

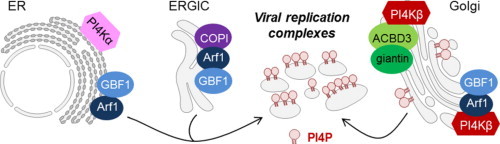
Graphical abstract
Keywords: PI4KIIIα, PI4KIIIβ, PI4P, Viral replication, Antiviral target
Abstract
Phosphoinositides (PI) are phospholipids that mediate signaling cascades in the cell by binding to effector proteins. Reversible phosphorylation of the inositol ring at positions 3, 4 and 5 results in the synthesis of seven different phosphoinositides. Each phosphoinositide has a unique subcellular distribution with a predominant localization in subsets of membranes. These lipids play a major role in recruiting and regulating the function of proteins at membrane interfaces [1]. Several bacteria and viruses modulate and exploit the host PI metabolism to ensure efficient replication and survival. Here, we focus on the roles of cellular phosphatidylinositol 4-phosphate (PI4P) and phosphatidylinositol 4-kinases (PI4Ks) during the replication cycle of various viruses. It has been well documented that phosphatidylinositol 4-kinase IIIβ (PI4KIIIβ, EC 2.7.1.67) is indispensable for viral RNA replication of several picornaviruses. Two recruitment strategies were reported: (i) binding and modulation of GBF1/Arf1 to enhance recruitment of PI4KIIIβ and (ii) interaction with ACBD3 for recruitment of PI4KIIIβ. PI4KIII has also been demonstrated to be crucial for hepatitis C virus (HCV) replication. PI4KIII appears to be directly recruited and activated by HCV NS5A protein to the replication complexes. In contrast to picornaviruses, it is still debated whether the α or the β isoform is the most important. PI4KIII can be explored as a target for inhibition of viral replication. The challenge will be to develop highly selective inhibitors for PI4KIIIα and/or β and to avoid off-target toxicity.
1. Introduction
Various viruses have an impact on the host's lipid metabolism, lipid/membrane transport and lipid mediated signal transduction. A key class of lipids involved in these cellular processes is the class of phosphoinositides (PIs). Phosphatidylinositol (PtdIns) is the basic scaffold of the PIs (Fig. 1 ). The inositol moiety of PtdIns can be reversible phosphorylated in three of the five free hydroxyl-groups at positions D3, D4 and D5. Differential phosphorylation can result into seven different species of PIs that are localized in distinct cellular compartments and, together with protein factors, define their identity: PtdIns 3-phosphate [PI3P] is found on early and late endosomes, PI4P at the Golgi complex (Fig. 2 ) and PI5P at several distinct subcellular locations [2]; PI(3,4)P2, PI(4,5)P2 (Fig. 2) and PI(3,4,5)P3 are (transiently) located at the plasma membrane [3], [4], and PI(3,5)P2 in the endosome–lysosome axis [5]. Besides the differential spatial distribution within the cell the PIs are also tightly regulated in time. Both spatial and temporal regulation is mediated by different PI kinases and phosphatases. Keys to the spatial regulation of PIs are the distinct substrate specificities and differential intracellular localizations of these enzymes. Hence, specific PI species are confined to similar membrane compartments as their concomitant kinases, resulting in a distinct spatial distribution of different PI species in the cell. The temporal regulation of PIs in the cell is ascertained by the fast catalytic rates of the PI kinases and phosphatases resulting in rapid synthesis or turnover of PIs in cellular membranes [1].
Fig. 1.
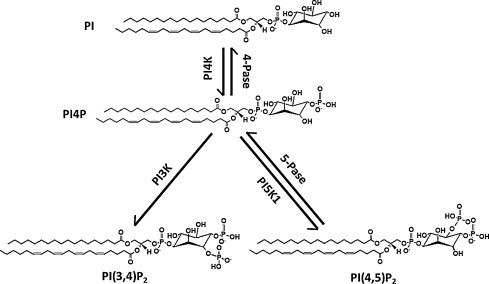
Metabolism reaction that leads to the synthesis of phosphatidylinositol 4-phosphate and related phosphoinositides. PI: phosphoinositol; PI4K: phosphatidylinositol 4-kinase; 4-Pase: phosphatidylinositol 4-phosphatase; PI3K: phosphatidylinositol 3-kinase; 5-Pase, phosphatidylinositol 5-phosphatase; PI5K1: phosphatidylinositol 5-kinase; PI4P, phosphatidylinositol 4-phosphate; PI(3,4)P2: phosphatidylinositol 3,4-biphosphate; PI(4,5)P2: phosphatidylinositol 4,5-biphosphate.
Fig. 2.
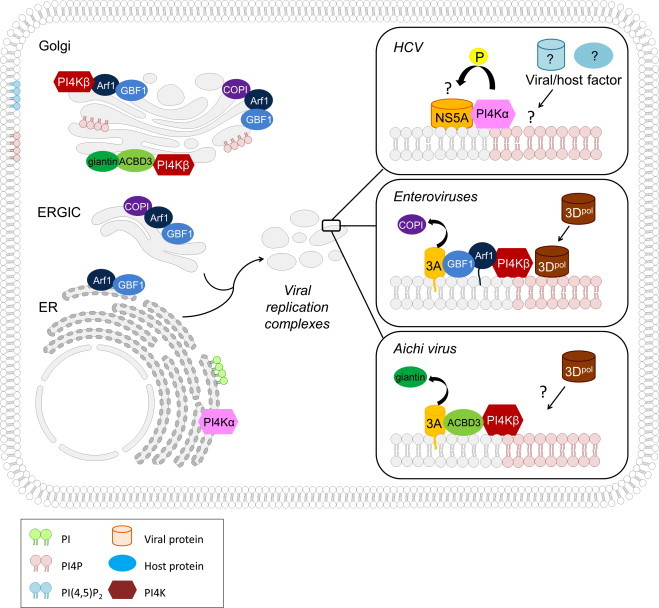
Overview of the different modes of recruitment of PI4KIIIs to the replication complexes of HCV, enteroviruses and the Aichi virus. PI: phosphoinositol; PI4P, phosphatidylinositol 4-phosphate; PI(4,5)P2: phosphatidylinositol 4,5-biphosphate; PI4K: phosphatidylinositol 4-kinase; ERGIC, ER-Golgi intermediate compartment.
PI lipids anchor to the cytoplasmatic leaflet of membranes. Together with small GTPases, PIs are able to recruit specific downstream effector proteins. In this way, the seven PIs species directly or indirectly modulate numerous cellular events such as processes (i) that help to define the identity of cellular organelles [6], (ii) that mediate signal transduction at the cell surface and (iii) that are involved in the regulation of membrane permeability and traffic. Furthermore, PIs are also involved in (iv) the remodeling of the actin cytoskeleton [6], (v) DNA repair, transcription regulation and RNA dynamics in the nucleus [7] and (vi) processes that aid cell migration [8]. In addition PIs also take part in (vii) the process of cellular polarization [6] and (viii) are important for the modulation of the sphingolipid metabolism [9].
Many pathogens exploit the PIs to harness the cell's resource for their replication. Many intracellular bacterial pathogens modulate and exploit PIs to ensure efficient replication and survival. Different strategies are employed to interfere with the host PI metabolism, including the production of (i) PI-metabolizing enzymes that directly affect PI levels in the host cell, (ii) PI-binding (effector) proteins that use PIs as a membrane anchor, and (iii) protein and lipid factors that recruit, activate or inactivate host cell PI metabolizing enzymes [10]. Modulation of the PI levels on a pathogen-induced intracellular vesicle can cause a change in the organelle identity, a process called ‘identity theft’ [11]. Also viruses have been shown to hijack the host PI metabolism for beneficial replication and survival. In this review, we will focus on the roles of cellular phosphatidylinositol 4-phosphate (PI4P) and phosphatidylinositol 4-kinases (PI4Ks) during the replication of a number of viruses and discuss PI4Ks as a potential target for inhibition of viral replication.
2. The biology of phosphatidylinositol 4-kinases (PI4K)
A number of synthetic paths can lead to the formation of PI4P (Fig. 1). Either PtdIns is phosphorylated by a phosphatidylinositol 4-kinase (PI4K) or by dephosphorylation of PI(3,4)P2 or PI(4,5)P2 by 5-Pase phosphatases [12]. Currently there are four PI4K isoforms identified in mammals, subdivided in type II or type III families based on their size and the catalytic properties of the enzymes [13]. The families are further subdivided in α/β isoforms based on their domain structure, i.e. type II PI4Kα and β and type III PI4Kα and β [14].
