E., Purcell R.H., Bukh J. A virus discovery method incorporating DNase treatment and its application to the identification of two bovine parvovirus species. Proc Natl Acad Sci U S A. 2001;98:11609–11614. doi: 10.1073/pnas.211424698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64••.Thurber R.V., Haynes M., Breitbart M., Wegley L., Rohwer F. Laboratory procedures to generate viral metagenomes. Nat Protoc. 2009;4:470–483. doi: 10.1038/nprot.2009.10. [DOI] [PubMed] [Google Scholar]; An excellent compilation of standard operating procedures to perform metagenomic analysis on different types of samples.
- 65.Angly F.E., Felts B., Breitbart M., Salamon P., Edwards R.A., Carlson C., Chan A.M., Haynes M., Kelley S., Liu H. The marine viromes of four oceanic regions. PLoS Biol. 2006;4:e368. doi: 10.1371/journal.pbio.0040368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Breitbart M., Rohwer F. Method for discovering novel DNA viruses in blood using viral particle selection and shotgun sequencing. Biotechniques. 2005;39:729–736. doi: 10.2144/000112019. [DOI] [PubMed] [Google Scholar]
- 67.Cann A.J., Fandrich S.E., Heaphy S. Analysis of the virus population present in equine faeces indicates the presence of hundreds of uncharacterized virus genomes. Virus Genes. 2005;30:151–156. doi: 10.1007/s11262-004-5624-3. [DOI] [PubMed] [Google Scholar]
- 68.Li L., Kapoor A., Slikas B., Bamidele O.S., Wang C., Shaukat S., Masroor M.A., Wilson M.L., Ndjango J.B., Peeters M. Multiple diverse circoviruses infect farm animals and are commonly found in human and chimpanzee feces. J Virol. 2009;84:1674–1682. doi: 10.1128/JVI.02109-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li L., Victoria J.G., Wang C., Jones M., Fellers G.M., Kunz T.H., Delwart E. Bat guano virome: predominance of dietary viruses from insects and plants plus novel mammalian viruses. J Virol. 2010;84:6955–6965. doi: 10.1128/JVI.00501-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Breitbart M., Haynes M., Kelley S., Angly F., Edwards R.A., Felts B., Mahaffy J.M., Mueller J., Nulton J., Rayhawk S. Viral diversity and dynamics in an infant gut. Res Microbiol. 2008;159:367–373. doi: 10.1016/j.resmic.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 71.Zhang T., Breitbart M., Lee W.H., Run J.Q., Wei C.L., Soh S.W., Hibberd M.L., Liu E.T., Rohwer F., Ruan Y. RNA viral community in human feces: prevalence of plant pathogenic viruses. PLoS Biol. 2006;4:e3. doi: 10.1371/journal.pbio.0040003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marhaver K.L., Edwards R.A., Rohwer F. Viral communities associated with healthy and bleaching corals. Environ Microbiol. 2008;10:2277–2286. doi: 10.1111/j.1462-2920.2008.01652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vega Thurber R., Willner-Hall D., Rodriguez-Mueller B., Desnues C., Edwards R.A., Angly F., Dinsdale E., Kelly L., Rohwer F. Metagenomic analysis of stressed coral holobionts. Environ Microbiol. 2009;11:2148–2163. doi: 10.1111/j.1462-2920.2009.01935.x. [DOI] [PubMed] [Google Scholar]
