

Abstract
Blood coagulation and inflammation are universal responses to infection and there is crosstalk between inflammation and coagulation that can either amplify or dampen the responses. Loss of appropriate interactions between these systems probably contributes to morbidity and mortality in infectious diseases. For instance, inflammatory cytokines and leukocyte elastase can downregulate natural anticoagulant proteins that help to maintain endothelial-cell integrity, control clotting, inhibit vasoactive peptides and dampen leukocyte infiltration into the vessel wall. This Review will summarize our current understanding of the mechanisms involved in the crosstalk between these two important systems.
When infectious agents, regardless of their nature, initiate the innate immune response, it in turn triggers blood coagulation [1]. For instance, as viral infections, such as severe acute respiratory syndrome (SARS), progress into severe sepsis, markers of blood coagulation are increased markedly [2]. Blood coagulation components are not simply bystanders but can either amplify or inhibit the inflammatory response. Blood clotting can be initiated when inflammatory cytokines and endotoxin induce the de novo synthesis of tissue factor on leukocytes [3]. Exposure of tissue factor to the blood then triggers the coagulation cascade. Complement activation can lead to the formation of plasma membrane surfaces enriched in negatively charged phospholipids that amplify the coagulation reactions 4, 5.
Opposing this coagulation cascade are the natural anticoagulant pathways. These pathways not only limit the coagulation response but also dampen the inflammatory response by minimizing leukocyte chemotaxis [6] and endothelial-cell interactions [7], and by suppressing apoptosis 8, 9 and reducing cytokine expression 10, 11, 12. However, the acute inflammatory response can lead to suppression of the natural anticoagulant pathways by consumption, proteolytic inactivation and downregulation of protein expression [13]. Loss of the coordinate control of the coagulation and inflammation pathways due to impaired function of the natural anticoagulant systems probably contributes to organ failure in severe sepsis.
The blood clotting process is extremely complex (the more biochemical details are reviewed in Refs 14, 15). Briefly, blood coagulation is initiated when tissue factor comes into contact with blood (Figure 1 ). Under normal circumstances, there is little tissue factor in the intravascular compartment. Most of the tissue factor is found constitutively expressed on extravascular cells, where it surrounds the vessels providing a mechanism to rapid seal breaches in vessel integrity [16]. Recent studies have indicated that there is a circulating form of tissue factor, probably localized on leukocyte-derived microparticles 17, 18, 19. This microparticle-bound tissue factor might be concentrated on P-selectin on activated platelets in forming thrombi through interaction with its ligand, PSGL 1 (P-selectin glycoprotein ligand 1), from the activated leukocytes. Inflammatory stimuli can increase this response, both by stimulating tissue factor synthesis and promoting microparticle formation. Microparticles express high levels of negatively charged phospholipids on their surface [5], thus promoting both the tissue factor–factor VIIa-mediated factor X and IX activation, and enabling the propagation of coagulation by the factor Xa–factor Va activation of prothrombin and the factor IXa–factor VIIIa activation of factor X. Potent platelet agonists, such as thrombin in combination with collagen or complement C5b-9, can further augment the availability of negatively charged phospholipid membrane surfaces on cells, particularly platelets, thus enabling further amplification of the coagulation system 5, 20. Once initiated, the coagulation pathway has the potential to generate ∼100 times more thrombin than is required to form a rapid, firm clot. Under most conditions, this is prevented by the natural anticoagulant mechanism. The major inhibitory mechanisms involve the protein C anticoagulant pathway, antithrombin–heparin, and the tissue factor pathway inhibitor (TFPI).
Figure 1.

The function of membranes and cofactors in blood coagulation. The enzymes associate with cofactors on membrane surfaces. Factor VIIa associates with tissue factor (TF) to activate either factor X or factor IX. Factor IXa associates with factor VIIIa to activate factor X. Factor Va associates with factor Xa to activate prothrombin (Pro). Thrombin (T) associates with thrombomodulin (TM) to activate protein C (PC) and the activated protein C (APC) complexes with protein S (S) to inactivate factors Va and VIIIa, thereby blocking the coagulation cascade.