Type II PI 4-kinase enzymes (EC 2.7.1.67) are 56-kDa proteins that are abundant in almost all cellular membranes; they are enriched in plasma membrane preparations. Palmitoylation of PI4KII not only tethers the enzyme to the membrane, but also modulates the catalytic activity [15], [16]. Moreover the catalytic activity of type II PI 4-kinases is further controlled by means of phosphorylation [16]. To date the functions of the type II PI4Ks still remains somewhat elusive. PI4KIIα is the main contributor to the cellular PI4P levels and has probably a role in post-trans-Golgi network trafficking [17], [18] and endocytic traffic [19]. Furthermore recent studies suggest that PI4KIIα takes part in the regulation of sphingolipid homeostasis through the lysosomal delivery of β-glucocerebrosidase [20] and is required for the activation of sphingomyelin synthesis by oxysterol binding protein [21]. A role for PI4KIIα in cancer [22] and in Akt-activation and apoptosis [23] was also demonstrated. PI4KIIβ is a cytosolic protein that is recruited to plasma membrane, endoplasmic reticulum, or the Golgi and may have no role in membrane traffic [24].
Both type III PI4Ks α and β (EC 2.7.1.67) are not directly bound to membranes as is the case for PI4KIIs. They share structural similarities with PI3-kinases and are inhibited by relative high concentrations of PI3K inhibitors (e.g. wortmannin) [13]. PI4KIIIβ has an apparent molecular weight of 110 kDa and is mainly localized in the Golgi [25] (Fig. 2), but has also been detected in the nucleus [26], in secretory granules [27] and in synaptic vesicles [28]. Recruitment of PI4KIIIβ to the Golgi is facilitated by the small GTP-binding protein Arf1 [29] but also by NCS-1 [30], [31]. To which extend NCS-1 contributes to the recruitment of PI4KIIIβ to the Golgi in mammalian cells remains unknown [13]. The enzymatic activity of PI4KIIIβ can also be modulated by phosphorylation of one or more of its multiple phosphorylation sites [32]. For example, phosphorylation of Ser258 and Ser266 was shown to be important for Golgi recruitment of PI4KIIIβ [14] and phosphorylation of PI4KIIIβ at Ser268 by protein kinase D (PKD) has shown to be essential for its kinase activity and its ability to support post-Golgi transport, but was not necessary for Golgi recruitment [33]. Besides PKD, other kinases can phosphorylate PI4KIIIβ such as proline directed cyclin-dependent protein kinases (like cdc2), cyclic monophosphate-dependent protein kinases (protein kinase A/G), Ca2+/calmodulin-dependent protein kinase, casein kinase II or cyclin-dependent kinases [14].
PI4K type III alpha (PI4KIIIα) resides predominantly in the ER in mammalian cells [25] (Fig. 2) but can also be found in the pericentriolar area over the Golgi [34] and in the nucleolus [14]. In the mammalian nervous system PI4KIIIα can be distributed to unique membranous clusters associated with various organelles such as multivesicular bodies, mitochondria and the ER [13]. The functional role of PI4KIIIα in the ER in mammalian cells is still not understood. Recently it was shown that PI4KIIIα is required for the recruitment of GBF1 to Golgi membranes [35]. The functional role of PI4KIIIα in the plasma membrane of mammalian cells is better understood. PI4KIIIα takes part in the signaling complex associated with the P2X7 ion channels; moreover, it may support the Ins(1,4,5)P3–Ca2+ signaling cascade and it is responsible for the production of a plasma membrane pool of PI4P [13]. Based on potential phosphorylation sites, the cAMP-dependent protein kinase (PKA), protein kinase C, casein kinase II and protein tyrosine kinases have been suggested to be involved in the regulation of PI4KIIIα through phosphorylation [14].
3. The physiology of phosphatidylinositol 4-phosphate (PI4P)
Key to the PIs physiological function is their inositol head group that protrudes from the cytosolic leaflet of cell membranes. Hence, this allows interaction with not only the PI kinases and phosphatases, but also with numerous proteins that contain a specific PI-binding domain that aids in tethering these proteins to the membranes [36]. In case of integral membrane proteins, PI-binding can induce conformational changes that result in allosteric regulation of these proteins [37]. There exist a myriad of PI-binding proteins belonging to all kinds of protein categories [38]. Specific PI4P-binding proteins are for example (i) clathrin adaptors (i.e. adaptor protein complex 1, AP1; gamma-ear-containing, ADP-ribosylation factor-binding proteins, GGAs; and epsinR), (ii) lipid-transfer proteins (the four-phosphate-adaptor proteins, FAPPs; the ceramide transport protein, CERT; and the oxysterol-binding protein, OSBP) and (iii) the GOLPH3 protein that modulates Golgi function according to the cell's metabolism [37]. The PI affinity of PI-binding proteins is however low; requiring other interactions with membrane-proteins or membrane-associated small GTPases of the RAB protein family or Arf1 in order to stabilize this interaction [39]. For example AP1, the GGAs, FAPP1 and FAPP2 bind in addition to PI4P also to the small GTPase Arf1. This dual recognition ensures the correct recruitment of these PI4P-binding proteins specifically to the trans compartments of the Golgi complex. For other cellular organelles and membrane domains similar combinatorial recognition mechanisms exist that determine their uniqueness or identity [11].
As reviewed in [37] PI4P is involved in (i) the architecture of the Golgi complex and its function, (ii) sorting of cargo at the trans Golgi network and (iii) consecutive transport from the Golgi to the plasma membrane. Furthermore PI4P is important in sphingomyelin synthesis [9]. De novo sphingolipid synthesis starts at the cytosolic leaflet of the ER where different ceramides are synthesized de novo, and finalizes at the Golgi network. To this end, newly synthesized ceramides are transported from the ER by vesicles or by the PI4P-binding protein CERT.
4. The role of PI4Ks in the life cycle of hepatitis C virus
Hepatitis C virus (HCV) is the sole member of the Hepacivirus genus within the Flaviviridae family, a group of enveloped, single-stranded positive-sense RNA viruses. With worldwide 170 million persons chronically infected that are at high risk of developing liver cirrhosis and cancer, HCV represents a major health burden. Several positive RNA viruses such as flaviviruses and picornaviruses drastically remodel intracellular membranes to generate specialized environments for RNA replication. For HCV, these remodeled replication membranes seem to be derived of the endoplasmic reticulum (ER) and are referred to as the ‘membranous web’ [40]. The formation of the membranous web is induced by the HCV NS4B protein. In addition, some host factors were shown to be indispensable for the integrity of the membranous web. Such an essential HCV host factor that has been identified across numerous studies is phosphatidylinositol kinase-4 α (PI4KIIIα) [41], [42], [43], [44], [45], [46], [47] (Table 1 , Fig. 2).
Table 1.
Overview of published studies on the role of PI4KIIIα and β in the HCV life cycle.
