

Résumé
L’accumulation d’une protéine spécifique sous forme agrégée est un phénomène commun aux maladies neurodégénératives humaines. Dans la maladie de Parkinson, cette protéine est l’α-synucléine qui est une protéine neuronale de 143 acides aminés. De conformation monomérique en solution, elle possède également une capacité naturelle à s’agréger en structures amyloïdes (dimères, oligomères, fibrilles puis corps ou neurites de Lewy). Elle détient donc les caractéristiques d’une protéine prion (différentes conformations, initiation et dissémination d’un processus transconformationnel). De nombreux arguments expérimentaux in vitro et in vivo sur des animaux transgéniques ou sauvages sont en faveur d’une progression prion-like de la maladie de Parkinson. La diffusion séquentielle et prédictive de l’α-synucléine mise en évidence par Braak et al. et sa corrélation avec les signes non moteurs vont tout à fait dans le sens de cette progression prion-like. Même si le facteur déclenchant à l’origine du mauvais repliement et de l’agrégation de la protéine cible reste inconnu, la maladie de Parkinson est un modèle très pertinent pour l’étude de ces mécanismes et aussi pour tester des traitements spécifiques ciblant les assemblages d’α-synucléine et leur propagation dès la phase pré-motrice de la maladie. Malgré cette progression prion-like, il n’existe actuellement aucun argument indiquant un risque de transmission interhumaine de la maladie de Parkinson.
Mots clés: Maladie de Parkinson, α-Synuclein, Prion-like
Abstract
The accumulation of a specific protein in aggregated form is a common phenomenon in human neurodegenerative diseases. In Parkinson's disease, this protein is α-synuclein which is a neuronal protein of 143 amino acids. With a monomeric conformation in solution, it also has a natural capacity to aggregate into amyloid structures (dimers, oligomers, fibrils and Lewy bodies or neurites). It therefore fulfils the characteristics of a prion protein (different conformations, seeding and spreading). In vitro and in vivo experimental evidence in transgenic and wild animals indicates a prion-like propagation of Parkinson's disease. The sequential and predictive distribution of α-synuclein demonstrated by Braak et al. and its correlation with non-motor signs are consistent with the prion-like progression. Although the triggering factor causing the misfolding and aggregation of the target protein is unknown, Parkinson's disease is a highly relevant model for the study of these mechanisms and also to test specific treatments targeting the assemblies of α-synuclein and propagation from pre-motor phase of the disease. Despite this prion-like progression, there is currently no argument indicating a risk of human transmission of Parkinson's disease.
Keywords: Parkinson's disease, α-Synuclein, Prion-like
1. Les maladies à prions, paradigme des maladies conformationnelles des protéines du système nerveux central
1.1. Qu’est-ce qu’un prion ?
L’hypothèse de la nature exclusivement protéique des agents transmissibles non conventionnels (ATNC) a été émise à la fin des années 1960 sur la base des propriétés physico-chimiques exceptionnelles de ces agents [1], [2]. Le terme prion a été créé par Stanley Prusiner en 1982 pour qualifier l’agent responsable de la transmission de la scrapie et des encéphalopathies subaiguës spongiformes transmissibles (ESST) [3]. Par définition, les prions sont des particules protéiques infectieuses de petites tailles (« proteinaceous infectious particle »), résistantes aux procédés d’inactivation efficaces pour modifier les acides nucléiques. Le terme prion souligne qu’une protéine dépourvue d’acide nucléique est nécessaire et suffisante à « l’infection ». Si les maladies à prions entrent bien dans la définition des maladies infectieuses proposée par l’OMS (« Les maladies infectieuses sont causées par des microorganismes pathogènes, tels que les bactéries, les virus, les parasites ou les champignons. Ces maladies peuvent se transmettre, directement ou indirectement, d’une personne à l’autre ») (http://www.who.int/topics/infectious_diseases/fr/il), il faut souligner que, chez l’homme et contrairement à certaines maladies animales comme la maladie du dépérissement chronique des cervidés, elles ne sont pas contagieuses. Les rares cas de transmission interhumaine, depuis la disparition du kuru, résultent de contaminations accidentelles en règle iatrogène [4]. Un prion est donc un agent transmissible accidentellement ou expérimentalement dont l’infectivité peut être titrée. Il faut bien les distinguer de ce qui a été appelé par facilité de langage « prions de levure » ou « prions physiologiques » en référence au mécanisme moléculaire gouvernant la réplication de ces agents et aux propriétés de la protéine prion.
En effet, la protéine prion a pour principales caractéristiques :
-
•
d’exister sous différentes conformations, une forme normale dont la structure secondaire est riche en hélice α et une forme agrégée enrichie en feuillets β qui peut exister sous la forme d’oligomères ou de fibrilles et qui s’accumule en cas de pathologie ;
-
•
de pouvoir initier, sous sa forme pathologique, l’agrégation et le mauvais repliement de la forme normale de la protéine (« seeding ») ;
-
•
de pouvoir être transférée de cellules à cellules sous sa forme agrégée, in vitro et in vivo, et d’induire dans la cellule ou le tissu hôte le processus de conversion (« spreading »).
Dans le cas des prions de mammifères, ces phénomènes sont associés au déclenchement d’une ESST.
Le terme de prion a été utilisé par la suite pour nommer des phénomènes biologiques très différents et bien distincts des « proteinaceous infectious particle » dont le point commun est la mise en jeu de protéines spécifiques partageant les caractéristiques de la protéine prion (deux conformations, initiation et dissémination d’un processus transconformationnel). À titre d’exemple, les « prions » de levure mettent en jeu des protéines notamment impliquées dans l’adaptation de ces organismes à leur environnement [5] ; l’agrégation auto-entretenue d’un facteur de traduction appelé CPEB, pour cytoplasmic polyadenylation element binding protein, interviendrait dans le mécanisme physiologique de maintien de la mémoire à long terme [6]. Du fait de ce glissement sémantique, le terme de prion recouvre donc deux phénomènes bien distincts : un mécanisme biochimique de transmission d’une information biologique, d’une part, (mécanisme de type prion, protéine de type prion, prions de levure, prions physiologiques, etc.) ; un agent transmissible responsable d’une encéphalopathie chez l’homme et l’animal d’autre part. Si les concepts mécanistiques et infectieux sont bien associés dans le cas des maladies à prions, il semble clair que cela ne vaut pas dans tout phénomène biologique où ces modifications conformationnelles sont observées.
PrPc et PrPsc
La forme normale de la protéine prion (PrP) est la protéine PrPc dont la structure primaire comprend, chez l’homme, 253 acides aminés avec un poids moléculaire qui varie de 33 à 35 kDa selon le niveau de glycosylation. Sa structure secondaire est constituée d’un taux élevé d’hélices α (42 %) et de peu de feuillets β (3 %). Sa structure tridimensionnelle a été établie par résonance magnétique nucléaire sur des protéines recombinantes [7]. Elle se compose d’une partie N-terminale, des acides aminés 23 à 121, qui a une structure très flexible pouvant adopter plusieurs conformations en fonction de l’environnement, d’une structure globulaire stable contenant 3 hélices α au niveau des acides aminés 144–154 ; 175–193 ; 200–219 et 2 courts feuillets β antiparallèles au niveau 128–131 ; 161–164 et d’une partie C-terminale possédant une ancre GPI permettant d’arrimer la PrP au feuillet externe de la membrane cellulaire. Si le rôle physiologique joué par la PrP cellulaire reste débattu, elle s’avère indispensable au développement de la maladie puisque les animaux dont le gène de la PrP a été invalidé (animaux « knockout » ou KO) ne sont pas susceptibles aux ATNC [8]. Son niveau d’expression a un rôle déterminant dans la durée de la période d’incubation qui est d’autant plus courte que le niveau d’expression est élevé. Enfin certains polymorphismes du gène PRNP codant la PrP et surtout le polymorphisme méthionine/valine au codon 129 jouent un rôle important dans la susceptibilité individuelle aux ESST. L’homologie de séquence entre la PrPsc de l’inoculum et la PrPc de l’hôte détermine en partie la barrière d’espèce lors d’une transmission interspécifique. Ainsi les souris sont résistantes aux souches de hamster sauf si l’on introduit dans leur génome une PrPc de hamster [9]. Ceci est vrai aussi pour les souches humaines dont la transmission nécessite en plus de l’introduction du gène humain l’invalidation du gène de la PrP murine. Dans certains modèles, l’expression d’une protéine PrP chimère dont les extrémités portent les séquences murines et le domaine central une séquence humaine facilite la transmission des souches humaines, particularité expliquée par l’action de cofacteurs associés spécifiques d’espèce encore non déterminés (protéine X) [10].
La forme pathologique de la PrP, la PrPsc (pour PrP scrapie) a la même structure primaire que la PrPc [11]. La digestion par la protéinase K utilisée lors de la purification de la protéine conduit à l’élimination de la portion N-terminale de la PrP générant une forme de 27 à 30 kDa. Comme la PrPc, la PrPsc possède une ancre GPI, et son profil de glycosylation est identique à celui de la PrPc. L’analyse de la structure secondaire de la PrPsc, bien qu’incomplète, a révélé d’importantes différences avec celle de la PrP cellulaire, suggérant un enrichissement marqué en feuillets β plissées [12], [13]. À ce jour, en raison des propriétés physico-chimiques de la PrPsc (pouvoir d’agrégation, insolubilité dans les détergents), aucune structure tertiaire n’a été obtenue par résonance magnétique ou par cristallographie aux rayons X. Les seules structures disponibles ont été établies par modélisation moléculaire.