The protein C pathway is triggered when thrombin binds to thrombomodulin on the endothelial-cell surface. This complex, in concert with protein C bound to the endothelial-cell protein C receptor (EPCR), generates activated protein C. Once activated, protein C dissociates from the EPCR and can bind to protein S on membrane surfaces. The activated protein C–protein S complex then inactivates factors Va and VIIIa. Without these cofactors, factor Xa and factor IXa have much less than 1% of the capacity to activate the downstream zymogen. Practically, this means that inactivation of the cofactors shuts off the coagulation system completely. In addition, relative to thrombin in solution, thrombin bound to thrombomodulin is inhibited much more rapidly by antithrombin and protein C inhibitor [21].
The antithrombin–heparin mechanism neutralizes factor Xa, thrombin and factor IXa. It also inactivates factor VIIa but only when the factor VIIa is bound to tissue factor 22, 23. Antithrombin inhibition of the factor VIIa–tissue factor complex, factor IXa, factor Xa and thrombin are all thought to be accelerated by vascular heparin-like proteoglycans [24].
TFPI inactivates factor VIIa bound to tissue factor using a unique mechanism. The inhibitor has two functional inhibitory Kunitz domains. The second domain binds to and inhibits factor Xa, which, as a result of its ability to bind negatively charged membranes, concentrates the TFPI–factor Xa complex on the membrane surface, at which time the first Kunitz domain of the inhibitor reversibly neutralizes factor VIIa.
The physiological significance of these pathways is demonstrated by gene experiments in mice, which result in embryonic or neonatal lethality when any single pathway is disrupted 25, 26, 27, 28, 29. In humans, complete deficiencies of protein C lead to neonatal microvascular thrombosis (purpura fulminans) and subsequent death unless treated [30]. Complete deficiencies of antithrombin and the TFPI in humans have not been described.
Impact of acute inflammatory responses on natural anticoagulant mechanisms
Inflammation has multiple effects on the coagulation system (Figure 2 ). Antithrombin is consumed and/or inactivated in sepsis and other acute inflammatory injury. Antithrombin inhibitory activity decreases markedly during severe sepsis, often to <50% of normal levels [31]. Because the rates of inhibition of the target proteases are strongly dependent on the antithrombin concentration, this decrease in inhibitor concentration would contribute to increased stability of the coagulation enzymes and hence favor intravascular coagulation.
Figure 2.

The impact of inflammatory mediators on the regulation of coagulation. Inflammatory mediators, such as TNF-α or endotoxin, can effect the changes indicated. An upward arrow indicates increases in levels and a downward arrow indicates decreases. Abbreviations: α1-AT, α1 antitrypsin; EPCR, endothelial-cell protein C receptor; PAI-1, plasminogen activator inhibitor-1; TNF-α, tumor necrosis factor-α.
There is evidence that the vascular heparin-like molecules are reduced by inflammatory cytokines and neutrophil activation products [32]. Clinically, in severe sepsis, these heparin-like molecules can be downregulated or degraded [33], further diminishing the natural anticoagulant potential, especially when the antithrombin level has been reduced by consumption.
The protein C pathway appears to be especially sensitive to downregulation by inflammatory responses. Interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α) and endotoxin can downregulate thrombomodulin and the EPCR by inhibiting gene transcription 34, 35, reducing the ability to generate activated protein C. Neutrophil elastase cleaves thrombomodulin from the endothelial-cell surface, generating a much less active form of thrombomodulin [36]. In septic patients, both the EPCR and thrombomodulin can be severely downregulated, as demonstrated both immunohistochemically [37] and by analysis of the ability of the patients to generate activated protein C [38]. In addition, protein C levels decrease dramatically in patients with severe sepsis. This is probably due to a combination of consumption and liver (the main site of protein C synthesis) dysfunction. The degree of protein C reduction correlates with a negative prognosis in septic patients [39].