| Authors | Year | PI4KIII isoform | HCV genotype | Methods and results |
|---|---|---|---|---|
| Trotard et al. | 2009 | PI4KIIIα and β | Replicon 1a, 1b | siRNA library screen. |
| HCVpp 1a, 1b, 2a | Genotype dependency: | |||
| HCVcc 2a | - Replication: PI4KIIIα required for 1a, 2a, PI4KIIIβ for 1a | |||
| - Entry: PI4KIIIα required for 1a, PI4KIIIβ for 1a, 1b. | ||||
| Berger et al. | 2009 | PI4KIIIα | Replicon 1b, 2a | siRNA library screen, validation by individual siRNAs and microscopy. |
| HCVcc 2a | PI4KIIIα is required for HCV replication but not for HCV entry. | |||
| Borawski et al. | 2009 | PI4KIIIα and β | Replicon 1a, 1b | siRNA library screen. |
| HCVcc 2a | Genotype dependency: | |||
| PI4KIIIα: 1a, 1b and 2a; PI4KIIIβ: 1a and 1b. | ||||
| Tai et al. | 2009 | PI4KIIIα | Replicon 1b | siRNA library screen, validation by individual siRNAs and microscopy. |
| HCVcc 2a | PI4KIIIα is required for HCV replication. | |||
| Vaillancourt et al. | 2009 | PI4KIIIα | Replicon 1a, 1b, 2a | shRNA library screen, validation by siRNA. |
| HCV replication is inhibited by PI4KIIIα silencing (genotype independent). | ||||
| Hsu et al. | 2010 | PI4KIIIβ | Replicon 2a | siRNA knockdown of PI4KIIIα/β. |
| Expression of a kinase-dead PI4KIIIβ inhibited HCV replication. | ||||
| Reiss et al. | 2011 | PI4KIIIα | Replicon 1b, 2a | siRNA library screen, validation by individual siRNAs and microscopy. NS5A recruits and activates PI4KIIIα. |
| HCVcc 2a | ||||
| Berger et al. | 2011 | PI4KIIIα | HCVcc 2a | siRNA trans-complementation assay, microscopy, coimmunoprecipitation. |
| NS5A enhances PI4KIIIα activity. | ||||
| Lim et al. | 2011 | PI4KIIIα | Replicon 1b | Coimmunoprecipitation (NS5A interacts with PI4KIIIα), siRNA knockdown. |
| HCVcc 2a | ||||
| Tai et al. | 2011 | PI4KIIIα and β | HCVcc 2a | shRNA knockdown, microscopy. |
| PI4KIIIβ silencing inhibits HCV infection but does not disturb HCV membranous web formation. | ||||
| Coller et al. | 2012 | PI4KIIIβ | HCVcc 2a | RNAi analysis of host factors combined with live cell imaging of HCV core trafficking. |
| PI4KIIIβ seems essential for HCV secretion, not for HCV replication. | ||||
| Zhang et al. | 2012 | PI4KIIIβ | HCVcc 2a | siRNA knockdown of PI4KIIIβ, Sac1 expression and microscopy. |
| PI4KIIIβ is required for HCV replication. | ||||
| Bianco et al. | 2012 | PI4KIIIα | Replicon 1b, 2a | AL-9, a specific inhibitor of PI4KIIIα, inhibits HCV replication. |
| HCVcc 2a | ||||
The requirement of PI4KIIIα kinase activity for efficient HCV replication was first observed in 2009 by several independent siRNA-based screenings [42], [43], [44], [45], [46], despite of differences in experimental set-up (different genotypes, replicon vs. infectious virus) and analysis. Later studies confirmed that PI4KIIIα is indispensable for HCV replication, more in particular for the structural integrity of the membranous HCV replication complex. Silencing of PI4KIIIα expression led to the formation of abnormal NS5A clusters. PI4KIIIα is actively recruited by HCV NS5A to the HCV replication complexes. This interaction occurs between domain I of NS5A and amino acids 400–600 of PI4KIIIα [48]. More recently, the required NS5A amino acids for interaction with PI4KIIIα were assigned to the C-terminal end of domain I [49]. The enzymatic activity of the lipid kinase is required for HCV replication, as shown by rescue experiments in PI4KIIIα knockdown cell lines with wild-type or a kinase-dead mutant of the enzyme [41]. Interestingly, the kinase activity of PI4KIIIα has also been shown to be enhanced by the interaction with NS5A [41]. At the level of the membranes of HCV replication complexes, PI4KIIIα induces a massive accumulation of PI4P lipids. This was confirmed in infected liver tissues obtained from HCV-infected patients [41].
At present however, conflicting reports exist as to which specific isoform of PI4KIII (α/β) is required for HCV replication. In most publications PI4KIIIα is designated as the essential isoform for HCV (Table 1). However, in some siRNA screens also PI4KIIIβ emerged as an indispensable host factor for HCV replication [45], [46]. This dependency on PI4KIIIβ seemed to be associated with HCV genotype 1. Nonetheless, recent reports indicate a role for PI4KIIIβ in HCV genotype 2 as well [50], [51], [52]. The question remains how essential this isoform is for HCV replication. Tai and colleagues demonstrated that PI4KIIIβ shRNAs inhibit JFH-1 replication in a dose-dependent manner but do not cause any alterations in membranous web morphology [51]. They therefore concluded that the PI4P enrichment at the membranous web is due to PI4KIIIα activity and does not require PI4KIIIβ. In contrast, in another recent study, PI4KIIIβ was found to be essential for JFH-1 HCV replication [52]. Moreover, the vesicular transport proteins ARF1 and GBF1 colocalized with PI4KIIIβ and were both required for HCV replication as shown by siRNA knockdown experiments. Interestingly, these transport proteins are also indispensable for enterovirus replication (cf. infra). More studies are obviously required to elucidate this matter.
The reason why HCV hijacks PI4KIIIα for its replication remains to be clarified. One hypothesis is that HCV recruits PI4KIIIα to generate a microenvironment of PI4P lipids at the replication complexes. The function of these PI4P lipids is not clear yet. It was shown that PI4P lipids can locally change membrane curvature [53]. PI4P lipid enriched membranes could therefore generate high-curvature membrane pockets to protect viral proteins and RNA from host defense [54]. On the other hand, PI4P lipids may provide binding sites to concentrate viral/host proteins at the membranous web for efficient viral RNA synthesis. Several PI4P binding host proteins such as OSBP1 and CERT were previously identified as host factors for HCV replication [55], [56]. Alternatively, HCV proteins may also be able to bind PI4P lipids. Although HCV proteins do not contain any canonical PIP-binding motifs, a number of non-predicted PIP-binding proteins have been identified experimentally, such as the RNA-dependent RNA polymerase of poliovirus [50], [57]. Further studies are therefore required to determine the importance of PI4P accumulation at the HCV replication complexes. Another recent hypothesis is that PI4KIIIα influences the modulation of HCV NS5A phosphorylation, by supporting p56 synthesis and thereby stimulating RNA replication [49] (Fig. 2). NS5A is modified by phosphorylation giving rise to a basally (NS5Ap56) and a hyperphosphorylated variant (NS5Ap58). It is believed that p56 and p58 are involved in the regulation of the viral replication cycle by favoring RNA replication or assembly, respectively. Loss of PI4KIIIα interaction and RNA replication correlated with increased levels of p58, suggesting that PI4KIIIα might be involved in the regulation of NS5A phosphorylation. Overexpression and knockdown of PI4KIIIα resulted in relatively increased or decreased portions of NS5Ap56, respectively [49]. It remains to be elucidated whether NS5A phosphorylation is directly or indirectly modulated by PI4KIIIα.
PI4KIIIs are not only involved in HCV replication, some studies indicate that they play a role in other steps in HCV infection. PI4KIIIα and β were suggested to be essential for the internalization step of HCV [45]. This dependency seemed to be influenced by the particular genotype of HCV pseudoparticle (HCVpp). Although PI4P is a precursor of PI(4,5)P2 that recruits proteins required for clathrin-mediated endocytosis (CME), the inhibition of HCV entry observed during PI4KIIIα and β knockdown seemed unrelated to a disruption of CME. Indeed the endocytosis of labeled transferrin was not inferred. Silencing of PI4KIIIα or β did not affect the surface expression of the CD-81 and SR-BI receptor, suggesting that transport of membrane proteins at the cell surface was not disrupted. Other studies showed however no effect on HCVpp entry in Huh 7.5 cells when PI4K genes were silenced with pooled or individual siRNAs [41], [42].
By a combined approach of RNAi analysis of host factors and live cell imaging of HCV core trafficking PI4KIIIβ was recently identified as a host factor that is both required for infectious HCV release and co-traffics with HCV core [58]. Also other components of the secretory pathway were identified by this strategy, such as Rab11A, SAR1A and others. Knockdown of PI4KIIIβ by siRNA resulted in 90% inhibition of HCV infection. By quantifying the effects on core movements in the presence of PIK93 the role of PI4KIIIβ in the exit from the Golgi in core trafficking was assessed. Treatment with 0.5 μM PIK93, a concentration specifically inhibiting PI4KIIIβ, reduced the average velocity of motile core puncta. Furthermore, silencing of PI4KIIIβ decreased localization of core with VAMP1 (vesicular associated membrane protein 1) [58], a cofactor in the release of infectious HCV particles also identified in the RNAi screen. These data suggest that PI4KIIIs can be involved in different steps of the viral life cycle.
5. The role of PI4Ks in picornavirus replication
The family of the Picornaviridae is a group of non-enveloped, single-stranded positive-sense RNA viruses. Picornaviruses include many important human pathogens as well as for animals, such as poliovirus, enterovirus 71, rhinoviruses [enteroviruses], hepatitis A virus [hepatovirus], and foot-and-mouth disease virus [aphtovirus]. Picornaviruses rely on host intracellular membranes for replication. These replication membranes appear to originate from the ER, as is the case for HCV. Cellular factors are believed to be essential for this membrane reorganization. Many studies have demonstrated the involvement of Arf1 (ADP-ribosylation factor 1) and GBF1 (Golgi-specific Brefeldin A resistance factor 1) in the replication of enteroviruses [59], [60]. Recently, host factor PI4KIIIβ was also shown to be required for viral RNA replication of multiple picornaviruses [50], [61], [62] (Fig. 2).