Les ATNC possèdent des propriétés peu communes de résistance aux procédés habituels d’inactivation des agents microbiologiques. Les ultrasons, les rayonnements ionisants et non ionisants, les nucléases, le formaldéhyde et les détergents sont peu efficaces, voire totalement inefficaces. Par ailleurs, ces agents présentent une forte résistance à l’inactivation par la chaleur sèche. De manière générale, l’infectivité résiste bien mieux aux procédés détruisant les acides nucléiques qu’aux procédés dégradant ou dénaturant les protéines ; ainsi, les ions chaotropes, le phénol ou les digestions par des protéases diminuent sensiblement le titre infectieux.
1.2. Avancées récentes en faveur de l’hypothèse du prion
La réplication des prions reposerait sur un mécanisme de conversion protéique dans lequel la présence d’une forme pathologique, en règle agrégée et riche en feuillets β-plissés, d’une protéine déclenche l’agrégation de la forme normale qui devient à son tour capable de promouvoir le recrutement et la polymérisation de la protéine normale [14]. Plusieurs modèles en ont été proposés, notamment :
-
•
le modèle de conversion (ou polymérisation) assisté par une amorce dans lequel la PrPsc serait plus stable que la PrPc mais cinétiquement inaccessible, la conversion passerait par un intermédiaire conformationnel équidistant thermodynamiquement de la PrPc et de la PrPsc, la liaison entre la PrPsc et cet intermédiaire favoriserait sa conversion en PrPsc (Fig. 1 A) ;
-
•
le modèle de nucléation-polymérisation dans lequel la transconformation de la PrPc en PrPsc monomérique serait réversible, la PrPsc étant moins stable que la PrPc la présence d’agrégats déplaçant l’équilibre vers la formation de PrPsc [15], [16]. La liaison entre la PrPsc et cet intermédiaire favoriserait sa conversion en PrPsc (Fig. 1B).
Fig. 1.
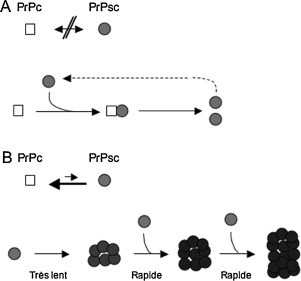
Modèles de la conversion de la PrPc en PrPsc [61], [62]. A. Modèle de polymérisation assistée par une amorce. B. Modèle de nucléation-polymérisation.
Dans ces modèles, une conversion spontanée, phénomène aléatoire et rare possiblement lié à une mutation somatique, conduirait à une forme sporadique. Dans les formes génétiques, la présence d’une mutation augmenterait la probabilité de l’événement de conversion. Dans les formes infectieuses, l’introduction de PrPsc dans un organisme hôte conduirait dans certaines conditions au recrutement et à la conversion de la PrPc endogène. La dissémination de cette anomalie conformationnelle serait assurée par la capacité qu’ont ces structures agrégées à passer d’une cellule à l’autre (« spreading ») selon des mécanismes non complètement élucidés à ce jour (exocytose/endocytose, exosomes, nanotubes…). Après avoir atteint leurs cellules cibles, les noyaux d’agrégation gardent leur activité et sont capables de recruter et de convertir la protéine de l’hôte.
La démonstration de la validité du modèle du prion passe par la production, à partir de PrPc, de PrPsc infectieuse dans un système expérimental contrôlé. Différentes approches ont été tentées dans le but de démontrer l’hypothèse du prion. Elles ont permis d’apporter des arguments en sa faveur et de valider l’hypothèse d’une conversion PrPsc-dépendante de la PrPc en PrP protéinase K résistante.
Très récemment, quatre avancées majeures ont apporté des arguments presque définitifs en faveur de l’hypothèse du prion, elles doivent tout de même être confirmées :
-
•
le groupe de Susan Lindquist au MIT a mis à profit la technique de la mutagenèse dirigée pour produire des lignées murines exprimant une PrP humaine D178N-M129 ou une PrP E200K. Alors que la lignée D178-M129 développait un phénotype clinico-pathologique très proche de l’IFF (insomnie, troubles neurovégétatifs et atrophie thalamique), les souris E200K présentaient une maladie proche de la MCJ [17], [18]. Les deux maladies étaient transmissibles et conservaient leurs caractéristiques lors des passages successifs ;
-
•
des équipes indépendantes ont montré que l’amplification de la PrPsc issue d’un sujet atteint et produite dans un tube à essai par la technique de Protein Misfolding Cyclic Amplification (PMCA) s’accompagnait d’une amplification de l’infectiosité [19], [20] ;
-
•
des agents infectieux ont pu être produits par la méthode de PMCA en utilisant des composants « prion-free » très simple : PrPc extraite de cerveau normal, poly-A, lipides ou PrP recombinante produite dans E. coli, ARN totaux extraits du foie de souris saine, lipide de synthèse [21], [22] ;
-
•
le groupe de Stanley Prusiner a pu produire un prion synthétique en modifiant la conformation et l’état d’agrégation d’une PrP recombinante tronquée [23], [24].
1.3. Phénomène de souches
Une diversité de souches a pu être mise en évidence par des expériences de transmission dans des lignées de souris ayant le même fond génétique. En pratique, pour une même dose infectante inoculée selon une voie donnée, chaque souche d’agent induit chez des souris syngéniques, une maladie caractérisée par :
-
•
un temps d’incubation bien déterminé ;
-
•
un profil lésionnel caractéristique dont les critères sont les sites d’accumulation dans le système nerveux central (SNC) de la PrPsc et de la spongiose.
Les caractéristiques d’une souche ne s’établissent qu’après 2 à 3 passages sériés : le temps d’incubation diminue pour se stabiliser et les profils lésionnels deviennent constants. On dit alors que la souche est « stabilisée ». Plus d’une vingtaine de souches différentes ont été ainsi isolées à partir d’ESST naturelles (tremblante du mouton, de la chèvre, encéphalopathie spongiforme bovine…). Le support moléculaire de la diversité des souches de prions reposerait sur la capacité de la PrP à adopter des conformations et à former des assemblages différents [25], [26].
1.4. Propagation périphérique dans les maladies à prions
Dans le cas d’une contamination par voie périphérique (voie orale ou intrapéritonéale par exemple), une étape de réplication dans les tissus lymphoïdes est nécessaire à la neuroinvasion. Chez l’homme, ce phénomène a été particulièrement bien mis en évidence dans la variante de la MCJ. L’agent est trouvé dans les structures lymphoïdes, notamment celles associées au tube digestif, au sein des cellules folliculaires dendritiques des follicules à centres clairs [27]. Il utilise ensuite l’abondante innervation sympathique de ces tissus pour gagner, via les ganglions cœliaques et stellaires, la mœlle épinière pour ensuite se propager à l’ensemble du SNC [28], [29]. Une prionémie est également associée à cette forme et quelques cas post-transfusionnels ont été observés au Royaume-Uni [30], [31].
2. L’α-synucléine
2.1. L’α-synucléine, une protéine intracellulaire, composant majeur des corps de Lewy
L’α-synucléine a été isolée et séquencée à partir de l’organe électrique du poisson torpille en 1988. Dans sa description initiale, l’équipe de Richard Scheller a montré que cette protéine neuronale de 143 acides aminés était localisée dans les terminaisons synaptiques et dans le noyau, d’où son nom (« syn » pour synapse et « nuclein » pour noyau) [32]. Une protéine homologue de 140 acides aminés a ensuite été identifiée dans le SNC du rat et de l’homme [33]. Cette découverte de l’α-synucléine n’a reçu initialement que peu d’écho. Il a fallu attendre la deuxième partie des années 1990 et l’identification d’une mutation du gène codant l’α-synucléine, dans une famille avec une maladie de Parkinson de transmission autosomique dominante, pour que l’α-synucléine passe sur le devant de la scène [34]. Quelques mois après la mise en évidence de cette première forme monogénique de maladie de Parkinson, une équipe de Cambridge a montré que les corps et prolongements de Lewy de patients atteints de formes sporadiques de la maladie étaient fortement immunoréactifs pour l’α -synucléine et qu’elle était donc un des principaux composants de la pathologie de Lewy [35]. En 2002, Fujiwara et al. ont par ailleurs montré que l’α-synucléine présente dans les corps et prolongements de Lewy était non seulement agrégée mais aussi phosphorylée sur un résidu sérine (Sérine 129) [36]. L’immunohistochimie de l’α-synucléine et de la phospho-α-synucléine est depuis lors devenue la technique de référence pour la mise en évidence des corps et prolongements de Lewy, cette approche étant plus sensible que les techniques qui étaient utilisées auparavant qu’elles soient histochimiques (marquage hématine/éosine) ou immunohistochimiques (anticorps anti-ubiquitine) [37]. Cette technique a permis une analyse plus précise de la distribution de la pathologie de Lewy. Elle a en effet montré qu’en dehors de la substance noire, certaines structures du SNC comme le bulbe olfactif et le noyau dorsal moteur du vague et des systèmes nerveux autonomes périphériques comme le système nerveux entérique (SNE) sont touchées par le processus pathologique chez la quasi-totalité des patients parkinsoniens [38], [39].