- 74.Schoenfeld T., Patterson M., Richardson P.M., Wommack K.E., Young M., Mead D. Assembly of viral metagenomes from yellowstone hot springs. Appl Environ Microbiol. 2008;74:4164–4174. doi: 10.1128/AEM.02598-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Edwards R.A., Rodriguez-Brito B., Wegley L., Haynes M., Breitbart M., Peterson D.M., Saar M.O., Alexander S., Alexander E.C., Jr, Rohwer F. Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics. 2006;7:57. doi: 10.1186/1471-2164-7-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Willner D., Furlan M., Schmieder R., Grasis J.A., Pride D.T., Relman D.A., Angly F.E., McDole T., Mariella R.P., Jr, Rohwer F. Microbes and health sackler colloquium: metagenomic detection of phage-encoded platelet-binding factors in the human oral cavity. Proc Natl Acad Sci U S A. 2010 doi: 10.1073/pnas.1000089107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lee S., Hallam S.J. Extraction of high molecular weight genomic DNA from soils and sediments. J Vis Exp. 2009:1569. doi: 10.3791/1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dean F.B., Nelson J.R., Giesler T.L., Lasken R.S. Rapid amplification of plasmid and phage DNA using Phi 29 DNA polymerase and multiply-primed rolling circle amplification. Genome Res. 2001;11:1095–1099. doi: 10.1101/gr.180501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pinard R., de Winter A., Sarkis G.J., Gerstein M.B., Tartaro K.R., Plant R.N., Egholm M., Rothberg J.M., Leamon J.H. Assessment of whole genome amplification-induced bias through high-throughput, massively parallel whole genome sequencing. BMC Genomics. 2006;7:216. doi: 10.1186/1471-2164-7-216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tomlins S.A., Mehra R., Rhodes D.R., Shah R.B., Rubin M.A., Bruening E., Makarov V., Chinnaiyan A.M. Whole transcriptome amplification for gene expression profiling and development of molecular archives. Neoplasia. 2006;8:153–162. doi: 10.1593/neo.05754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Allen L.Z., Ishoey T., Novotny M.A., McLean J.S., Lasken R.S., Williamson S.J. Single virus genomics: a new tool for virus discovery. PLoS ONE. 2011;6:e17722. doi: 10.1371/journal.pone.0017722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brussaard C.P., Marie D., Bratbak G. Flow cytometric detection of viruses. J Virol Methods. 2000;85:175–182. doi: 10.1016/s0166-0934(99)00167-6. [DOI] [PubMed] [Google Scholar]
- 83.Sanger F., Nicklen S., Coulson A.R. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kreuze J.F., Perez A., Untiveros M., Quispe D., Fuentes S., Barker I., Simon R. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: a generic method for diagnosis, discovery and sequencing of viruses. Virology. 2009;388:1–7. doi: 10.1016/j.virol.2009.03.024. [DOI] [PubMed] [Google Scholar]
- 85.Bishop-Lilly K.A., Turell M.J., Willner K.M., Butani A., Nolan N.M., Lentz S.M., Akmal A., Mateczun A., Brahmbhatt T.N., Sozhamannan S. Arbovirus detection in insect vectors by rapid, high-throughput pyrosequencing. PLoS Negl Trop Dis. 2010;4:e878. doi: 10.1371/journal.pntd.0000878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Valles S.M., Hashimoto Y. Isolation and characterization of Solenopsis invicta virus 3, a new positive-strand RNA virus infecting the red imported fire ant, Solenopsis invicta. Virology. 2009;388:354–361. doi: 10.1016/j.virol.2009.03.028. [DOI] [PubMed] [Google Scholar]
- 87.Palacios G., Druce J., Du L., Tran T., Birch C., Briese T., Conlan S., Quan P.L., Hui J., Marshall J. A new arenavirus in a cluster of fatal transplant-associated diseases. N Engl J Med. 2008;358:991–998. doi: 10.1056/NEJMoa073785. [DOI] [PubMed] [Google Scholar]