In 2010, Hsu et al. demonstrated in an elegant study how enteroviruses (such as poliovirus and Coxsackievirus) remodel the host secretory pathway to generate replication organelles [50] (Fig. 2). As previously mentioned, GBF1 and Arf1 are known host factors for enteroviral replication. In uninfected mammalian cells, these host proteins are both localized to the ER and the Golgi apparatus. A major effector of Arf1 at these sites includes PI4KIIIβ. Immunofluorescence experiments revealed that PI4KIIIβ remained colocalized with Arf1 during enteroviral infection, in contrast to other Arf1 effectors. This colocalization was dependent on GBF1/Arf1 localization and activity. Furthermore, PI4KIIIβ was shown to associate with enteroviral proteins 3A, 3AB, 3CD and 3D. Expression of 3A alone enhanced the membrane recruitment of PI4KIIIβ, but not that of PI4KIIIα. In contrast to HCV, for which PI4KIIIα was shown to interact with NS5A [41], [57], no direct interaction between 3A and PI4KIIIβ was observed [63]. 3A was however shown to bind directly with GBF1. As also observed for HCV, enterovirus infection stimulates the synthesis of PI4P lipids at the level of the replication complexes. A substantial fraction of these PI4P lipids were a product of PI4KIIIβ activity. Interestingly, protein–lipid overlay studies suggested that the enteroviral 3Dpol RNA polymerase can preferentially bind to PI4P lipids [50]. Based on these findings, a model was proposed for the reorganization of the secretory pathway in enterovirus infections. Membrane-bound 3A proteins bind and modulate GBF1/Arf1 to enhance the recruitment of PI4KIIIβ to membranes. There the synthesis of PI4P lipids is catalyzed, resulting in a PI4P-enriched membrane microenvironment. This PI4P lipid-rich microenvironment will, in turn, enhance the recruitment of the RNA-dependent RNA polymerase 3Dpol. 3Dpol will then, as a part of the replication complex, initiate RNA synthesis at these membranes [50].
More recently, it was shown that the Aichi virus, another picornavirus belonging to the genus of Kobuviruses, also requires PI4KIIIβ for its RNA replication [61], [62] (Fig. 2). Intriguingly, the Aichi virus adopts a different strategy to recruit PI4KIIIβ to the viral replication sites from that used by other enteroviruses. The recruitment of PI4KIIIβ to the Aichi virus replication complexes was not dependent on GBF1/Arf1, but relied on interaction with the Golgi protein ACBD3 (acyl-coenzyme A binding domain protein 3). The ACBD3 protein is involved in the maintenance of the Golgi apparatus structure. Its localization to the Golgi occurs through interaction with giantin [64]. The Aichi virus non-structural proteins 2B, 2BC, 2C, 3A and 3AB were shown to interact with ACBD3 [61]. The viral protein-binding domain of ACBD3 overlaps with the binding domain for giantin. In addition, it was observed that ACBD3 interacts with PI4KIIIβ. No direct interaction between PI4KIIIβ and the viral proteins was observed. Interestingly, this study demonstrated that Aichi virus replication is, in contrast to enterovirus replication, insensitive to brefeldin A (BFA). BFA inactivates GBF1 and therefore inhibits the binding of GBF1/Arf1 to viral protein 3A. The difference in sensitivity to BFA between Aichi virus and enteroviruses thus corroborates the observation that these viruses adopt a different PI4KIIIβ recruitment strategy. Knockdown of ACBD3 or PI4KIIIβ by siRNA inhibited viral RNA replication by 70% or 99% respectively. PI4KIIIβ was shown to colocalize with ABCD3 and Aichi virus non-structural proteins at the level of the replication complexes in the cells. As is the case for enteroviruses and HCV, PI4P lipids were shown to accumulate in Aichi virus replications sites [61]. Based on these results, the current hypothesis is that the Aichi virus membrane proteins compete with giantin for binding to ACBD3 at the Golgi to form the viral protein/ACBD3/PI4KIIIβ complex. ACBD3 has a double function in this complex. On the one hand, it is able to recruit viral proteins which results in an increased concentration of the viral proteins at the Golgi. On the other hand, ACBD3 efficiently recruits PI4KIIIβ to the replication sites which results in a PI4P lipid-enriched environment [61]. Whether PI4P lipids are able to bind the 3D RNA polymerase of Aichi virus remains to be elucidated.
The ACBD3-mediated strategy of picornaviruses to recruit PI4KIIIβ was confirmed by Greninger et al. by using a mass spectrometry-based proteomic approach [62] (Fig. 2). They demonstrated that the 3A protein from multiple picornaviruses [including Aichi virus, bovine kobuvirus, CVB, HRV14, poliovirus], associates with PI4KIIIβ and ACBD3. The 3A of some picornaviruses did not appear to associate with ACBD3, namely the cardioviruses and EV71. Surprisingly, despite its insensitivity to BFA, knockdown of GBF1 abolished Aichi viral replication. It is however possible that the replication inhibition is due to a loss of PI4KIIIβ indirectly caused by a loss of GBF1. Knockdown of ACBD3 did not only inhibit Aichi virus replication, but also polio virus replication.
Since so far, two different PI4KIIIβ recruitment strategies were observed in the Picornaviridae family, it will be of interest to explore whether all picornaviruses require PI4KIIIβ for replication and, if so, how PI4KIIIβ is recruited to the replication complexes.
6. PI4Ks as host factor for other viruses
The first reports on altered PI4K activities by virus infections were published in the 1980s. Elevated phosphatidylinositol kinase activity was observed in Rous sarcoma virus (an alpharetrovirus)-transformed cells [65]. This elevation was indirectly enhanced by the transforming oncoprotein of the virus [66]. Similarly, PI4K activities were increased when human B cells were infected with Epstein–Barr virus (a DNA herpesvirus) [67]. The mechanism for the observed increase remained unclear. Also HIV-1 hijacks the phosphoinositide signaling system [68], [69], [70]. Interestingly, PI(4,5)P2 seems the be the major phosphoinositide involved. PI(4,5)P2 plays a critical role in targeting Gag to the plasma membrane. Depletion of PI(4,5)P2 compromises virus assembly and leads to accumulation of Gag at the membranes of late endosomes and multiple vesicular bodies [68]. Furthermore, it was found to be largely enriched in HIV-1 virions [69]. Recent studies demonstrate that PI(4,5)P2 binds directly to the matrix domain of the Gag protein, thereby regulating Gag localization and assembly at the plasma membrane [70]. In contrast, related phosphoinositides like PI4P, PI3P, PI5P and PI(3,5)P2 do not bind the matrix domain with substantial affinity. However, a monoclonal antibody specific to PI4P inhibited infection by two HIV-1 primary isolates in neutralization assays [71]. This antibody does not directly bind to the target cell prior to binding of HIV-1, as shown by fluorescence-activated cell sorter analysis. Notwithstanding, it was hypothesized that this effect is possibly due to a reaction of the antibody with PI(4,5)P2 during viral assembly and budding. In contrast to HIV-1, the Rous Sarcoma virus Gag has no specific requirement for PI(4,5)P2 for plasma membrane association [72]. Depletion of PI(4,5)P2 did not affect the release of virus-like particles nor the membrane localization of Gag. Moreover, liposome flotation experiments demonstrated that Rous Sarcoma virus Gag required acidic lipids for binding but showed no specificity for PI(4,5)P2.
The replication of West Nile virus (WNV) (genus flavivirus within the family of Flaviviridae to which also HCV belongs) is independent of PI4P [73]. This is in agreement with the observation that PI4KIIIα silencing has no effect on Dengue virus (another flavivirus) replication [41]. WNV infection did not alter the localization of PI4P lipids and no colocalization of PI4P was observed with dsRNA. Furthermore, no reduction in WNV RNA replication was observed upon PIK93 treatment. Instead, fatty acids seemed to be required for viral replication, since inhibition of the fatty acid synthase (FASN) reduced WNV production and FASN was shown to be localized close to WNV replication complexes in infected cells.
PI4KIIIβ was shown to be involved in the entry of SARS coronavirus (SARS CoV) [74]. Silencing of PI4KIIIβ, but not of PI4KIIIα strongly inhibited SARS CoV spike-mediated entry. Transient transfection of Sac1, a PI phosphatase that converts PI4P lipids back to PI, reduced SARS CoV entry, indicating that PI4P lipids were essential for viral entry. Furthermore, it was shown that PI4KIIIβ is not required for virus binding and internalization, but plays a role at, or before, virus fusion with the late endosomes.
7. PI4KIIIs: promising targets for therapeutic intervention?
As PI4KIIIs are involved in different steps of the life cycle of several viruses, these lipid kinases represent potential promising targets for the development of inhibitors of viral replication. Targeting host factors instead of viral proteins is an attractive yet controversial strategy for antiviral intervention. On the one hand, the barrier to resistance for a host-targeting antiviral (HTA) is apparently higher. On the other hand, inhibition of host factors may have a higher chance of resulting in toxicity/adverse effects. Moreover, polymorphisms in a host factor and variant expression levels between patients may be complicating factors. Feasibility of such antiviral strategies will likely depend on which host factor is targeted and how the virus and the cell depend on its function(s). Extensive mouse genetic studies were performed by Boehringer Ingelheim to estimate the potential effect of PI4KIIIα inhibition in vivo and thus to assess the safety of PI4KIIIα inhibitors [75]. PI4KIIIα knockdown resulted in a lethal phenotype within 5 days in all mice. Next, a conditional knock-in line was generated in which a well-folded but enzymatically inactive kinase was induced. Induction of animals homozygous for the knock-in gene displayed a lethal phenotype similar to, but less dramatic than observed for the inducible knockdown model. Heterozygous animals in both models displayed a less severe phenotype [75]. Follow-up on these results is needed to define the potential role of PI4KIIIα as a target for therapeutic intervention.