Depuis 1997, de nombreux travaux se sont intéressés aux rôles physiologique et physiopathologique de l’α-synucléine. L’α-synucléine humaine est une petite protéine de 140 acides aminés (14,5 kDa) divisée d’un point de vue fonctionnel en 3 domaines (Fig. 2 A). La partie amino-terminale est fortement conservée entre les 3 isoformes de synucléine et est impliquée dans la liaison de l’α-synucléine aux lipides et aux membranes cellulaires. Le domaine central joue un rôle essentiel dans les capacités d’agrégation de la protéine alors que l’extrémité carboxy-terminale serait responsable de l’activité chaperonne et contient les principaux sites de phosphorylation. Bien que l’α-synucléine présente une conformation monomérique en solution, elle a une capacité naturelle à s’agréger en structures amyloïdes grâce à son domaine central selon la progression suivante : dimères, oligomères, protofibrilles et fibrilles puis enfin corps de Lewy (Fig. 2B). L’agrégation de l’α-synucléine est plus importante pour les formes tronquées de la protéine et pour les formes mutées responsables de maladie de Parkinson génétique. L’introduction d’oligomères ou de protofibrilles préformés dans des neurones en culture primaire ou in vivo provoque une agrégation de l’α-synucléine endogène, un phénomène appelé « nucléation » (revue dans [40]).
Fig. 2.

A. Structure primaire de l’α-synucléine humaine. L’α-synucléine humaine est une petite protéine de 140 acides aminés divisée d’un point de vue fonctionnel en 3 domaines. La partie amino-terminale (résidus 1 à 60) contient 6 domaines de répétition imparfaite (R1 à R6) contenant des résidus lysine, impliqués dans la liaison aux lipides et aux membranes cellulaires. Le domaine central (résidus 61 à 95) joue un rôle essentiel dans les capacités d’agrégation de la protéine alors que l’extrémité carboxy-terminale (résidus 96–140) serait responsable de l’activité et contient le principal sites de phosphorylation sur le résidu sérine 129. B. Agrégation de l’α-synucléine. Des études in vitro ont montré que l’α-synucléine est en équilibre dynamique. La forme monomérique a une capacité naturelle à s’agréger en structures amyloïdes grâce à son domaine central selon la progression suivante : dimères, oligomères, protofibrilles et fibrilles puis enfin corps de Lewy.
2.2. L’α-synucléine est aussi une protéine extracellulaire
Bien que l’α-synucléine ait longtemps été considérée comme une protéine purement intracellulaire, la présence de la protéine dans le liquide céphalo-rachidien et le plasma a logiquement conduit certains groupes à étudier si elle pouvait être sécrétée. Plusieurs équipes indépendantes ont montré que l’α-synucléine était en effet sécrétée puisque mise en évidence dans le milieu de culture de neurones du SNC ou du système nerveux périphérique [41], [42]. Cette sécrétion d’α-synucléine se produit en condition physiologique mais est plus importante en conditions pathologiques par exemple lors d’un stress oxydant, d’une dysfonction mitochondriale ou du système ubiquitine-protéasome. Dans ces conditions pathologiques, l’α-synucléine est essentiellement libérée sous forme oligomérique [43]. L’α-synucléine serait sécrétée par différents mécanismes impliquant des mécanismes conventionnels ou non conventionnels d’exocytose ainsi que la voie des exosomes [41], [42], [44]. Il est probable que les voies de sécrétion mises en jeu dépendent du type de neurone impliqué, périphérique ou central.
3. Arguments en faveur d’une progression « prion-like » au cours de la maladie de Parkinson
3.1. Corps de Lewy dans des neurones embryonnaires transplantés
L’observation de corps de Lewy (CL) et de neurites de Lewy (NL), dans certains neurones embryonnaires transplantés 11 à 16 ans auparavant dans le striatum de patients parkinsoniens a été le premier argument en faveur d’une transmission de cellules à cellules de dépôts constitués majoritairement d’α-synucléine [45], [46]. En effet, la présence de ces inclusions est anormale dans des neurones jeunes. Les inclusions sont constituées des mêmes éléments (α-synucléine fibrillaire phosphorylée en Sérine 129, ubiquitine) que les CL observés dans la substantia nigra des patients parkinsoniens [45]. Ces inclusions sont marquées par la thioflavine S indiquant la présence de feuillets β. Il existe parallèlement une diminution du transporteur de la dopamine et de la tyrosine hydroxylase [46]. Cependant, l’apparition des inclusions ne concerne pas tous les neurones chez un patient donné [45] et l’analyse post-mortem du cerveau de patients greffés 9 à 14 ans auparavant peut révéler des neurones greffés fonctionnels et sans inclusions pathologiques [47]. L’étude de Kurowska et al. a montré que les greffes les plus âgées (plus de 20 ans) ont une expression réduite de la tyrosine hydroxylase et du transporteur de la dopamine et une pathologie de Lewy plus marquée [48]. Une des hypothèses pouvant expliquer ces observations est celle d’une propagation de l’anomalie conformationnelle du tissu hôte vers le greffon à la manière des prions. Prusiner et Olanow l’ont formalisé en proposant que la maladie de Parkinson pourrait être une maladie à prion [49], résultant directement d’une augmentation de la production ou d’une diminution de la clearance de l’α-synucléine favorisant son repliement anormal et la formation d’oligomères neurotoxiques. L’α-synucléine pourrait être un analogue de la protéine prion capable d’auto-agrégation et de dissémination à des cellules saines.
3.2. Expérimentations in vitro en faveur des phénomènes de « seeding and spreading »
Les propriétés d’agrégation de l’α-synucléine ont bien été documentées in vitro. L’α-synucléine monomérique sauvage ou mutée forme à 37 °C des fibrilles ayant des caractéristiques similaires à celles observées dans les CL [50], [51]. Bien qu’elles aient la même structure native, l’α, la β- et la γ-synucléine ont un pouvoir intrinsèque de fibrillation différent qui est nettement plus élevé pour l’α-synucléine que pour les deux autres formes. Les fibrilles peuvent constituer une amorce (« seeds ») et accélérer la formation de fibrilles à partir d’α-synucléine monomérique. La mise en contact d’α-synucléine monomérique sauvage avec des fibrilles d’α-synucléine porteuse de la mutation A30P entraîne la formation de fibrilles porteuses des caractéristiques associées à la protéine A30P, argument en faveur d’un changement de conformation de l’α-synucléine sauvage [52].
Les assemblages d’α-synucléine peuvent être transférés de neurones à neurones. Une co-culture de neurones sur-exprimant l’α-synucléine et de neurones ne sur-exprimant pas l’α-synucléine permet d’obtenir au bout de 24 heures, la diffusion de l’α-synucléine des premiers aux seconds avec formation d’inclusions dans les neurones « receveurs » [53]. Un phénomène identique est observé en cas d’incubation des neurones « receveurs » dans un milieu acellulaire contenant l’α-synucléine issue des neurones sur-exprimant l’α-synucléine signifiant que le contact cellule à cellule n’est pas obligatoire. Le mode de transport de l’α-synucléine d’un neurone à l’autre n’est pas connu actuellement. Divers mécanismes pourraient être en cause :
-
•
le relargage de l’α-synucléine libre dans le milieu extracellulaire ;
-
•
l’accès direct aux neurones voisins par voie transmembranaire ;
-
•
un phénomène d’exocytose–endocytose ;
-
•
un mécanisme de libération et captation via les exosomes ;
-
•
un transfert via des nanotubes membranaires intercellulaires ;
-
•
une transmission par contact synaptique direct.
Au-delà de la transmission de neurones à neurones, une transmission de l’α-synucléine des neurones vers les astrocytes selon des mécanismes d’exo- et d’endocytose est également possible [54]. Les astrocytes accumulant de l’α-synucléine vont libérer cytokines et chemokines contribuant ainsi au déclenchement d’une réaction inflammatoire.
3.3. Expérimentations in vivo en faveur des phénomènes de « seeding and spreading »
3.3.1. Transfert de l’α-synucléine agrégée à des cellules neurales greffées
Après la constatation que de l’α-synucléine et des CL pouvaient apparaître dans des cellules neurales greffées à des patients parkinsoniens, plusieurs équipes ont mis en évidence expérimentalement la transmission directe, in situ, d’agrégats d’α-synucléine à de telles cellules neurales greffées. Desplats et al. ont rapporté, en 2009, que la greffe de cellules souches dans l’hippocampe de souris transgéniques sur-exprimant l’α-synucléine humaine entraînait l’accumulation d’α-synucléine dans les cellules greffées sans toutefois formation de fibrilles ou de CL [53]. Hansen et al. ont montré, en 2011, que la greffe de neurones embryonnaires mésencéphaliques murins dans le striatum de souris transgéniques sur-exprimant l’α-synucléine humaine déclenchait 6 mois après la greffe, l’accumulation dans les cellules greffées d’α-synucléine monomérique mais aussi oligomérique et même fibrillaire [55]. D’autre part, chez des rats dont les neurones de la substantia nigra expriment l’α-synucléine humaine, il a été mis en évidence l’accumulation d’α-synucléine humaine dans les terminaisons striatales. Lorsque, dans ce même modèle, une greffe de cellules embryonnaires de rat est réalisée dans le striatum, le greffon durant les premières semaines ne va pas accumuler d’α-synucléine puis les semaines suivantes, une accumulation d’abord d’α-synucléine humaine puis d’α-synucléine de rat va être observée, suggérant un transfert de cellule à cellule de l’α-synucléine le long de la voie nigro-striatale et un processus d’amorçage au sein du greffon [56].