- 88.Palacios G., Savji N., Hui J., Travassos da Rosa A., Popov V., Briese T., Tesh R., Lipkin W.I. Genomic and phylogenetic characterization of Merino Walk virus, a novel arenavirus isolated in South Africa. J Gen Virol. 2008;91:1315–1324. doi: 10.1099/vir.0.017798-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Meyer M., Stenzel U., Hofreiter M. Parallel tagged sequencing on the 454 platform. Nat Protoc. 2008;3:267–278. doi: 10.1038/nprot.2007.520. [DOI] [PubMed] [Google Scholar]
- 90.Meyer M., Stenzel U., Myles S., Prufer K., Hofreiter M. Targeted high-throughput sequencing of tagged nucleic acid samples. Nucleic Acids Res. 2007;35:e97. doi: 10.1093/nar/gkm566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nyren P. The history of pyrosequencing. Methods Mol Biol. 2007;373:1–14. doi: 10.1385/1-59745-377-3:1. [DOI] [PubMed] [Google Scholar]
- 92.Hyman E.D. A new method of sequencing DNA. Anal Biochem. 1988;174:423–436. doi: 10.1016/0003-2697(88)90041-3. [DOI] [PubMed] [Google Scholar]
- 93.Brussow H. The not so universal tree of life or the place of viruses in the living world. Philos Trans R Soc Lond B Biol Sci. 2009;364:2263–2274. doi: 10.1098/rstb.2009.0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hegde N.R., Maddur M.S., Kaveri S.V., Bayry J. Reasons to include viruses in the tree of life. Nat Rev Microbiol. 2009;7:615. doi: 10.1038/nrmicro2108-c1. author reply 615. [DOI] [PubMed] [Google Scholar]
- 95.Ludmir E.B., Enquist L.W. Viral genomes are part of the phylogenetic tree of life. Nat Rev Microbiol. 2009;7:615. doi: 10.1038/nrmicro2108-c4. author reply 615. [DOI] [PubMed] [Google Scholar]
- 96.Raoult D. There is no such thing as a tree of life (and of course viruses are out!) Nat Rev Microbiol. 2009;7:615. doi: 10.1038/nrmicro2108-c6. author reply 615. [DOI] [PubMed] [Google Scholar]
- 97.Schmieder R., Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–864. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schmieder R., Edwards R. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS ONE. 2011;6:e17288. doi: 10.1371/journal.pone.0017288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Schmieder R., Lim Y.W., Rohwer F., Edwards R. TagCleaner: identification and removal of tag sequences from genomic and metagenomic datasets. BMC Bioinformatics. 2010;11:341. doi: 10.1186/1471-2105-11-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Benson D.A., Karsch-Mizrachi I., Lipman D.J., Ostell J., Sayers E.W. GenBank. Nucleic Acids Res. 2011;38:D46–D51. doi: 10.1093/nar/gkp1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 102.Field D., Garrity G., Gray T., Morrison N., Selengut J., Sterk P., Tatusova T., Thomson N., Allen M.J., Angiuoli S.V. The minimum information about a genome sequence (MIGS) specification. Nat Biotechnol. 2008;26:541–547. doi: 10.1038/nbt1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Raes J., Foerstner K.U., Bork P. Get the most out of your metagenome: computational analysis of environmental sequence data. Curr Opin Microbiol. 2007;10:490–498. doi: 10.1016/j.mib.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 104.Allander T., Tammi M.T., Eriksson M., Bjerkner A., Tiveljung-Lindell A., Andersson B. Cloning of a human parvovirus by molecular screening of respiratory tract samples. Proc Natl Acad Sci U S A. 2005;102:12891–12896. doi: 10.1073/pnas.0504666102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Culley A.I., Lang A.S., Suttle C.A. Metagenomic analysis of coastal RNA virus communities. Science. 2006;312:1795–1798. doi: 10.1126/science.1127404. [DOI] [PubMed] [Google Scholar]