Another major challenge in the development of PI4KIII inhibitors will be to achieve selective inhibition of PI4KIIIs or even of the isoforms α or β. A commonly used specific inhibitor of PI4KIIIs is PIK93, a phenylthiazole (Fig. 3 ). PIK93 was originally developed as an inhibitor of class I PI3Ks [76]. In in vitro kinase assays PIK93 was found to be about 100-fold more potent against PI4KIIIβ than against PI4KIIIα and is ineffective against type II PI4Ks (Table 2 ) [77]. However, PIK93 at higher concentrations also seems to inhibit other PI3Ks. PIK93 was shown to inhibit in vitro picornavirus and HCV replication [46], [78], [79]. Another inhibitor of PI4KIIIβ and α is enviroxime [2-amino-1-(isopropylsulfonyl)-6-benzimidazole phenyl ketone oxime] (Fig. 3). Enviroxime is since long known as an inhibitor of the in vitro replication of rhinoviruses and enteroviruses (Table 2). Recently, we demonstrated that enviroxime inhibits HCV subgenomic replicon replication [79]. Picornaviruses that are resistant to enviroxime carry mutations in protein 3A [80]. The picornavirus protein 3A is a membrane associated protein that is involved in the formation of the membranous web. Direct interaction of enviroxime with the 3A protein has not been reported [81]. Clinical trials in natural and experimentally induced rhinovirus infections in the 1980s produced disappointing results and the drug was not further developed [82], [83]. For decades, it was not clear what the mechanism was by which enviroxime inhibits the replication of picornaviruses. Recently Arita and colleagues demonstrated that enviroxime inhibits RNA replication of picornaviruses by inhibiting PI4KIIIβ [78].
Fig. 3.
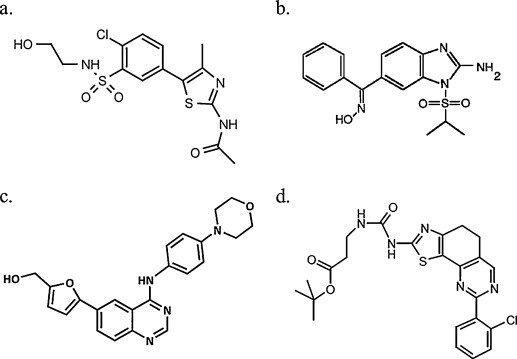
Structural formulae of PI4KIII inhibitors. (a) Phenylthiazole PIK93, (b) enviroxime, (c) 4-anilino quinazoline AL-9, (d) PI4KIIIα inhibitor of Boehringer Ingelheim.
Table 2.
Summary of the antiviral potency and selectivity of PI4KIII inhibitors.
| Molecule | PIK inhibition (IC50, μM) |
Viral potency in vitro (EC50, μM) |
|||
|---|---|---|---|---|---|
| PI4KIIIα | PI4KIIIβ | Other PIKs | Picornavirus | HCV | |
| PIK93 | 1.1 | 0.019 | Active against some PI3Ks of class I, II and III | 0.14 (PV) | 0.17 (GT 1b replicon) |
| Enviroxime | 1.4 | 0.12 | nd | 0.7 (CVB3), 0.19 (PV1), 0.11 (HRV14) | 0.22 (GT 1b replicon) |
| Cpd 6 (Novartis) | nd | 0.024 | nd | nd | 0.13 (GT 1b replicon) |
| AL-9 | 0.57 | 3.08 | PI3K p110α: 1.1 | nd | 0.29 (GT 1b replicon) |
| Inhibitor A (BI) | 0.45 | nd | nd | nd | 0.3 (GT 1b replicon) |
nd: not determined; GT: genotype; CVB3: coxsackievirus B3; PV1: poliovirus 1; HRV14: human rhinovirus 14; Cpd: compound; BI: Boehringer Ingelheim.
In order to identify highly specific PI4KIIIβ inhibitors, Novartis carried out a small molecule high throughput screen. Several different chemical molecule classes were identified as selective inhibitors of PI4KIIIβ [84]. Interestingly, these molecules also strongly inhibited HCV subgenomic replicons (genotype 1a and 1b) and the JFH-1 virus (genotype 2a) in vitro (Table 2). A high barrier to resistance was suggested by the observation of low levels of resistance following resistance selection with a representative molecule (compound 6) of more than five weeks. Although other cellular functions of PI4KIIIβ (such as insulin secretion and regulation of ion channels) seemed to be not perturbed, an unexpected anti-proliferative effect in lymphocytes impeded the further development of these PI4KIIIβ inhibitors as antiviral agents for HCV.
Another new class of PI4KIII inhibitors is the class of 4-anilino quinazolines (Fig. 3) that were first identified as inhibitors of in vitro HCV replication [85]. The target of this class of molecules was thought to be the viral protein NS5A. However, a molecule prototypical for this class, AL-9, was recently shown to inhibit purified PI4KIIIα and, to a lesser extent, PI4KIIIβ (IC50 of 0.57 μM vs. 3.08 μM) (Table 2) [86]. Treatment of Huh 7.5 cells with AL-9 resulted in a gradual decrease of PI4P levels in the plasma membrane, indicating that this agent inhibits PI4KIIIα in intact cells. On the other hand, AL-9 did not influence the PI4P levels in the Golgi membrane, suggesting that PI4KIIIβ is not inhibited. In cells expressing HCV proteins, treatment with AL-9 induced abnormally large clusters of NS5A. It was therefore proposed that the antiviral effect of 4-anilino quinazolines results from the inhibition of PI4KIIIα and the consequent depletion of PI4P lipids. Of note, AL-9 was also shown to inhibit class I PI3K p110α (IC50 of 1.1 μM) and, to a lesser extent, PI3K p110β (IC50 >10 μM).
Elaborating on the results of their siRNA screen which identified PI4KIIIα as a host factor for HCV [44], Boehringer Ingelheim performed an enzymatic screening in search of specific PI4KIIIα inhibitors [75]. Cell culture studies with these inhibitors demonstrated that the lipid kinase activity of PI4KIIIα, rather than protein-protein interactions alone, is essential for HCV RNA replication (Fig. 3). Of note, these inhibitors exhibited also activity against certain other lipid kinases (not specified). HCV resistance to this class of inhibitors was mapped to the C-terminal end of NS4B and the N-terminal domain of NS5A. Because of the mouse genetic studies mentioned above, work on PI4KIIIα as an anti-HCV drug target was terminated.
8. Discussion
Several viruses are able to hijack the host cell PI4KIIIs in different ways. Viruses can produce either proteins that recruit and activate PI4KIIIs to the replication complexes or effector proteins that use PI4P as a membrane anchor. The role of PI4KIIIβ in picornavirus replication has been relatively well described. The characterization of the role of PI4KIIIα/β in HCV replication and the identification of HCV/host PI4P-interacting factors provide future challenges. As pointed out by Lohmann and colleagues it cannot be excluded that the increased PI4P levels in HCV-infected cells are coincidental and not relevant for HCV replication. Since a tentative role of PI4KIII in NS5A phosphorylation is suggested (Fig. 2), it could well be that the NS5A, simultaneously with PI4KIII, recruits protein kinases (cf. supra) that can phosphorylate NS5A or that both proteins compete for the same protein kinases. This could result in a modulation of NS5A phosphorylation and the concomitant assembly/dissociation of HCV replicase complexes [87]. However, protein kinase D, known to phosphorylate PI4KIIIβ, is a negative regulator of HCV virus particle egress and this effect was mediated through OSB and CERT [55]. The latter proteins are, as mentioned, essential for the de novo sphingomyelin synthesis, which was shown to be required for HCV replication [88]. More in particular virion-associated sphingomyelin is essential for the infectivity of HCV virions [89]. Recently it was shown that, depending on the genotype, sphingomyelin can activate the HCV RNA-dependent RNA polymerase [90]. Thus HCV might exploit PI4KIIIα to curb the sphingomyelin metabolism for its own benefit. As suggested by Lindenbach, HCV creates an unctuous home to replicate [91]. This home might well resemble unique membranous clusters associated with various organelles such as multivesicular bodies, mitochondria and the ER in the mammalian nervous system [13]. The manipulation of the PI metabolism and PI distribution by HCV might not be limited to PI4P and PI4KIII. Findings that show an essential role for Rab5, Rab7 and PI3K [92] suggested an even more extensive manipulation of this system by HCV.