3.3.2. Transmission et propagation de l’α-synucléine de cellule à cellule et recrutement de l’α-synucléine endogène chez des souris transgéniques
L’inoculation intracérébrale d’homogénats de cerveau de souris transgéniques (TgM83) âgées et cliniquement malades exprimant l’α-synucléine humaine mutée A53T insoluble et phosphorylée déclenche chez de jeunes souris TgM83 une maladie motrice typique et précoce [57], [58]. Cette maladie s’accompagne de l’accumulation intracérébrale d’α-synucléine insoluble et phosphorylée en Sérine 129 détectée en Western Blot et par immunocytochimie. Les souris témoins c’est-à-dire TgM83 non inoculées ou inoculées à partir d’homogénats de cerveaux de souris TgM83 jeunes et non malades ont une durée de survie identique et significativement plus longue que les souris inoculées à partir d’homogénats de cerveau de souris âgées et malades [57]. Dans le cerveau des souris inoculées avec une solution sans homogénats de cerveau, il n’y a pas d’accumulation d’α-synucléine [58]. De plus, les souris KO n’exprimant pas le gène de l’α-synucléine ne développent pas la maladie et ont une longue survie indiquant l’importance de l’α-synucléine de l’hôte pour la transmission de la maladie. L’α-synucléine anormale se propage à partir des sites d’injection (striatum ou cortex) à l’ensemble du SNC avec une atteinte préférentielle des régions cérébrales les plus connectées avec les régions inoculées [58]. Des résultats identiques sont obtenus à partir non plus d’homogénats de cerveaux mais de fibrilles synthétiques d’α-synucléine humaine [59].
3.3.3. Accumulation d’α-synucléine et dégénérescence de la voie dopaminergique nigro-striatale chez des souris non transgéniques
Le déclenchement possible d’une maladie liée à l’atteinte de la voie nigro-striatale, chez des souris sauvages non transgéniques, après inoculation d’α-synucléine constitue un argument important en faveur du rôle de l’α-synucléine dans la dégénérescence de cette voie nigro-striatale [59]. C’est ainsi que, 30 jours après injection unilatérale intra-striatale d’α-synucléine synthétique sous forme fibrillaire dans le striatum dorsal de souris sauvages, des dépôts d’α-synucléine hyperphosphorylée sont visibles au site d’injection, des LN et des CL sont présents dans des zones ipsilatérales connectées au striatum comme les couches IV–V du cortex ou le bulbe olfactif confirmant la conversion de l’α-synucléine endogène de ces souris sauvages. L’accumulation des CL est aussi observée de manière bilatérale dans les zones de projection bilatérales du striatum comme les amygdales suggérant une transmission de cellule à cellule le long des voies de connections inter-neuronales. L’accumulation d’α-synucléine augmente de j90 à j180 et va intéresser de nombreuses régions du SNC. L’accumulation d’α-synucléine dans la substantia nigra pars compacta s’accompagne d’une diminution unilatérale des neurones dopaminergiques et d’une atteinte clinique de la coordination et de l’équilibre des animaux.
3.3.4. Expériences de transmission des synucléinopathies humaines chez l’animal
L’équipe de Prusiner et al. a mis en évidence que l’inoculation intracérébrale d’homogénats de cerveau issus de deux patients décédés d’atrophie multisystématisée (MSA) déclenchait une maladie neurologique chez des souris transgéniques hétérozygotes pour la mutation A53T du gène de l’α-synucléine [60]. Contrairement aux souris homozygotes pour cette mutation, les souris hétérozygotes ne déclarent pas de maladie naturelle au cours du vieillissement. Dans le cerveau de ces souris inoculées, il est mis en évidence une gliose astrocytaire avec une activation microgliale et des dépôts diffus d’α-synucléine phosphorylée sensible à la protéinase K et insoluble dans les détergents. Les dépôts, abondants surtout dans les régions sous-corticales, sont situés dans les neurones ou leurs prolongements. Ces données sont en faveur d’une possible transmissibilité de la MSA.
L’injection de préparations enrichies en α-synucléine insoluble issues du cerveau de patients atteints de démence à corps de Lewy à des souris sauvage est également capable d’induire une pathologie de type Lewy, mais avec une efficacité moindre que celle observée avec des fibrilles synthétiques [61].
Plus récemment, il a été montré que l’inoculation de fractions enrichies en CL provenant de la substantia nigra de patients décédés de maladie de Parkinson idiopathique à des souris sauvages C57BL/6 entraîne, à 4 mois de l’inoculation, une dégénérescence de la voie nigro-striée et une accumulation d’α-synucléine résistante à la protéinase K et phosphorylée dans la substantia nigra, le striatum et le cortex [62]. Les souris présentent des anomalies motrices mises en évidence par le test du rotarod. La dégénérescence de la voie nigro-striée n’est pas observée en cas d’inoculation de ces préparations à des souris KO pour le gène de l’α-synucléine indiquant que l’accumulation se fait à partir de l’α-synucléine endogène. L’inoculation de fractions cérébrales provenant des mêmes patients mais dépourvues de CL n’occasionne pas d’accumulation d’α-synucléine. L’inoculation de CL provenant de la substantia nigra des mêmes patients parkinsoniens dans la substantia nigra de macaques entraîne à partir de 3 mois une diminution de l’innervation striatale mise en évidence sur le DaTSCAN. Lorsque les animaux sont sacrifiés précocement, 14 mois après l’inoculation, il est mis en évidence une accumulation d’α-synucléine dans plusieurs régions cérébrales connectées à la substantia nigra et une dégénérescence modérée de la voie nigro-striée compatible avec l’absence de signes cliniques [62].
3.3.5. Existe-t-il un phénomène de souches ?
L’existence de différentes souches de ce que l’on pourrait appeler par facilité de langage des prions d’α-synucléine a été proposée par les prionologues dès l’émergence de l’hypothèse prion dans les synucléinopathies. La diversité des souches pourrait, au moins en partie, expliquer la diversité des formes de synucléinopathies (maladie de Parkinson, démence à corps de Lewy, atrophie multisystématisée) et la diversité phénotypique au sein de chacune de ces formes. Au-delà des aspects fondamentaux, cette question pourrait avoir des conséquences opérationnelles si l’on se souvient que l’efficacité des procédures de décontamination et des composés anti-prion développés à des fins thérapeutiques varient selon les souches. L’équipe de Lee et al. a montré que des fibres d’α-synucléine recombinante ayant des propriétés biochimiques différentes n’avaient pas la même capacité d’induire une tauopathie in vitro et in vivo, suggérant un lien entre structure des assemblages d’α-synucléine et pouvoir pathogène évocateur d’un effet souche [63]. Cette hypothèse a été récemment confirmée par un travail franco-belge montrant que l’injection intracérébrale d’assemblages d’α-synucléine de structures différentes induisait des phénotypes distincts chez le rat [64]. À ce jour, les différentes souches présentes chez l’homme n’ont pas été clairement isolées et les assemblages d’α-synucléine responsables de la maladie de Parkinson, de la démence à corps de Lewy et des différentes MSA ne sont pas connus.
4. Est-il possible d’intégrer l’hypothèse prion-like avec la progression neuropathologique de la maladie de Parkinson ?
En faisant le constat que certaines structures cérébrales sont toujours touchées par la pathologie de Lewy quand d’autres sont intègres, Braak et al. ont proposé un scénario de progression temporo-spatiale de la pathologie de Lewy [38]. Grossièrement ascendante, elle est classée en 6 stades (Tableau 1 ) : les premières lésions encéphaliques sont observées dans le noyau dorsal du vague, le bulbe olfactif et les noyaux olfactifs antérieurs au stade 1. Des agrégats d’α-synucléine sont alors déjà présents dans les centres sympathiques médullaires, dans les axones des efférences vagales (noyau dorsal du vague) et au niveau du SNE. Au stade 2, les lésions gagnent les noyaux du raphé et le locus cœruleus. C’est à partir du stade 3 que le mésencéphale et la substance noire sont touchés conjointement à certains noyaux de la base (noyau basal de Meynert) et à l’amygdale. L’atteinte de l’amygdale est massive au stade 4, et s’accompagne de lésions du mésocortex temporal et de la corne d’Amon. Puis l’atteinte corticale gagne l’insula et le cortex cingulaire (stade 5) et le néocortex dans son ensemble (stade 6).
Tableau 1.
Corrélation anatomo-clinique de la progression temporo-spatiale de la pathologie de Lewy selon le modèle de Braak.
| Stade de Braak | Région anatomique | Corrélations cliniques putatives |
|---|---|---|
| 1 | Bulbe olfactif, noyaux olfactifs antérieurs | Hyposmie, anosmie |
| Noyau dorsal du vague, système nerveux entérique | Constipation, gastroparésie | |
| Neurones sympathiques pré- et post-ganglionnaires | Troubles génito-urinaires, hypotension orthostatique | |
| Corne dorsale de la mœlle épinière | Douleurs | |
| 2 | Complexe cœruleus/subcœruleus, noyaux réticulaires | Troubles du sommeil paradoxal, dépression |
| 3 | Substance noire | Akinésie, bradykinésie, rigidité |
| Amygdale (noyau central), noyau pédiculopontin, aire tegmentale ventrale noyaux cholinergiques du diencéphale | Dysautonomie, syndrome dysexécutif | |
| 4 | Mésocortex temporal | Syndrome dysexécutif, apathie, troubles mnésiques |
| Amygdale (noyau basolatéral) | Troubles émotionnels | |
| 5 | Isocortex associatif multimodal (préfrontal notamment) | Agnosie, apraxie |
| 6 | Isocortex associatif unimodal, isocortex primaire | Dysfonctions sensorimotrices |
Ce modèle de progression séquentielle est conforté par des arguments cliniques et anatomiques. Des études épidémiologiques prospectives ont montré que l’anosmie, la constipation et les troubles du comportement en sommeil paradoxal constituent des signes précurseurs de la maladie de Parkinson (revue dans [65]). L’atteinte des centres autonomes et olfactifs au stade 1 et du locus subcœruleus au stade 2 rendrait compte, respectivement, de ces signes cliniques (Tableau 1). Prenant exemple sur la transmission du prion, Braak et al. posent l’hypothèse d’un pathogène neurotoxique qui provoquerait une atteinte initiale des structures nerveuses entériques en déclenchant in situ une agrégation de l’α-synucléine. Le processus pathologique serait véhiculé par transport axonal rétrograde et par voie trans-synaptique. Il gagnerait le SNC et plus précisément le noyau dorsal moteur du vague en suivant l’innervation vagale comme cela a été décrit pour l’agent bovin qui se propage via le système nerveux autonome associé au tube digestif dans la variante de la maladie de Creutzfeldt-Jakob [29] ou pour des traceurs neuronaux [66]. Dans ce modèle, la substance noire n’est en définitive que l’un des maillons de cette chaîne d’évènements propagatifs.