- 106.Allander T., Andreasson K., Gupta S., Bjerkner A., Bogdanovic G., Persson M.A., Dalianis T., Ramqvist T., Andersson B. Identification of a third human polyomavirus. J Virol. 2007;81:4130–4136. doi: 10.1128/JVI.00028-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bench S.R., Hanson T.E., Williamson K.E., Ghosh D., Radosovich M., Wang K., Wommack K.E. Metagenomic characterization of Chesapeake Bay virioplankton. Appl Environ Microbiol. 2007;73:7629–7641. doi: 10.1128/AEM.00938-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cox-Foster D.L., Conlan S., Holmes E.C., Palacios G., Evans J.D., Moran N.A., Quan P.L., Briese T., Hornig M., Geiser D.M. A metagenomic survey of microbes in honey bee colony collapse disorder. Science. 2007;318:283–287. doi: 10.1126/science.1146498. [DOI] [PubMed] [Google Scholar]
- 109.Fierer N., Breitbart M., Nulton J., Salamon P., Lozupone C., Jones R., Robeson M., Edwards R.A., Felts B., Rayhawk S. Metagenomic and small-subunit rRNA analyses reveal the genetic diversity of bacteria, archaea, fungi, and viruses in soil. Appl Environ Microbiol. 2007;73:7059–7066. doi: 10.1128/AEM.00358-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Desnues C., Rodriguez-Brito B., Rayhawk S., Kelley S., Tran T., Haynes M., Liu H., Furlan M., Wegley L., Chau B. Biodiversity and biogeography of phages in modern stromatolites and thrombolites. Nature. 2008;452:340–343. doi: 10.1038/nature06735. [DOI] [PubMed] [Google Scholar]
- 111.Djikeng A., Halpin R., Kuzmickas R., Depasse J., Feldblyum J., Sengamalay N., Afonso C., Zhang X., Anderson N.G., Ghedin E. Viral genome sequencing by random priming methods. BMC Genomics. 2008;9:5. doi: 10.1186/1471-2164-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Finkbeiner S.R., Allred A.F., Tarr P.I., Klein E.J., Kirkwood C.D., Wang D. Metagenomic analysis of human diarrhea: viral detection and discovery. PLoS Pathog. 2008;4:e1000011. doi: 10.1371/journal.ppat.1000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Honkavuori K.S., Shivaprasad H.L., Williams B.L., Quan P.L., Hornig M., Street C., Palacios G., Hutchison S.K., Franca M., Egholm M. Novel borna virus in psittacine birds with proventricular dilatation disease. Emerg Infect Dis. 2008;14:1883–1886. doi: 10.3201/eid1412.080984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Victoria J.G., Kapoor A., Dupuis K., Schnurr D.P., Delwart E.L. Rapid identification of known and new RNA viruses from animal tissues. PLoS Pathog. 2008;4:e1000163. doi: 10.1371/journal.ppat.1000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Adams I.P., Glover R.H., Monger W.A., Mumford R., Jackeviciene E., Navalinskiene M., Samuitiene M., Boonham N. Next-generation sequencing and metagenomic analysis: a universal diagnostic tool in plant virology. Mol Plant Pathol. 2009;10:537–545. doi: 10.1111/j.1364-3703.2009.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Briese T., Paweska J.T., McMullan L.K., Hutchison S.K., Street C., Palacios G., Khristova M.L., Weyer J., Swanepoel R., Egholm M. Genetic detection and characterization of Lujo virus, a new hemorrhagic fever-associated arenavirus from southern Africa. PLoS Pathog. 2009;5:e1000455. doi: 10.1371/journal.ppat.1000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Greninger A.L., Runckel C., Chiu C.Y., Haggerty T., Parsonnet J., Ganem D., DeRisi J.L. The complete genome of klassevirus — a novel picornavirus in pediatric stool. Virol J. 2009;6:82. doi: 10.1186/1743-422X-6-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ng T.F., Manire C., Borrowman K., Langer T., Ehrhart L., Breitbart M. Discovery of a novel single-stranded DNA virus from a sea turtle fibropapilloma by using viral metagenomics. J Virol. 2009;83:2500–2509. doi: 10.1128/JVI.01946-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ng T.F., Suedmeyer W.K., Wheeler E., Gulland F., Breitbart M. Novel anellovirus discovered from a mortality event of captive California sea lions. J Gen Virol. 2009;90:1256–1261. doi: 10.1099/vir.0.008987-0. [DOI] [PubMed] [Google Scholar]