Viruses related to HCV, such as the flaviviruses DENV and WNV do not depend on PI4P for their replication. For other viruses such as retroviruses, PI4P is clearly linked to the need for PI(4,5)P2 to target Gag to the plasma membrane. Also EBV seems to require increased PI4K activities. Surprisingly PI4KIIIβ is required for SARS-CoV entry. It is most interesting that entirely unrelated viruses, such as HCV, picornaviruses, EBV, retroviruses and coronaviruses each exploit PI4K by apparently largely unrelated mechanisms. The question remains as to whether PI4K is a good target for inhibition of viral replication in patients. Knockdown of PI4KIIIα in mice suggest at least that the enzyme is critical and may thus have liabilities as an antiviral target. At present, the isoform and target specificity of the known PI4K inhibitors is far from optimal. It will be of interest to study whether PI4K inhibitors highly specific for one isoform can be developed and whether these have a sufficient therapeutic window. Furthermore alternative targeting strategies could be envisioned that do not interfere with the enzymatic activity of PI4K, possibly resulting in less or no adverse side effects. Indeed the interface at which PI4K interacts with viral factors such as the picornavirus 3A may be potentially drugable.
Acknowledgments
This work was supported by a postdoctoral fellowship from the KU Leuven to Leen Delang and from the Research Foundation Flanders-FWO to Jan Paeshuyse, the IWT-SBO project #100042 and by grant G.0728.09N of the Research Foundation Flanders-FWO.
References
- 1.Di Paolo G., De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- 2.Grainger D.L., Tavelis C., Ryan A.J., Hinchliffe K.A. The emerging role of PtdIns5P: another signalling phosphoinositide takes its place. Biochem Soc Trans. 2012;40:257–261. doi: 10.1042/BST20110617. [DOI] [PubMed] [Google Scholar]
- 3.Kerr W.G., Colucci F. Inositol phospholipid signaling and the biology of natural killer cells. J Innate Immun. 2011;3:249–257. doi: 10.1159/000323920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bunney T.D., Katan M. Phosphoinositide signalling in cancer: beyond PI3K and PTEN. Nat Rev Cancer. 2010;10:342–352. doi: 10.1038/nrc2842. [DOI] [PubMed] [Google Scholar]
- 5.Michell R.H., Heath V.L., Lemmon M.A., Dove S.K. Phosphatidylinositol 3,5-bisphosphate: metabolism and cellular functions. Trends Biochem Sci. 2006;31:52–63. doi: 10.1016/j.tibs.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 6.Shewan A., Eastburn D.J., Mostov K. Phosphoinositides in cell architecture. Cold Spring Harb Perspect Biol. 2011;3:a004796. doi: 10.1101/cshperspect.a004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hammond G., Thomas C.L., Schiavo G. Nuclear phosphoinositides and their functions. Curr Top Microbiol Immunol. 2004;282:177–206. doi: 10.1007/978-3-642-18805-3_7. [DOI] [PubMed] [Google Scholar]
- 8.Cain R.J., Ridley A.J. Phosphoinositide 3-kinases in cell migration. Biol Cell. 2009;101:13–29. doi: 10.1042/BC20080079. [DOI] [PubMed] [Google Scholar]
- 9.Gault C.R., Obeid L.M., Hannun Y.A. An overview of sphingolipid metabolism: from synthesis to breakdown. Adv Exp Med Biol. 2010;688:1–23. doi: 10.1007/978-1-4419-6741-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weber S.S., Ragaz C., Hilbi H. Pathogen trafficking pathways and host phosphoinositide metabolism. Mol Microbiol. 2009;71:1341–1352. doi: 10.1111/j.1365-2958.2009.06608.x. [DOI] [PubMed] [Google Scholar]
- 11.Behnia R., Munro S. Organelle identity and the signposts for membrane traffic. Nature. 2005;438:597–604. doi: 10.1038/nature04397. [DOI] [PubMed] [Google Scholar]
- 12.Roth M.G. Phosphoinositides in constitutive membrane traffic. Physiol Rev. 2004;84:699–730. doi: 10.1152/physrev.00033.2003. [DOI] [PubMed] [Google Scholar]
- 13.Balla A., Balla T. Phosphatidylinositol 4-kinases: old enzymes with emerging functions. Trends Cell Biol. 2006;16:351–361. doi: 10.1016/j.tcb.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 14.Heilmeyer L.M., Jr., Vereb G., Jr., Vereb G., Kakuk A., Szivak I. Mammalian phosphatidylinositol 4-kinases. IUBMB Life. 2003;55:59–65. doi: 10.1002/tbmb.718540873. [DOI] [PubMed] [Google Scholar]
- 15.Barylko B., Mao Y.S., Wlodarski P., Jung G., Binns D.D., Sun H.Q. Palmitoylation controls the catalytic activity and subcellular distribution of phosphatidylinositol 4-kinase II{alpha} J Biol Chem. 2009;284:9994–10003. doi: 10.1074/jbc.M900724200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jung G., Wang J., Wlodarski P., Barylko B., Binns D.D., Shu H. Molecular determinants of activation and membrane targeting of phosphoinositol 4-kinase IIbeta. Biochem J. 2008;409:501–509. doi: 10.1042/BJ20070821. [DOI] [PubMed] [Google Scholar]
- 17.Wang J., Sun H.Q., Macia E., Kirchhausen T., Watson H., Bonifacino J.S. PI4P promotes the recruitment of the GGA adaptor proteins to the trans-Golgi network and regulates their recognition of the ubiquitin sorting signal. Mol Biol Cell. 2007;18:2646–2655. doi: 10.1091/mbc.E06-10-0897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Minogue S., Chu K.M., Westover E.J., Covey D.F., Hsuan J.J., Waugh M.G. Relationship between phosphatidylinositol 4-phosphate synthesis, membrane organization, and lateral diffusion of PI4KIIalpha at the trans-Golgi network. J Lipid Res. 2010;51:2314–2324. doi: 10.1194/jlr.M005751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Craige B., Salazar G., Faundez V. Phosphatidylinositol-4-kinase type II alpha contains an AP-3-sorting motif and a kinase domain that are both required for endosome traffic. Mol Biol Cell. 2008;19:1415–1426. doi: 10.1091/mbc.E07-12-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jovic M., Kean M.J., Szentpetery Z., Polevoy G., Gingras A.C., Brill J.A. Two phosphatidylinositol 4-kinases control lysosomal delivery of the Gaucher disease enzyme, beta-glucocerebrosidase. Mol Biol Cell. 2012;23:1533–1545. doi: 10.1091/mbc.E11-06-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Banerji S., Ngo M., Lane C.F., Robinson C.A., Minogue S., Ridgway N.D. Oxysterol binding protein-dependent activation of sphingomyelin synthesis in the Golgi apparatus requires phosphatidylinositol 4-kinase IIalpha. Mol Biol Cell. 2010;21:4141–4150. doi: 10.1091/mbc.E10-05-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li J., Lu Y., Zhang J., Kang H., Qin Z., Chen C. PI4KIIalpha is a novel regulator of tumor growth by its action on angiogenesis and HIF-1alpha regulation. Oncogene. 2010;29:2550–2559. doi: 10.1038/onc.2010.14. [DOI] [PubMed] [Google Scholar]
- 23.Chu K.M., Minogue S., Hsuan J.J., Waugh M.G. Differential effects of the phosphatidylinositol 4-kinases, PI4KIIalpha and PI4KIIIbeta, on Akt activation and apoptosis. Cell Death Dis. 2010;1:e106. doi: 10.1038/cddis.2010.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei Y.J., Sun H.Q., Yamamoto M., Wlodarski P., Kunii K., Martinez M. Type II phosphatidylinositol 4-kinase beta is a cytosolic and peripheral membrane protein that is recruited to the plasma membrane and activated by Rac-GTP. J Biol Chem. 2002;277:46586–46593. doi: 10.1074/jbc.M206860200. [DOI] [PubMed] [Google Scholar]
- 25.Wong K., Meyers ddR, Cantley L.C. Subcellular locations of phosphatidylinositol 4-kinase isoforms. J Biol Chem. 1997;272:13236–13241. doi: 10.1074/jbc.272.20.13236. [DOI] [PubMed] [Google Scholar]
- 26.de Graaf P., Klapisz E.E., Schulz T.K., Cremers A.F., Verkleij A.J., van Bergen en Henegouwen P.M. Nuclear localization of phosphatidylinositol 4-kinase beta. J Cell Sci. 2002;115:1769–1775. doi: 10.1242/jcs.115.8.1769. [DOI] [PubMed] [Google Scholar]
- 27.Czech M.P. Dynamics of phosphoinositides in membrane retrieval and insertion. Annu Rev Physiol. 2003;65:791–815. doi: 10.1146/annurev.physiol.65.092101.142522. [DOI] [PubMed] [Google Scholar]
- 28.Wenk M.R., De C.P. Protein–lipid interactions and phosphoinositide metabolism in membrane traffic: insights from vesicle recycling in nerve terminals. Proc Natl Acad Sci USA. 2004;101:8262–8269. doi: 10.1073/pnas.0401874101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Godi A., Pertile P., Meyers R., Marra P., Di T.G., Iurisci C. ARF mediates recruitment of PtdIns-4-OH kinase-beta and stimulates synthesis of PtdIns(4,5)P2 on the Golgi complex. Nat Cell Biol. 1999;1:280–287. doi: 10.1038/12993. [DOI] [PubMed] [Google Scholar]