Les propriétés biologiques de l’α-synucléine (capacité à s’agréger, sécrétion, recapture) viennent logiquement s’intégrer dans l’hypothèse de Braak. Il est tentant d’imaginer qu’une fois la formation d’agrégats d’α-synucléine initiée dans le SNE ou les voies olfactives, l’α-synucléine pathologique se propage de proche en proche le long de voies anatomiques bien définies reproduisant la progression temporo-spatiale proposée par Braak et al. (Fig. 3 ). Plusieurs données expérimentales viennent renforcer cette hypothèse. Dans une étude anatomique très précise réalisée chez le rat, Phillips et al. ont montré que les efférences vagales, qui trouvent leur origine dans le noyau dorsal du vague, expriment fortement l’α-synucléine et qu’elles ne font synapse qu’avec des neurones entériques eux aussi riches en α-synucléine. Ces données suggèrent l’existence d’une voie anatomique contenant beaucoup d’α-synucléine entre l’intestin et le cerveau qui pourrait propager le processus pathologique du SNC au SNE et vice versa [67]. Ainsi des expériences d’injection locale d’α-synucléine dans différentes région du SNC et du SNE ont permis de montrer que l’α-synucléine pathologique pouvait diffuser in vivo dans des régions connectées anatomiquement :
-
•
l’injection stéréotaxique de formes fibrillaires d’α-synucléine humaine dans le striatum dorsal de souris provoque, comme on l’a vu plus haut, non seulement la formation de corps et de prolongements de Lewy au site d’injection mais aussi, après 3 à 6 mois, dans l’amygdale, la substance noire pars compacta, le bulbe olfactif et les couches IV et V du néocortex ipsilatéraux [59] ;
-
•
l’α-synucléine surexprimée dans le noyau dorsal moteur du vague à l’aide d’un vecteur viral gagne progressivement le complexe locus cœruleus/subcœruleus, l’hypothalamus et l’amygdale homolatéraux et à un moindre degré contralatéraux [68] ;
-
•
des formes fibrillaires d’α-synucléine injectées dans la paroi de l’estomac et du duodénum sont transportées par le nerf vague jusqu’au noyau dorsal moteur du vague [69] ;
-
•
une injection dans le bulbe olfactif de formes oligomériques et fibrillaires entraîne à terme la présence d’inclusions d’α-synucleine dans le cortex frontal [70].
Fig. 3.

Représentation schématique des possibles facteurs responsables de l’agrégation et de la propagation de l’α-synucléine selon un mécanisme prion-like. 1 – Entrée d’un facteur exogène (virus, neurotoxique, agrégats d’α-synucléine) par voie orale ou nasale, éventuellement favorisée par un phénomène inflammatoire. 2 – Dissémination des agrégats d’α-synucléine ingérés, inhalés ou formés, in situ, sous l’influence du virus ou du neurotoxique à partir du système nerveux périphérique. L’hypothèse alternative serait la propagation directe d’un virus dont la réplication périphérique ou centrale entraînerait l’agrégation de l’α-synucléine le long de son trajet. 3 – Accumulation de l’α-synucléine dans le tronc cérébral (voie digestive) ou dans le lobe temporal (voie olfactive). 4 – Propagation des agrégats d’α-synucléine à l’ensemble du cortex.
5. Quel pourrait-être le facteur déclenchant ?
Si l’on admet que la progression de la maladie de Parkinson se fait selon le modèle de Braak par un mécanisme « prion-like » allant de la périphérie vers le SNC, il reste à déterminer quel pourrait être le facteur déclenchant (Fig. 3).
Pour Braak, il pourrait s’agir d’un agent pathogène neurotrope, possiblement un virus qui progresserait de la muqueuse olfactive vers le lobe temporal ou qui contaminerait la salive puis l’estomac à partir des sécrétions nasales et à partir du tube digestif gagnerait le tronc cérébral via le SNE. Parmi les virus qui pourraient être en cause, notons que l’influenza A est un virus à l’origine de phénomènes auto-immuns qui a un tropisme particulier pour la substantia nigra, le cervelet ou l’hippocampe. L’implication d’autres virus comme l’herpès simplex ou le coronavirus a été également évoquée [71].
Des arguments expérimentaux plaident, depuis longtemps, pour une origine inflammatoire de la maladie de Parkinson avec l’observation dans les cerveaux de patients parkinsoniens d’une augmentation du nombre de cellules microgliales activées et de l’expression de cytokines pro-inflammatoires [72], [73]. Des arguments épidémiologiques plaident aussi en faveur de cette possible origine inflammatoire. Ainsi, une étude rétrospective portant sur 196 patients parkinsoniens appariés sur le sexe et l’âge à 196 témoins de population générale a montré que les parkinsoniens avaient deux fois plus d’antécédents d’allergie de type asthme, rhinites allergiques ou saisonnières que les témoins [74]. Cependant, le caractère rétrospectif et l’effectif limité incitent à considérer les résultats de cette étude avec prudence. Plus récemment, il a été montré que la consommation d’anti-inflammatoires non stéroïdiens pourrait diminuer le risque de développer la maladie de Parkinson [75].
L’implication d’une substance neurotoxique comme cause de la maladie de Parkinson a connu un regain d’intérêt à la fin des années 1970 et au début des années 1980 par l’observation de cas de syndromes parkinsoniens aigus après injection de MPTP. En 1976, un jeune toxicomane américain a développé un syndrome parkinsonien quelques jours après s’être injecté un opioïde de synthèse contenant des traces de MPTP. Son décès d’une overdose deux ans plus tard a permis de mettre en évidence, à l’autopsie, une destruction des neurones pigmentés dopaminergiques de la substantia nigra avec même des inclusions pouvant ressembler à des corps de Lewy [76]. L’observation de quatre autres cas quelques années après a permis de confirmer le lien de cause à effet entre l’injection du MPTP et l’émergence de ce syndrome parkinsonien sévère répondant au traitement dopaminergique [77]. Le MPTP a été ensuite utilisé, comme la 6-OHDA, pour générer des modèles de syndromes parkinsoniens chez l’animal de laboratoire. Cependant, ces neurotoxiques sont à l’origine de syndromes parkinsoniens aigus ne reproduisant pas l’apparition lente et progressive des symptômes chez l’homme. De nombreuses études épidémiologiques ont montré que l’exposition professionnelle et même non professionnelle à des pesticides augmentait le risque de développer une maladie de Parkinson [78]. Cette association, mise en évidence avec un grand nombre de pesticides, présente dans certains cas une relation effet-dose [79]. Le stress oxydatif et le dysfonctionnement mitochondrial sont deux mécanismes physiopathologiques impliqués dans les modèles expérimentaux et dans les formes génétiques de maladie de Parkinson. Une étude cas-témoin a rapporté une association entre l’utilisation de pesticides à l’origine d’un stress oxydatif comme le paraquat ou inhibant le complexe I mitochondrial comme la roténone [80]. Avec ce dernier pesticide, un modèle animal reproduisant la progression « prion-like » de la maladie de Parkinson selon le modèle de Braak a pu être mis au point [81]. L’administration par sonde gastrique de 5 mg/kg/jour de roténone chez la souris sauvage C57BL/6 permet d’observer, après 1 mois et demi de traitement, des agrégats d’α-synucléine, d’α-synucléine phosphorylée et une gliose dans le système nerveux entérique. Après 3 mois, le nombre de ces agrégats va diminuer avec l’apparition d’inclusions de plus grande taille. De manière concomitante, une accumulation d’α-synucléine est observée dans la colonne inter-mediolatérale de la mœlle-épinière et dans le noyau dorso-médial du vague sans perte cellulaire puis, après 3 mois de traitement, une accumulation d’α-synucléine et une perte des neurones dopaminergiques sont mis en évidence dans la substantia nigra. La perte neuronale et l’accumulation d’α-synucléine ne sont pas liées à un effet systémique, sont séquentielles, mises en évidence uniquement dans les régions connectées, accompagnées de stigmates d’inflammation et se traduisant par une atteinte clinique motrice qui apparaît seulement au bout de 3 mois.
Enfin, un autre facteur déclenchant pourrait être une contamination par l’agent causal lui-même qui serait constitué d’assemblage d’α-synucléine présent dans l’environnement. La contamination pourrait être interhumaine lors de soins médicaux ou lors de procédures chirurgicales comme cela a été observé dans les ESST. Cependant, aucun argument épidémiologique n’existe actuellement en faveur d’une telle hypothèse. Une autre possibilité serait une contamination à partir d’animaux atteints mais il n’est pas certain qu’il existe parmi les animaux consommés de cas naturels de maladie de Parkinson. De plus, les mesures mises en place depuis le milieu des années 1990 après les premiers cas de variante de la MCJ (retrait des matériaux à risques spécifiés comme le SNC et les intestins chez les ruminants) ont grandement limité l’exposition à des pathogènes associés aux tissus nerveux y compris le système nerveux autonome.