- 120.Rosario K., Nilsson C., Lim Y.W., Ruan Y., Breitbart M. Metagenomic analysis of viruses in reclaimed water. Environ Microbiol. 2009;11:2806–2820. doi: 10.1111/j.1462-2920.2009.01964.x. [DOI] [PubMed] [Google Scholar]
- 121.Blinkova O., Victoria J., Li Y., Keele B.F., Sanz C., Ndjango J.B., Peeters M., Travis D., Lonsdorf E.V., Wilson M.L. Novel circular DNA viruses in stool samples of wild-living chimpanzees. J Gen Virol. 2010;91:74–86. doi: 10.1099/vir.0.015446-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Honkavuori K.S., Shivaprasad H.L., Briese T., Street C., Hirschberg D.L., Hutchison S.K., Lipkin W.I. Novel picornavirus in Turkey poults with hepatitis, California, USA. Emerg Infect Dis. 2011;17:480–487. doi: 10.3201/eid1703.101410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shan T., Li L., Simmonds P., Wang C., Moeser A., Delwart E. The fecal virome of pigs on a high-density farm. J Virol. 2011;85:11697–11708. doi: 10.1128/JVI.05217-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Gomez-Alvarez V., Teal T.K., Schmidt T.M. Systematic artifacts in metagenomes from complex microbial communities. ISME J. 2009;3:1314–1317. doi: 10.1038/ismej.2009.72. [DOI] [PubMed] [Google Scholar]
- 125.Wooley J.C., Ye Y. Metagenomics: facts and artifacts, and computational challenges. J Comput Sci Technol. 2009;25:71–81. doi: 10.1007/s11390-010-9306-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Angly F., Rodriguez-Brito B., Bangor D., McNairnie P., Breitbart M., Salamon P., Felts B., Nulton J., Mahaffy J., Rohwer F. PHACCS, an online tool for estimating the structure and diversity of uncultured viral communities using metagenomic information. BMC Bioinformatics. 2005;6:41. doi: 10.1186/1471-2105-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Deschavanne P., DuBow M.S., Regeard C. The use of genomic signature distance between bacteriophages and their hosts displays evolutionary relationships and phage growth cycle determination. Virol J. 2010;7:163. doi: 10.1186/1743-422X-7-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Willner D., Thurber R.V., Rohwer F. Metagenomic signatures of 86 microbial and viral metagenomes. Environ Microbiol. 2009;11:1752–1766. doi: 10.1111/j.1462-2920.2009.01901.x. [DOI] [PubMed] [Google Scholar]
- 129.Babkin I.V., Babkina I.N. Molecular dating in the evolution of vertebrate poxviruses. Intervirology. 2011;54:253–260. doi: 10.1159/000320964. [DOI] [PubMed] [Google Scholar]
- 130.Gilbert C., Feschotte C. Genomic fossils calibrate the long-term evolution of hepadnaviruses. PLoS Biol. 2010:8. doi: 10.1371/journal.pbio.1000495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Heinemann J., Maaty W.S., Gauss G.H., Akkaladevi N., Brumfield S.K., Rayaprolu V., Young M.J., Lawrence C.M., Bothner B. Fossil record of an archaeal HK97-like provirus. Virology. 2011;417:362–368. doi: 10.1016/j.virol.2011.06.019. [DOI] [PubMed] [Google Scholar]
- 132.Sanderson M.J. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics. 2003;19:301–302. doi: 10.1093/bioinformatics/19.2.301. [DOI] [PubMed] [Google Scholar]