- 30.Weisz O.A., Gibson G.A., Leung S.M., Roder J., Jeromin A. Overexpression of frequenin, a modulator of phosphatidylinositol 4-kinase, inhibits biosynthetic delivery of an apical protein in polarized Madin–Darby canine kidney cells. J Biol Chem. 2000;275:24341–24347. doi: 10.1074/jbc.M000671200. [DOI] [PubMed] [Google Scholar]
- 31.Haynes L.P., Thomas G.M., Burgoyne R.D. Interaction of neuronal calcium sensor-1 and ADP-ribosylation factor 1 allows bidirectional control of phosphatidylinositol 4-kinase beta and trans-Golgi network-plasma membrane traffic. J Biol Chem. 2005;280:6047–6054. doi: 10.1074/jbc.M413090200. [DOI] [PubMed] [Google Scholar]
- 32.Suer S., Sickmann A., Meyer H.E., Herberg F.W., Heilmeyer L.M., Jr. Human phosphatidylinositol 4-kinase isoform PI4K92. Expression of the recombinant enzyme and determination of multiple phosphorylation sites. Eur J Biochem. 2001;268:2099–2106. doi: 10.1046/j.1432-1327.2001.02089.x. [DOI] [PubMed] [Google Scholar]
- 33.Hausser A., Storz P., Martens S., Link G., Toker A., Pfizenmaier K. Protein kinase D regulates vesicular transport by phosphorylating and activating phosphatidylinositol-4 kinase IIIbeta at the Golgi complex. Nat Cell Biol. 2005;7:880–886. doi: 10.1038/ncb1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakagawa T., Goto K., Kondo H. Cloning, expression, and localization of 230-kDa phosphatidylinositol 4-kinase. J Biol Chem. 1996;271:12088–12094. doi: 10.1074/jbc.271.20.12088. [DOI] [PubMed] [Google Scholar]
- 35.Dumaresq-Doiron K., Savard M.F., Akam S., Costantino S., Lefrancois S. The phosphatidylinositol 4-kinase PI4KIIIalpha is required for the recruitment of GBF1 to Golgi membranes. J Cell Sci. 2010;123:2273–2280. doi: 10.1242/jcs.055798. [DOI] [PubMed] [Google Scholar]
- 36.Downes C.P., Gray A., Lucocq J.M. Probing phosphoinositide functions in signaling and membrane trafficking. Trends Cell Biol. 2005;15:259–268. doi: 10.1016/j.tcb.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 37.Santiago-Tirado F.H., Bretscher A. Membrane-trafficking sorting hubs: cooperation between PI4P and small GTPases at the trans-Golgi network. Trends Cell Biol. 2011;21:515–525. doi: 10.1016/j.tcb.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vicinanza M., D’Angelo G., Di C.A., De Matteis M.A. Function and dysfunction of the PI system in membrane trafficking. EMBO J. 2008;27:2457–2470. doi: 10.1038/emboj.2008.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hutagalung A.H., Novick P.J. Role of Rab GTPases in membrane traffic and cell physiology. Physiol Rev. 2011;91:119–149. doi: 10.1152/physrev.00059.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gosert R., Egger D., Lohmann V., Bartenschlager R., Blum H.E., Bienz K. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J Virol. 2003;77:5487–5492. doi: 10.1128/JVI.77.9.5487-5492.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reiss S., Rebhan I., Backes P., Romero-Brey I., Erfle H., Matula P. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe. 2011;9:32–45. doi: 10.1016/j.chom.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Berger K.L., Cooper J.D., Heaton N.S., Yoon R., Oakland T.E., Jordan T.X. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc Natl Acad Sci USA. 2009;106:7577–7582. doi: 10.1073/pnas.0902693106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tai A.W., Benita Y., Peng L.F., Kim S.S., Sakamoto N., Xavier R.J. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe. 2009;5:298–307. doi: 10.1016/j.chom.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vaillancourt F.H., Pilote L., Cartier M., Lippens J., Liuzzi M., Bethell R.C. Identification of a lipid kinase as a host factor involved in hepatitis C virus RNA replication. Virology. 2009;387:5–10. doi: 10.1016/j.virol.2009.02.039. [DOI] [PubMed] [Google Scholar]
- 45.Trotard M., Lepere-Douard C., Regeard M., Piquet-Pellorce C., Lavillette D., Cosset F.L. Kinases required in hepatitis C virus entry and replication highlighted by small interference RNA screening. FASEB J. 2009;23:3780–3789. doi: 10.1096/fj.09-131920. [DOI] [PubMed] [Google Scholar]
- 46.Borawski J., Troke P., Puyang X., Gibaja V., Zhao S., Mickanin C. Class III phosphatidylinositol 4-kinase alpha and beta are novel host factor regulators of hepatitis C virus replication. J Virol. 2009;83:10058–10074. doi: 10.1128/JVI.02418-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Q., Brass A.L., Ng A., Hu Z., Xavier R.J., Liang T.J. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc Natl Acad Sci USA. 2009;106:16410–16415. doi: 10.1073/pnas.0907439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lim Y.S., Hwang S.B. Hepatitis C virus NS5A protein interacts with phosphatidylinositol 4-kinase type III{alpha} and regulates viral propagation. J Biol Chem. 2011;286:11290–11298. doi: 10.1074/jbc.M110.194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reiss S., Harak C., Romero-Brey I., Rebhan I., Radujkovic D., Bartenschlager R. 47th annual meeting of the European association for the study of the liver. 2012. Modulation of phosphorylation of hepatitis C virus nonstructural protein 5A by the lipid kinase PI4KIIIalpha. [Google Scholar]
- 50.Hsu N.Y., Ilnytska O., Belov G., Santiana M., Chen Y.H., Takvorian P.M. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell. 2010;141:799–811. doi: 10.1016/j.cell.2010.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tai A.W., Salloum S. The role of the phosphatidylinositol 4-kinase PI4KA in hepatitis C virus-induced host membrane rearrangement. PLoS ONE. 2011;6:e26300. doi: 10.1371/journal.pone.0026300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang L., Hong Z., Lin W., Shao R.X., Goto K., Hsu V.W. ARF1 and GBF1 generate a PI4P-enriched environment supportive of hepatitis C virus replication. PLoS ONE. 2012;7:e32135. doi: 10.1371/journal.pone.0032135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McMahon H.T., Gallop J.L. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature. 2005;438:590–596. doi: 10.1038/nature04396. [DOI] [PubMed] [Google Scholar]
- 54.Stapleford K.A., Miller D.J. Role of cellular lipids in positive-sense RNA virus replication complex assembly and function. Viruses. 2010;2:1055–1068. doi: 10.3390/v2051055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Amako Y., Syed G.H., Siddiqui A. Protein kinase D negatively regulates hepatitis C virus secretion through phosphorylation of oxysterol-binding protein and ceramide transfer protein. J Biol Chem. 2011;286:11265–11274. doi: 10.1074/jbc.M110.182097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Amako Y., Sarkeshik A., Hotta H., Yates J., III, Siddiqui A. Role of oxysterol binding protein in hepatitis C virus infection. J Virol. 2009;83:9237–9246. doi: 10.1128/JVI.00958-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Berger K.L., Kelly S.M., Jordan T.X., Tartell M.A., Randall G. Hepatitis C virus stimulates the phosphatidylinositol 4-kinase III alpha-dependent phosphatidylinositol 4-phosphate production that is essential for its replication. J Virol. 2011;85:8870–8883. doi: 10.1128/JVI.00059-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Coller K.E., Heaton N.S., Berger K.L., Cooper J.D., Saunders J.L., Randall G. Molecular determinants and dynamics of hepatitis C virus secretion. PLoS Pathog. 2012;8:e1002466. doi: 10.1371/journal.ppat.1002466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Belov G.A., Altan-Bonnet N., Kovtunovych G., Jackson C.L., Lippincott-Schwartz J., Ehrenfeld E. Hijacking components of the cellular secretory pathway for replication of poliovirus RNA. J Virol. 2007;81:558–567. doi: 10.1128/JVI.01820-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lanke K.H., van der Schaar H.M., Belov G.A., Feng Q., Duijsings D., Jackson C.L. GBF1, a guanine nucleotide exchange factor for Arf, is crucial for coxsackievirus B3 RNA replication. J Virol. 2009;83:11940–11949. doi: 10.1128/JVI.01244-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sasaki J., Ishikawa K., Arita M., Taniguchi K. ACBD3-mediated recruitment of PI4KB to picornavirus RNA replication sites. EMBO J. 2012;31:754–766. doi: 10.1038/emboj.2011.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Greninger A.L., Knudsen G.M., Betegon M., Burlingame A.L., Derisi J.L. The 3A protein from multiple picornaviruses utilizes the Golgi adaptor protein ACBD3 to recruit PI4KIIIbeta. J Virol. 2012;86:3605–3616. doi: 10.1128/JVI.06778-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Teterina N.L., Pinto Y., Weaver J.D., Jensen K.S., Ehrenfeld E. Analysis of poliovirus protein 3A interactions with viral and cellular proteins in infected cells. J Virol. 2011;85:4284–4296. doi: 10.1128/JVI.02398-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sohda M., Misumi Y., Yamamoto A., Yano A., Nakamura N., Ikehara Y. Identification and characterization of a novel Golgi protein, GCP60, that interacts with the integral membrane protein giantin. J Biol Chem. 2001;276:45298–45306. doi: 10.1074/jbc.M108961200. [DOI] [PubMed] [Google Scholar]