6. Arguments en défaveur d’une progression « prion-like » au cours de la maladie de Parkinson
Le scénario de progression de la maladie de Parkinson décrit par Braak et al., clair et stéréotypé avec un début par le tube digestif et une progression trans-synaptique du processus pathologique jusqu’au SNC, est encore débattu. Si la qualité et l’exhaustivité des marquages immunohistochimiques ne font que peu de doute, la sélection des cas et l’interprétation des données peuvent être discutées. Ainsi, dans son étude princeps publiée en 2003, Braak et al. rapportent que la totalité des 41 patients atteints de maladie de Parkinson dont le cerveau a été analysé ont des lésions dans le noyau dorsal moteur du vague [38]. Ceci combiné à la présence de corps de Lewy dans le noyau dorsal moteur du vague chez 69/69 des sujets indemnes de maladie de Parkinson, l’a amené à conclure que le noyau dorsal moteur du vague était un passage obligé du processus pathologique. Cependant, les critères de sélection de ces 69 sujets parmi une large série de cerveaux de sujets sans signes moteurs évocateurs de syndrome parkinsonien, mais qui présentaient des corps de Lewy à l’analyse microscopique ne sont pas mentionnés. Un biais de sélection ne peut donc pas être formellement éliminé. Il en va de même pour l’article publié 3 ans plus tard par la même équipe sur l’atteinte précoce du SNE chez les patients parkinsoniens qui reposait sur la description pathologique précise du SNE et du SNC chez 5 sujets sélectionnés [82]. Depuis ces publications des séries neuropathologiques ont montré qu’une proportion non négligeables de cas ne suivaient pas la prédiction de progression rostro-caudale proposée par Braak. Sur 71 cas analysés, Kalaitzakis et al. ont montré que 22 sujets avaient une atteinte sévère de la substantia nigra avec peu ou pas d’atteinte du noyau dorsal moteur vague [83]. Ces données ont été confirmées par une étude indépendante publiée la même année [84].
Les deux publications originales des équipes de Brundin et Kordower montrant la présence de corps de Lewy (CL) et de neurites de Lewy (NL) dans certains neurones embryonnaires transplantés ont reçu un large écho. Il est toutefois important de contrebalancer les résultats de ces études par ceux de l’équipe d’Ole Isacson qui est plus circonspecte quant au rôle éventuel d’un passage de neurone à neurone de l’α-synucléine dans la diffusion de la maladie. En analysant les cerveaux de 5 patients parkinsoniens transplantés 9 à 14 ans auparavant, ils n’ont pas mis en évidence de signes de neurodégénérescence ni de dépôts d’α-synucléine dans les neurones greffés [47]. Bien qu’ils n’excluent pas que des corps de Lewy puissent être trouvés dans les neurones greffés, ils incitent à la prudence en suggérant qu’il s’agit d’un phénomène rare et qui ne leur semble pas être impliqué dans la diffusion du processus pathologique.
Nous l’avons vu, de nombreuses expériences de biologie cellulaire sont venues appuyer l’hypothèse d’un passage de neurone à neurone de l’α-synucléine. Il convient là aussi de garder un œil critique car même si les expériences proposées sont globalement convaincantes, elles ne sont pas exemptes de critiques. Cet aspect a été récemment discuté dans une excellente revue qui souligne les limites de ces expériences dont certaines ont été réalisées avec des agents de lipofection pour favoriser le passage transmembranaire de l’α-synucléine ou chez des souris transgéniques qui sur-expriment massivement l’α-synucléine humaine et qui sont bien loin de ce qui peut être observé en physiopathologie humaine (revue dans [85]).
7. Implications thérapeutiques et diagnostiques
L’hypothèse selon laquelle l’α-synucléine agrégée se propagerait selon un mode prion-like permet d’envisager de nouvelles possibilités thérapeutiques pour la maladie de Parkinson qui cibleraient directement l’α-synucléine. Il a été ainsi proposé qu’une diminution de l’expression et/ou une augmentation de l’élimination de l’α-synucléine pourraient permettre de ralentir l’évolution de la maladie. Une autre stratégie « neuroprotectrice » consisterait à diminuer le niveau de phosphorylation de l’α-synucléine et par la même sa capacité à s’agréger. Ces approches ont été testées dans des modèles animaux de la maladie et se sont montrées globalement efficaces (revue dans [86]). Il est toutefois important d’attendre les résultats chez l’homme, les études en cours n’en étant pour le moment qu’à une phase précoce [86].
L’approche qui a reçu le plus d’écho est l’utilisation d’anticorps anti-α-synucléine, comme cela a été fait pour le peptide β-amyloïde dans la maladie d’Alzheimer. En effet, le mécanisme de propagation implique un passage extracellulaire des assemblages pathologiques qui serait alors plus facilement accessibles. Plusieurs groupes de recherche ont montré que l’immunisation active ou passive contre l’α-synucléine permettait de diminuer la charge lésionnelle et améliorait les performances motrices dans des modèles de souris transgéniques de la maladie [87], [88], [89]. Une étude pilote par immunisation active contre l’α-synucléine a été proposé à 32 patients parkinsoniens par une Société de biotechnologie autrichienne (Protocole PD01 AFFITOPE, Clinical Trials NCT01568099). Cette première étape a permis de montrer que cette immunisation était bien supportée et qu’aucun des patients n’avait développé d’effets secondaires notables. D’autres protocoles sont en cours avec une évaluation à plus long terme sur l’évolution de la maladie.
Enfin, les méthodes d’amplification du mauvais repliement des protéines, qui ont montré leur intérêt dans le diagnostic des maladies à prions [90], [91], pourraient être adaptées à l’alpha-synucléine et fournir prochainement à la communauté neurologique des outils innovants et très performants pour le diagnostic et le suivi des alpha-synucleinopathies.
8. Conclusion
L’implication de mécanismes de type prion dans la survenue et l’aggravation progressive des maladies conformationnelles des protéines du SNC et notamment de la maladie de Parkinson est une avancée conceptuelle importante dans la compréhension de la physiopathologie de ces maladies. Cependant, la cause initiale, et il pourrait s’agir d’un ensemble de facteurs (environnementaux, génétiques, cellulaires, liés au vieillissement…), entraînant le mauvais repliement et l’agrégation de la protéine cible demeure inconnue. Il en est de même des mécanismes de dissémination des protéines agrégées. La corrélation entre les signes non moteurs pré-symptomatiques et la diffusion séquentielle et prédictive de l’α-synucléine, selon le modèle de Braak, font de la maladie de Parkinson un modèle très pertinent, non seulement pour l’étude de ces mécanismes, mais aussi pour tester des traitements spécifiques ciblant les assemblages d’α-synucléine et leur propagation dès la phase pré-motrice de la maladie. Il faut enfin insister sur le fait qu’il n’existe actuellement aucun argument permettant de suspecter un risque de transmission interhumaine de la maladie de Parkinson.
Déclaration de liens d’intérêts
Les auteurs déclarent ne pas avoir de liens d’intérêts.