- 133.Simmonds P., Davidson F., Lycett C., Prescott L.E., MacDonald D.M., Ellender J., Yap P.L., Ludlam C.A., Haydon G.H., Gillon J. Detection of a novel DNA virus (TTV) in blood donors and blood products. Lancet. 1998;352:191–195. doi: 10.1016/s0140-6736(98)03056-6. [DOI] [PubMed] [Google Scholar]
- 134.Tanaka M., Nishiguchi S., Tanaka T., Enomoto M., Fukuda K., Takeda T., Nakajima S., Shiomi S., Kuroki T., Monna T. Prevalence of GBV-C and hepatitis G virus variants in patients with fulminant hepatic failure in Japan. J Hepatol. 1997;27:966–972. doi: 10.1016/s0168-8278(97)80138-9. [DOI] [PubMed] [Google Scholar]
- 135.Wang J.T., Tsai F.C., Lee C.Z., Chen P.J., Sheu J.C., Wang T.H., Chen D.S. A prospective study of transfusion-transmitted GB virus C infection: similar frequency but different clinical presentation compared with hepatitis C virus. Blood. 1996;88:1881–1886. [PubMed] [Google Scholar]
- 136.Bernardin F., Operskalski E., Busch M., Delwart E. Transfusion transmission of highly prevalent commensal human viruses. Transfusion. 2010;50:2474–2483. doi: 10.1111/j.1537-2995.2010.02699.x. [DOI] [PubMed] [Google Scholar]
- 137.Tang S., Lai K.N. Chronic viral hepatitis in hemodialysis patients. Hemodial Int. 2005;9:169–179. doi: 10.1111/j.1492-7535.2005.01129.x. [DOI] [PubMed] [Google Scholar]
- 138.Willner D., Furlan M., Haynes M., Schmieder R., Angly F.E., Silva J., Tammadoni S., Nosrat B., Conrad D., Rohwer F. Metagenomic analysis of respiratory tract DNA viral communities in cystic fibrosis and non-cystic fibrosis individuals. PLoS ONE. 2009;4:e7370. doi: 10.1371/journal.pone.0007370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.George S.L. Persistent GB virus C infection is associated with decreased HIV-1 disease progression in the Amsterdam Cohort Study. J Infect Dis. 2005;191:2156–2157. doi: 10.1086/430504. author reply 2158–2160. [DOI] [PubMed] [Google Scholar]
- 140.Lefrere J.J., Ferec C., Roudot-Thoraval F., Loiseau P., Cantaloube J.F., Biagini P., Mariotti M., LeGac G., Mercier B. GBV-C/hepatitis G virus (HGV) RNA load in immunodeficient individuals and in immunocompetent individuals. J Med Virol. 1999;59:32–37. doi: 10.1002/(sici)1096-9071(199909)59:1<32::aid-jmv6>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 141.Watt G., Kantipong P., Jongsakul K. Decrease in human immunodeficiency virus type 1 load during acute dengue fever. Clin Infect Dis. 2003;36:1067–1069. doi: 10.1086/374600. [DOI] [PubMed] [Google Scholar]
- 142.Rivers T.M. Viruses and Koch's postulates. J Bacteriol. 1937;33:1–12. doi: 10.1128/jb.33.1.1-12.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Fredericks D.N., Relman D.A. Sequence-based identification of microbial pathogens: a reconsideration of Koch's postulates. Clin Microbiol Rev. 1996;9:18–33. doi: 10.1128/cmr.9.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Feng H., Shuda M., Chang Y., Moore P.S. Clonal integration of a polyomavirus in human Merkel cell carcinoma. Science. 2008;319:1096–1100. doi: 10.1126/science.1152586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145••.Falkow S. Molecular Koch's postulates applied to microbial pathogenicity. Rev Infect Dis. 1988;10(Suppl 2):S274–S276. doi: 10.1093/cid/10.supplement_2.s274. [DOI] [PubMed] [Google Scholar]; Significant paradigm change and a challenge to the existing Koch's postulates and the proposal to use molecular methods to assign etiology to infectious agents.