Once tight control of thrombin and other coagulation enzymes is lost, they can participate in promoting the inflammatory response (Figure 3 ). The enhanced inflammatory response enhances the cell-associated coagulation activities as described earlier. Therefore, anticoagulants that dampen the cellular inflammatory response also dampen the coagulant response [40].
Figure 3.

Thrombin is a multifunctional enzyme. Thrombin generates procoagulant, anticoagulant, inflammatory and mitogenic responses. These responses shift the hemostatic balance. Abbreviations: CD40L, CD40 ligand; EC, endothelial cell; IL-6, interleukin-6; MCP-1, macrophage chemotactic protein-1; PAF, platelet activating factor; PDGF, platelet-derived growth factor; PMNs, polymorphonucleocytes; TGF-β, transforming growth factor-β. Reproduced with permission from Ref. [77].
Impact of natural anticoagulant mechanisms on the inflammatory response and cellular apoptosis
The antithrombin–heparin pathway can modulate inflammatory responses not only by inhibiting coagulant enzyme-mediated cell signaling through the protease activated receptors but also by modulating cellular responses [12]. Antithrombin can downregulate the expression of CD11b/CD18 on leukocytes. Because factor X binding to this receptor augments factor X activation 41, 42, the downregulation of this cellular factor X receptor decreases both leukocyte adhesion and coagulation. The addition of antithrombin to endothelial cells in culture increases prostacyclin formation [43] and decreases NF-κB signaling [10]. By modulating cellular responses to endotoxin, antithrombin appears to decrease both tissue factor and IL-6 expression in monocytes and endothelium [40]. Antithrombin binding to syndecan 4, a proteoglycan on neutrophils, inhibits chemokine-induced neutrophil migration [44]. Interestingly, heparin blocks this effect. In addition, administration of high levels of antithrombin to septic experimental animals and animals undergoing ischemia reperfusion injury reverses leukocyte recruitment [45] (Figure 4 ).
Figure 4.

Antithrombin binds to heparin-like glycosaminoglycans, such as those on syndecan 4. Binding induces a conformational chance in antithrombin (AT). Thrombin (T) and some other coagulation enzymes react preferentially with the bound AT. In the case of T this requires T binding to the glycosaminoglycan (syndecan 4). Once T binds to the inhibitor, major conformational changes occur that lead to the release of the complex [78]. In the absence of T, AT binding to the glycosaminoglycan leads to cell signaling, increasing prostacyclin (PGI2) formation and decreasing NF-κB activation.
The protein C anticoagulant pathway has several components that reduce the inflammatory response. Thrombomodulin not only increases protein C activation but also prevents thrombin from activating protease-activated receptors (PARs) 46, 47. This is accomplished at least in part because the site on thrombin (anion binding exosite 1) responsible for binding PAR1 and thrombomodulin overlap (Figure 5 ). In addition, thrombin bound to thrombomodulin gains the ability to activate a plasma procarboxypeptidase R [also known as thrombin activatable fibrinolysis inhibitor (TAFI)] [48]. Recent studies have shown that his carboxypeptidase is a very potent inhibitor of the complement anaphylatoxin, C5a 49, 50, and bradykinin [50]. By inhibiting these vasoactive substances, the generation of this carboxypeptidase, especially in the microcirculation [51], probably helps to prevent severe drops in blood pressure and microvascular injury, and subsequent edema, in animals with severe acute inflammatory injuries.
Figure 5.
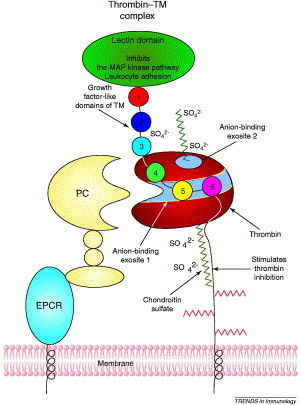
Thrombin binding to TM involves anion-binding exosite 1 on thrombin (shown as a strip through the middle of thrombin) and EGF domains 4 to 6 on TM. A chondroitin sulfate moiety on TM increases the affinity for thrombin but is not required for function. This chondroitin sulfate interacts with anion-binding exosite 2 on thrombin, a second, basic area near the heparin-binding site. The lectin domain of TM inhibits leukocyte adhesion and the MAP kinase pathway. Other functions of TM require the presence of different EGF domains. Domains 1–6 stimulate fibroblast growth; domains 3–6 are required for TAFI activation; domains 4–6 are required for PC activation; and thrombin-clotting activity is blocked by domains 5–6. The activation of PC to APC by the thrombin–TM complex is enhanced by binding of PC to EPCR through its gla domain [60]. Abbreviations: APC, activated protein C; EGF, epidermal growth factor; EPCR, endothelial-cell protein C receptor; MAP, mitogen-activated protein; PC, protein C; TAFI, thrombin activatable fibrinolysis inhibitor; TM, thrombomodulin.