- 65.Tones M.A., Kellie S., Hawthorne J.N. Elevated phosphatidylinositol kinase activity in Rous sarcoma virus-transformed cells. Lack of evidence for enzyme translocation. Biochim Biophys Acta. 1987;931:165–169. doi: 10.1016/0167-4889(87)90202-3. [DOI] [PubMed] [Google Scholar]
- 66.Fukui Y., Hanafusa H. Phosphatidylinositol kinase activity associates with viral p60src protein. Mol Cell Biol. 1989;9:1651–1658. doi: 10.1128/mcb.9.4.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Suzuki Y., Ohsugi K., Ono Y. EBV increases phosphoinositide kinase activities in human B cells. J Immunol. 1992;149:207–213. [PubMed] [Google Scholar]
- 68.Ono A., Ablan S.D., Lockett S.J., Nagashima K., Freed E.O. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc Natl Acad Sci USA. 2004;101:14889–14894. doi: 10.1073/pnas.0405596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chan R., Uchil P.D., Jin J., Shui G., Ott D.E., Mothes W. Retroviruses human immunodeficiency virus and murine leukemia virus are enriched in phosphoinositides. J Virol. 2008;82:11228–11238. doi: 10.1128/JVI.00981-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saad J.S., Miller J., Tai J., Kim A., Ghanam R.H., Summers M.F. Structural basis for targeting HIV-1 Gag proteins to the plasma membrane for virus assembly. Proc Natl Acad Sci USA. 2006;103:11364–11369. doi: 10.1073/pnas.0602818103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brown B.K., Karasavvas N., Beck Z., Matyas G.R., Birx D.L., Polonis V.R. Monoclonal antibodies to phosphatidylinositol phosphate neutralize human immunodeficiency virus type 1: role of phosphate-binding subsites. J Virol. 2007;81:2087–2091. doi: 10.1128/JVI.02011-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chan J., Dick R.A., Vogt V.M. Rous sarcoma virus gag has no specific requirement for phosphatidylinositol-(4,5)-bisphosphate for plasma membrane association in vivo or for liposome interaction in vitro. J Virol. 2011;85:10851–10860. doi: 10.1128/JVI.00760-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martin-Acebes M.A., Blazquez A.B., Jimenez de O.N., Escribano-Romero E., Saiz J.C. West Nile virus replication requires fatty acid synthesis but is independent on phosphatidylinositol-4-phosphate lipids. PLoS ONE. 2011;6:e24970. doi: 10.1371/journal.pone.0024970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang N., Ma P., Lang J., Zhang Y., Deng J., Ju X. Phosphatidylinositol 4-kinase IIIbeta is required for severe acute respiratory syndrome coronavirus spike-mediated cell entry. J Biol Chem. 2012;287:8457–8467. doi: 10.1074/jbc.M111.312561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vaillancourt F.H., Brault M., Pilote L., Uyttersprot N., Gaillard E., Stoltz J. Viruses and cells Gordon research conference. 2011. Evaluation of the lipid kinase PI4KIIIalpha as an anti-HCV drug target. [Google Scholar]
- 76.Knight Z.A., Gonzalez B., Feldman M.E., Zunder E.R., Goldenberg D.D., Williams O. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell. 2006;125:733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Balla A., Tuymetova G., Toth B., Szentpetery Z., Zhao X., Knight Z.A. Design of drug-resistant alleles of type-III phosphatidylinositol 4-kinases using mutagenesis and molecular modeling. Biochemistry. 2008;47:1599–1607. doi: 10.1021/bi7017927. [DOI] [PubMed] [Google Scholar]
- 78.Arita M., Kojima H., Nagano T., Okabe T., Wakita T., Shimizu H. Phosphatidylinositol 4-kinase III beta is a target of enviroxime-like compounds for antipoliovirus activity. J Virol. 2011;85:2364–2372. doi: 10.1128/JVI.02249-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Delang L., Leyssen P., Neyts J. 18th international symposium on hepatitis C virus and related viruses. 2011. The picornavirus inhibitor enviroxime inhibits HCV RNA replication in vitro. [Google Scholar]
- 80.Heinz B.A., Vance L.M. The antiviral compound enviroxime targets the 3A coding region of rhinovirus and poliovirus. J Virol. 1995;69:4189–4197. doi: 10.1128/jvi.69.7.4189-4197.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brown-Augsburger P., Vance L.M., Malcolm S.K., Hsiung H., Smith D.P., Heinz B.A. Evidence that enviroxime targets multiple components of the rhinovirus 14 replication complex. Arch Virol. 1999;144:1569–1585. doi: 10.1007/s007050050611. [DOI] [PubMed] [Google Scholar]
- 82.Miller F.D., Monto A.S., DeLong D.C., Exelby A., Bryan E.R., Srivastava S. Controlled trial of enviroxime against natural rhinovirus infections in a community. Antimicrob Agents Chemother. 1985;27:102–106. doi: 10.1128/aac.27.1.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Phillpotts R.J., Wallace J., Tyrrell D.A., Tagart V.B. Therapeutic activity of enviroxime against rhinovirus infection in volunteers. Antimicrob Agents Chemother. 1983;23:671–675. doi: 10.1128/aac.23.5.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schmitz U., Tan S.L. NS5A—from obscurity to new target for HCV therapy. Recent Pat Antiinfect Drug Discov. 2008;3:77–92. doi: 10.2174/157489108784746597. [DOI] [PubMed] [Google Scholar]
- 85.Bianco A., Reghellin V., Donnici L., Fenu S., Alvarez R., Baruffa C. Metabolism of phosphatidylinositol 4-kinase IIIalpha-dependent PI4P is subverted by HCV and is targeted by a 4-anilino quinazoline with antiviral activity. PLoS Pathog. 2012;8:e1002576. doi: 10.1371/journal.ppat.1002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Evans M.J., Rice C.M., Goff S.P. Phosphorylation of hepatitis C virus nonstructural protein 5A modulates its protein interactions and viral RNA replication. Proc Natl Acad Sci USA. 2004;101:13038–13043. doi: 10.1073/pnas.0405152101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sakamoto H., Okamoto K., Aoki M., Kato H., Katsume A., Ohta A. Host sphingolipid biosynthesis as a target for hepatitis C virus therapy. Nat Chem Biol. 2005;1:333–337. doi: 10.1038/nchembio742. [DOI] [PubMed] [Google Scholar]
- 88.Aizaki H., Morikawa K., Fukasawa M., Hara H., Inoue Y., Tani H. Critical role of virion-associated cholesterol and sphingolipid in hepatitis C virus infection. J Virol. 2008;82:5715–5724. doi: 10.1128/JVI.02530-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Weng L., Hirata Y., Arai M., Kohara M., Wakita T., Watashi K. Sphingomyelin activates hepatitis C virus RNA polymerase in a genotype-specific manner. J Virol. 2010;84:11761–11770. doi: 10.1128/JVI.00638-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lindenbach B.D. Understanding how hepatitis C virus builds its unctuous home. Cell Host Microbe. 2011;9:1–2. doi: 10.1016/j.chom.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Su W.C., Chao T.C., Huang Y.L., Weng S.C., Jeng K.S., Lai M.M. Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J Virol. 2011;85:10561–10571. doi: 10.1128/JVI.00173-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Manna D., Aligo J., Xu C., Park W.S., Koc H., Heo W.D. Endocytic Rab proteins are required for hepatitis C virus replication complex formation. Virology. 2010;398:21–37. doi: 10.1016/j.virol.2009.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]