- 146.Wensel D.L., Li W., Cunningham J.M. A virus-virus interaction circumvents the virus receptor requirement for infection by pathogenic retroviruses. J Virol. 2003;77:3460–3469. doi: 10.1128/JVI.77.6.3460-3469.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Olweny C.L. IARC Sci Publ; 1984. Etiology of Endemic Burkitt's Lymphoma. 647–653. [PubMed] [Google Scholar]
- 148.Al Rwahnih M., Daubert S., Golino D., Rowhani A. Deep sequencing analysis of RNAs from a grapevine showing Syrah decline symptoms reveals a multiple virus infection that includes a novel virus. Virology. 2009;387:395–401. doi: 10.1016/j.virol.2009.02.028. [DOI] [PubMed] [Google Scholar]
- 149.Metzger M.J., Holguin C.J., Mendoza R., Miller A.D. The prostate cancer-associated human retrovirus XMRV lacks direct transforming activity but can induce low rates of transformation in cultured cells. J Virol. 2010;84:1874–1880. doi: 10.1128/JVI.01941-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Holtz L.R., Finkbeiner S.R., Kirkwood C.D., Wang D. Identification of a novel picornavirus related to cosaviruses in a child with acute diarrhea. Virol J. 2008;5:159. doi: 10.1186/1743-422X-5-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Greninger A.L., Chen E.C., Sittler T., Scheinerman A., Roubinian N., Yu G., Kim E., Pillai D.R., Guyard C., Mazzulli T. A metagenomic analysis of pandemic influenza A (2009 H1N1) infection in patients from North America. PLoS ONE. 2010;5:e13381. doi: 10.1371/journal.pone.0013381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Towner J.S., Sealy T.K., Khristova M.L., Albarino C.G., Conlan S., Reeder S.A., Quan P.L., Lipkin W.I., Downing R., Tappero J.W. Newly discovered ebola virus associated with hemorrhagic fever outbreak in Uganda. PLoS Pathog. 2008;4:e1000212. doi: 10.1371/journal.ppat.1000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Taylor L.H., Latham S.M., Woolhouse M.E. Risk factors for human disease emergence. Philos Trans R Soc Lond B Biol Sci. 2001;356:983–989. doi: 10.1098/rstb.2001.0888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Nayar A. Emerging disease: looking for trouble. Nature. 2009;462:717–719. doi: 10.1038/462717a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wolfe N.D., Daszak P., Kilpatrick A.M., Burke D.S. Bushmeat hunting, deforestation, and prediction of zoonoses emergence. Emerg Infect Dis. 2005;11:1822–1827. doi: 10.3201/eid1112.040789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wolfe N.D., Heneine W., Carr J.K., Garcia A.D., Shanmugam V., Tamoufe U., Torimiro J.N., Prosser A.T., Lebreton M., Mpoudi-Ngole E. Emergence of unique primate T-lymphotropic viruses among central African bushmeat hunters. Proc Natl Acad Sci U S A. 2005;102:7994–7999. doi: 10.1073/pnas.0501734102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wolfe N.D., Switzer W.M., Carr J.K., Bhullar V.B., Shanmugam V., Tamoufe U., Prosser A.T., Torimiro J.N., Wright A., Mpoudi-Ngole E. Naturally acquired simian retrovirus infections in central African hunters. Lancet. 2004;363:932–937. doi: 10.1016/S0140-6736(04)15787-5. [DOI] [PubMed] [Google Scholar]
- 158.Reperant L.A. Applying the theory of island biogeography to emerging pathogens: toward predicting the sources of future emerging zoonotic and vector-borne diseases. Vector Borne Zoonotic Dis. 2010;10:105–110. doi: 10.1089/vbz.2008.0208. [DOI] [PubMed] [Google Scholar]
- 159.Dinsdale E.A., Edwards R.A., Hall D., Angly F., Breitbart M., Brulc J.M., Furlan M., Desnues C., Haynes M., Li L. Functional metagenomic profiling of nine biomes. Nature. 2008;452:629–632. doi: 10.1038/nature06810. [DOI] [PubMed] [Google Scholar]
- 160.Rodriguez-Brito B., Rohwer F., Edwards R.A. An application of statistics to comparative metagenomics. BMC Bioinformatics. 2006;7:162. doi: 10.1186/1471-2105-7-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Jones M.S., Kapoor A., Lukashov V.V., Simmonds P., Hecht F., Delwart E. New DNA viruses identified in patients with acute viral infection syndrome. J Virol. 2005;79:8230–8236. doi: 10.1128/JVI.79.13.8230-8236.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lauck M., Hyeroba D., Tumukunde A., Weny G., Lank S.M., Chapman C.A., O’Connor D.H., Friedrich T.C., Goldberg T.L. Novel, divergent simian hemorrhagic fever viruses in a wild Ugandan red colobus monkey discovered using direct pyrosequencing. PLoS ONE. 2011;6:e19056. doi: 10.1371/journal.pone.0019056. [DOI] [PMC free article] [PubMed] [Google Scholar]