Références
- 1.Latarget R., Muel B., Haig D.A., Clarke M.C., Alper T. Inactivation of the scrapie agent by near monochromatic ultraviolet light. Nature. 1970;227:1341–1343. doi: 10.1038/2271341a0. [DOI] [PubMed] [Google Scholar]
- 2.Alper T. Special lecture – The scrapie enigma: insights from radiation experiments (Reprinted from Radiation Research 1993;135:283–92) Int J Radiat Biol. 1997;71:759–768. [PubMed] [Google Scholar]
- 3.Prusiner S.B. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 4.Haik S., Brandel J.P. Infectious prion diseases in humans: cannibalism, iatrogenicity and zoonoses. Infect Genet Evol. 2014;26C:303–312. doi: 10.1016/j.meegid.2014.06.010. [DOI] [PubMed] [Google Scholar]
- 5.Chernova T.A., Wilkinson K.D., Chernoff Y.O. Physiological and environmental control of yeast prions. FEMS Microbiol Rev. 2014;38:326–344. doi: 10.1111/1574-6976.12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Si K., Choi Y.B., White-Grindley E., Majumdar A., Kandel E.R. Aplysia CPEB can form prion-like multimers in sensory neurons that contribute to long-term facilitation. Cell. 2010;140:421–435. doi: 10.1016/j.cell.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 7.Zahn R., Liu A., Luhrs T., Riek R., von Schroetter C., Lopez Garcia F. NMR solution structure of the human prion protein. Proc Natl Acad Sci U S A. 2000;97:145–150. doi: 10.1073/pnas.97.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Büeler H., Aguzzi A., Sailer A., Greiner R.A., Autenried P., Aguet M. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73:1339–1347. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- 9.Prusiner S.B., Scott M., Foster D., Pan K.M., Groth D., Mirenda C. Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell. 1990;63:673–686. doi: 10.1016/0092-8674(90)90134-z. [DOI] [PubMed] [Google Scholar]
- 10.Telling G.C., Scott M., Mastrianni J., Gabizon R., Torchia M., Cohen F.E. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell. 1995;83:79–90. doi: 10.1016/0092-8674(95)90236-8. [DOI] [PubMed] [Google Scholar]
- 11.Hope J., Morton L.J.D., Farquhar C.F., Multhaup G., Beyreuther K., Kimberlin R.H. The major polypeptide of scrapie-associated fibrils (SAF) has the same size, charge distribution and N-terminal protein sequence as predicted for the normal brain protein. EMBO J. 1986;5:2591–2597. doi: 10.1002/j.1460-2075.1986.tb04539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen J.T., Inouye H., Baldwin M.A., Fletterick R.J., Cohen F.E., Prusiner S.B. X-ray diffraction of scrapie prion rods and PrP peptides. J Mol Biol. 1995;252:412–422. doi: 10.1006/jmbi.1995.0507. [DOI] [PubMed] [Google Scholar]
- 13.Huang Z.W., Prusiner S.B., Cohen F.E. Scrapie prions – a 3-dimensional model of an infectious fragment (vol. 1, pg 13, 1996) Folding Design. 1996;1:406. doi: 10.1016/S1359-0278(96)00007-7. [DOI] [PubMed] [Google Scholar]
- 14.Prusiner S.B. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weissmann C. The Ninth Datta Lecture. Molecular biology of transmissible spongiform encephalopathies. FEBS Lett. 1996;389:3–11. doi: 10.1016/0014-5793(96)00610-2. [DOI] [PubMed] [Google Scholar]
- 16.Horwich A.L., Weissman J.S. Deadly conformations – protein misfolding in prion disease. Cell. 1997;89:499–510. doi: 10.1016/s0092-8674(00)80232-9. [DOI] [PubMed] [Google Scholar]
- 17.Jackson W.S., Borkowski A.W., Faas H., Steele A.D., King O.D., Watson N. Spontaneous generation of prion infectivity in fatal familial insomnia knockin mice. Neuron. 2009;63:438–450. doi: 10.1016/j.neuron.2009.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jackson W.S., Borkowski A.W., Watson N.E., King O.D., Faas H., Jasanoff A. Profoundly different prion diseases in knock-in mice carrying single PrP codon substitutions associated with human diseases. Proc Natl Acad Sci U S A. 2013;110:14759–14764. doi: 10.1073/pnas.1312006110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Castilla J., Saa P., Hetz C., Soto C. In vitro generation of infectious scrapie prions. Cell. 2005;121:195–206. doi: 10.1016/j.cell.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 20.Weber P., Giese A., Piening N., Mitteregger G., Thomzig A., Beekes M. Cell-free formation of misfolded prion protein with authentic prion infectivity. Proc Natl Acad Sci U S A. 2006;103:15818–15823. doi: 10.1073/pnas.0605608103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deleault N.R., Harris B.T., Rees J.R., Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci U S A. 2007;104:9741–9746. doi: 10.1073/pnas.0702662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim J.I., Cali I., Surewicz K., Kong Q., Raymond G.J., Atarashi R. Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J Biol Chem. 2010;285:14083–14087. doi: 10.1074/jbc.C110.113464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Legname G., Baskakov I.V., Nguyen H.O., Riesner D., Cohen F.E., DeArmond S.J. Synthetic mammalian prions. Science. 2004;305:673–676. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 24.Legname G., Nguyen H.O., Baskakov I.V., Cohen F.E., Dearmond S.J., Prusiner S.B. Strain-specified characteristics of mouse synthetic prions. Proc Natl Acad Sci U S A. 2005;102:2168–2173. doi: 10.1073/pnas.0409079102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Collinge J., Clarke A.R. A general model of prion strains and their pathogenicity. Science. 2007;318:930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 26.Haik S., Brandel J.P. Biochemical and strain properties of CJD prions: complexity versus simplicity. J Neurochem. 2011;119:251–261. doi: 10.1111/j.1471-4159.2011.07399.x. [DOI] [PubMed] [Google Scholar]
- 27.Brandel J.P., Heath C.A., Head M.W., Levavasseur E., Knight R., Laplanche J.L. Variant Creutzfeldt-Jakob disease in France and the United Kingdom: evidence for the same agent strain. Ann Neurol. 2009;65:249–256. doi: 10.1002/ana.21583. [DOI] [PubMed] [Google Scholar]
- 28.Haik S., Faucheux B.A., Hauw J.J. Brain targeting through the autonomous nervous system: lessons from prion diseases. Trends Mol Med. 2004;10:107–112. doi: 10.1016/j.molmed.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 29.Haik S., Faucheux B.A., Sazdovitch V., Privat N., Kemeny J.L., Perret-Liaudet A. The sympathetic nervous system is involved in variant Creutzfeldt-Jakob disease. Nat Med. 2003;9:1121–1123. doi: 10.1038/nm922. [DOI] [PubMed] [Google Scholar]
- 30.Llewelyn C.A., Hewitt P.E., Knight R.S., Amar K., Cousens S., Mackenzie J. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet. 2004;363:417–421. doi: 10.1016/S0140-6736(04)15486-X. [DOI] [PubMed] [Google Scholar]
- 31.Ironside J.W. Variant Creutzfeldt-Jakob disease: risk of transmission by blood transfusion and blood therapies. Haemophilia. 2006;12(Suppl. 1):8–15. doi: 10.1111/j.1365-2516.2006.01195.x. [discussion 26-8] [DOI] [PubMed] [Google Scholar]
- 32.Maroteaux L., Campanelli J.T., Scheller R.H. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 1988;8:2804–2815. doi: 10.1523/JNEUROSCI.08-08-02804.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ueda K., Fukushima H., Masliah E., Xia Y., Iwai A., Yoshimoto M. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:11282–11286. doi: 10.1073/pnas.90.23.11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Polymeropoulos M.H., Lavedan C., Leroy E., Ide S.E., Dehejia A., Dutra A. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 35.Spillantini M.G., Schmidt M.L., Lee V.M., Trojanowski J.Q., Jakes R., Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 36.Fujiwara H., Hasegawa M., Dohmae N., Kawashima A., Masliah E., Goldberg M.S. Alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- 37.Beach T.G., White C.L., Hamilton R.L., Duda J.E., Iwatsubo T., Dickson D.W. Evaluation of alpha-synuclein immunohistochemical methods used by invited experts. Acta Neuropathol. 2008;116:277–288. doi: 10.1007/s00401-008-0409-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Braak H., Del Tredici K., Rub U., de Vos R.A., Jansen Steur E.N., Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 39.Beach T.G., Adler C.H., Sue L.I., Vedders L., Lue L., White Iii C.L. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010;119:689–702. doi: 10.1007/s00401-010-0664-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waxman E.A., Giasson B.I. Molecular mechanisms of alpha-synuclein neurodegeneration. Biochim Biophys Acta. 2009;1792:616–624. doi: 10.1016/j.bbadis.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee H.J., Patel S., Lee S.J. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci. 2005;25:6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Emmanouilidou E., Melachroinou K., Roumeliotis T., Garbis S.D., Ntzouni M., Margaritis L.H. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci. 2010;30:6838–6851. doi: 10.1523/JNEUROSCI.5699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jang A., Lee H.J., Suk J.E., Jung J.W., Kim K.P., Lee S.J. Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J Neurochem. 2010;113:1263–1274. doi: 10.1111/j.1471-4159.2010.06695.x. [DOI] [PubMed] [Google Scholar]
- 44.Paillusson S., Clairembault T., Biraud M., Neunlist M., Derkinderen P. Activity-dependent secretion of alpha-synuclein by enteric neurons. J Neurochem. 2013;125:512–517. doi: 10.1111/jnc.12131. [DOI] [PubMed] [Google Scholar]
- 45.Li J.Y., Englund E., Holton J.L., Soulet D., Hagell P., Lees A.J. Lewy bodies in grafted neurons in subjects with Parkinson's disease suggest host-to-graft disease propagation. Nat Med. 2008;14:501–503. doi: 10.1038/nm1746. [DOI] [PubMed] [Google Scholar]
- 46.Kordower J.H., Chu Y., Hauser R.A., Freeman T.B., Olanow C.W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson's disease. Nat Med. 2008;14:504–506. doi: 10.1038/nm1747. [DOI] [PubMed] [Google Scholar]
- 47.Mendez I., Vinuela A., Astradsson A., Mukhida K., Hallett P., Robertson H. Dopamine neurons implanted into people with Parkinson's disease survive without pathology for 14 years. Nat Med. 2008;14:507–509. doi: 10.1038/nm1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kurowska Z., Englund E., Widner H., Lindvall O., Li J.Y., Brundin P. Signs of degeneration in 12–22-year old grafts of mesencephalic dopamine neurons in patients with Parkinson's disease. J Parkinsons Dis. 2011;1:83–92. doi: 10.3233/JPD-2011-11004. [DOI] [PubMed] [Google Scholar]
- 49.Olanow C.W., Prusiner S.B. Is Parkinson's disease a prion disorder? Proc Natl Acad Sci U S A. 2009;106:12571–12572. doi: 10.1073/pnas.0906759106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Serpell L.C., Berriman J., Jakes R., Goedert M., Crowther R.A. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc Natl Acad Sci U S A. 2000;97:4897–4902. doi: 10.1073/pnas.97.9.4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Biere A.L., Wood S.J., Wypych J., Steavenson S., Jiang Y., Anafi D. Parkinson's disease-associated alpha-synuclein is more fibrillogenic than beta- and gamma-synuclein and cannot cross-seed its homologs. J Biol Chem. 2000;275:34574–34579. doi: 10.1074/jbc.M005514200. [DOI] [PubMed] [Google Scholar]
- 52.Yonetani M., Nonaka T., Masuda M., Inukai Y., Oikawa T., Hisanaga S. Conversion of wild-type alpha-synuclein into mutant-type fibrils and its propagation in the presence of A30P mutant. J Biol Chem. 2009;284:7940–7950. doi: 10.1074/jbc.M807482200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Desplats P., Lee H.J., Bae E.J., Patrick C., Rockenstein E., Crews L. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009;106:13010–13015. doi: 10.1073/pnas.0903691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lee H.J., Kim C., Lee S.J. Alpha-synuclein stimulation of astrocytes: potential role for neuroinflammation and neuroprotection. Oxid Med Cell Longev. 2010;3:283–287. doi: 10.4161/oxim.3.4.12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hansen C., Angot E., Bergstrom A.L., Steiner J.A., Pieri L., Paul G. alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest. 2011;121:715–725. doi: 10.1172/JCI43366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Angot E., Steiner J.A., Lema Tome C.M., Ekstrom P., Mattsson B., Bjorklund A. Alpha-synuclein cell-to-cell transfer and seeding in grafted dopaminergic neurons in vivo. PLoS One. 2012;7:e39465. doi: 10.1371/journal.pone.0039465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mougenot A.L., Nicot S., Bencsik A., Morignat E., Verchere J., Lakhdar L. Prion-like acceleration of a synucleinopathy in a transgenic mouse model. Neurobiol Aging. 2012;33:2225–2228. doi: 10.1016/j.neurobiolaging.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 58.Luk K.C., Kehm V.M., Zhang B., O’Brien P., Trojanowski J.Q., Lee V.M. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012;209:975–986. doi: 10.1084/jem.20112457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Luk K.C., Kehm V., Carroll J., Zhang B., O’Brien P., Trojanowski J.Q. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–953. doi: 10.1126/science.1227157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Watts J.C., Giles K., Oehler A., Middleton L., Dexter D.T., Gentleman S.M. Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci U S A. 2013;110:19555–19560. doi: 10.1073/pnas.1318268110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Masuda-Suzukake M., Nonaka T., Hosokawa M., Oikawa T., Arai T., Akiyama H. Prion-like spreading of pathological alpha-synuclein in brain. Brain. 2013;136:1128–1138. doi: 10.1093/brain/awt037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Recasens A., Dehay B., Bove J., Carballo-Carbajal I., Dovero S., Perez-Villalba A. Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol. 2014;75:351–362. doi: 10.1002/ana.24066. [DOI] [PubMed] [Google Scholar]
- 63.Guo J.L., Covell D.J., Daniels J.P., Iba M., Stieber A., Zhang B. Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell. 2013;154:103–117. doi: 10.1016/j.cell.2013.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peelaerts W., Bousset L., Van der Perren A., Moskalyuk A., Pulizzi R., Giugliano M. alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 2015;522:340–344. doi: 10.1038/nature14547. [DOI] [PubMed] [Google Scholar]
- 65.Savica R., Rocca W.A., Ahlskog J.E. When does Parkinson disease start? Arch Neurol. 2010;67:798–801. doi: 10.1001/archneurol.2010.135. [DOI] [PubMed] [Google Scholar]
- 66.Powley T.L., Fox E.A., Berthoud H.R. Retrograde tracer technique for assessment of selective and total subdiaphragmatic vagotomies. Am J Physiol. 1987;253:R361–R370. doi: 10.1152/ajpregu.1987.253.2.R361. [DOI] [PubMed] [Google Scholar]
- 67.Phillips R.J., Walter G.C., Wilder S.L., Baronowsky E.A., Powley T.L. Alpha-synuclein-immunopositive myenteric neurons and vagal preganglionic terminals: autonomic pathway implicated in Parkinson's disease? Neuroscience. 2008;153:733–750. doi: 10.1016/j.neuroscience.2008.02.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ulusoy A., Rusconi R., Perez-Revuelta B.I., Musgrove R.E., Helwig M., Winzen-Reichert B. Caudo-rostral brain spreading of alpha-synuclein through vagal connections. EMBO Mol Med. 2013;5:1051–1059. doi: 10.1002/emmm.201302475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Holmqvist S., Chutna O., Bousset L., Aldrin-Kirk P., Li W., Bjorklund T. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014;128:805–820. doi: 10.1007/s00401-014-1343-6. [DOI] [PubMed] [Google Scholar]
- 70.Rey N.L., Petit G.H., Bousset L., Melki R., Brundin P. Transfer of human alpha-synuclein from the olfactory bulb to interconnected brain regions in mice. Acta Neuropathol. 2013;126:555–573. doi: 10.1007/s00401-013-1160-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hawkes C.H., Del Tredici K., Braak H. Parkinson's disease: a dual-hit hypothesis. Neuropathol Appl Neurobiol. 2007;33:599–614. doi: 10.1111/j.1365-2990.2007.00874.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McGeer P.L., Itagaki S., Boyes B.E., McGeer E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 73.Mogi M., Harada M., Kondo T., Riederer P., Inagaki H., Minami M. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci Lett. 1994;180:147–150. doi: 10.1016/0304-3940(94)90508-8. [DOI] [PubMed] [Google Scholar]
- 74.Bower J.H., Maraganore D.M., Peterson B.J., Ahlskog J.E., Rocca W.A. Immunologic diseases, anti-inflammatory drugs, and Parkinson disease: a case-control study. Neurology. 2006;67:494–496. doi: 10.1212/01.wnl.0000227906.99570.cc. [DOI] [PubMed] [Google Scholar]
- 75.Gagne J.J., Power M.C. Anti-inflammatory drugs and risk of Parkinson disease: a meta-analysis. Neurology. 2010;74:995–1002. doi: 10.1212/WNL.0b013e3181d5a4a3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Davis G.C., Williams A.C., Markey S.P., Ebert M.H., Caine E.D., Reichert C.M. Chronic Parkinsonism secondary to intravenous injection of meperidine analogues. Psychiatry Res. 1979;1:249–254. doi: 10.1016/0165-1781(79)90006-4. [DOI] [PubMed] [Google Scholar]
- 77.Langston J.W., Ballard P., Tetrud J.W., Irwin I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- 78.van der Mark M., Brouwer M., Kromhout H., Nijssen P., Huss A., Vermeulen R. Is pesticide use related to Parkinson disease? Some clues to heterogeneity in study results. Environ Health Perspect. 2012;120:340–347. doi: 10.1289/ehp.1103881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Elbaz A., Clavel J., Rathouz P.J., Moisan F., Galanaud J.P., Delemotte B. Professional exposure to pesticides and Parkinson disease. Ann Neurol. 2009;66:494–504. doi: 10.1002/ana.21717. [DOI] [PubMed] [Google Scholar]
- 80.Tanner C.M., Kamel F., Ross G.W., Hoppin J.A., Goldman S.M., Korell M. Rotenone, paraquat, and Parkinson's disease. Environ Health Perspect. 2011;119:866–872. doi: 10.1289/ehp.1002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pan-Montojo F., Schwarz M., Winkler C., Arnhold M., O'Sullivan G.A., Pal A. Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci Rep. 2012;2:898. doi: 10.1038/srep00898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Braak H., de Vos R.A., Bohl J., Del Tredici K. Gastric alpha-synuclein immunoreactive inclusions in Meissner's and Auerbach's plexuses in cases staged for Parkinson's disease-related brain pathology. Neurosci Lett. 2006;396:67–72. doi: 10.1016/j.neulet.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 83.Kalaitzakis M.E., Graeber M.B., Gentleman S.M., Pearce R.K. The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson's disease: a critical analysis of alpha-synuclein staging. Neuropathol Appl Neurobiol. 2008;34:284–295. doi: 10.1111/j.1365-2990.2007.00923.x. [DOI] [PubMed] [Google Scholar]
- 84.Attems J., Jellinger K.A. The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson's disease. Neuropathol Appl Neurobiol. 2008;34:466–467. doi: 10.1111/j.1365-2990.2008.00937.x. [DOI] [PubMed] [Google Scholar]
- 85.Visanji N.P., Brooks P.L., Hazrati L.N., Lang A.E. The prion hypothesis in Parkinson's disease: braak to the future. Acta Neuropathol Commun. 2013;1:2. doi: 10.1186/2051-5960-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dehay B., Bourdenx M., Gorry P., Przedborski S., Vila M., Hunot S. Targeting alpha-synuclein for treatment of Parkinson's disease: mechanistic and therapeutic considerations. Lancet Neurol. 2015;14:855–866. doi: 10.1016/S1474-4422(15)00006-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Masliah E., Rockenstein E., Adame A., Alford M., Crews L., Hashimoto M. Effects of alpha-synuclein immunization in a mouse model of Parkinson's disease. Neuron. 2005;46:857–868. doi: 10.1016/j.neuron.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 88.Sanchez-Guajardo V., Annibali A., Jensen P.H., Romero-Ramos M. alpha-Synuclein vaccination prevents the accumulation of parkinson disease-like pathologic inclusions in striatum in association with regulatory T cell recruitment in a rat model. J Neuropathol Exp Neurol. 2013;72:624–645. doi: 10.1097/NEN.0b013e31829768d2. [DOI] [PubMed] [Google Scholar]
- 89.Games D., Valera E., Spencer B., Rockenstein E., Mante M., Adame A. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson's disease-like models. J Neurosci. 2014;34:9441–9454. doi: 10.1523/JNEUROSCI.5314-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Saa P., Castilla J., Soto C. Presymptomatic detection of prions in blood. Science. 2006;313:92–94. doi: 10.1126/science.1129051. [DOI] [PubMed] [Google Scholar]
- 91.Atarashi R., Satoh K., Sano K., Fuse T., Yamaguchi N., Ishibashi Det al. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat Med. 2011;17:175–178. doi: 10.1038/nm.2294. [DOI] [PubMed] [Google Scholar]