In addition to these functions, recent studies have shown that thrombomodulin has direct anti-inflammatory activity on the endothelium. Thrombomodulin is a multi-domain molecule. When the N-terminal lectin domain is deleted, protein C and carboxypeptidase R activation proceed normally. However, when this mutant is used to replace the normal thrombomodulin gene in mice, the mice recruit leukocytes much more avidly than wild-type mice in response to inflammatory stimuli. Infusion of the isolated lectin domain can reverse this response [52]. Cell culture studies demonstrated that this domain dampened the mitogen activated kinase and NF-κB responses in endothelium [51].
Activated protein C dampens NF-κB signaling in monocytes 53, 54, 55. It also decreases inflammatory mediator-initiated generation of tissue factor on leukocytic cell lines 12, 56, 57 in an EPCR dependent fashion. In addition, activated protein C can inhibit tight neutrophil adhesion to the endothelium [6]. Activated protein C can also decrease endothelial-cell apoptosis [9]. This function is dependent on the EPCR and apparently involves activation of PAR1 [58]. In mouse models of stroke, activated protein C minimizes damage at least in part by inhibiting apoptosis through downregulation of P53 [59]. This inhibition of P53 expression was also dependent on EPCR and PAR1. How activating PAR1 generates anti-inflammatory activities remains an area of active investigation.
The recent determination of the EPCR structure suggests additional potential roles of the protein C pathway in regulating the immune response. EPCR shares considerable sequence identity to the MHC I–CD1 family of molecules [35]. The crystal structure of EPCR reveals that, like the CD1 family, EPCR has a tightly bound lipid, in this case phospholipid, located in a region virtually identical to the antigen-presenting groove in the CD1 family [60] (Figure 6 ). In CD1 proteins, glycolipids bind in this grove and can have a major role in the immune response to bacterial infection [61]. In addition, deficiency of CD1d in mice leads to autoimmune disease [62]. From the structural similarities, it is probable, but as yet unproven, that EPCR might have roles similar to those of CD1 molecules in the regulation of inflammation. Autoantibodies to EPCR and a correlation with autoimmune fetal loss have recently been demonstrated [63], consistent with its involvement in immune regulation.
Figure 6.

The EPCR molecule with a portion of the PC Gla domain and a lipid molecule. In the EPCR (yellow ribbon), two α-helices and an eight-stranded β-sheet create a groove that is filled with phospholipid (the space filling blue balls in the center). Binding of Ca2+ ions (magenta spheres) to the PC Gla domain (green ribbon) exposes the N-terminal ‘omega’ loop, which in the absence of EPCR interacts with the phospholipid surfaces on the membrane. There do not appear to be direct interactions between the PC Gla domain and the lipid molecule located in the groove of the EPCR. The model of the complex consists of residues 7–177 of the rsEPCR and the first 33 residues of the PC Gla domain. Abbreviations: EPCR, endothelial-cell protein C receptor; PC, protein C.
Platelet contributions to inflammatory responses
Platelets are also involved in the linking of inflammation and coagulation. Inflammatory mediators, such as IL-6, not only increase platelet production, but the platelets that are generated are more thrombogenic, demonstrating an increased sensitivity to platelet agonists, such as thrombin [64]. Platelets contain high concentrations of the proinflammatory mediator CD40 ligand. On platelet activation, CD40 ligand is released. This protein then induces tissue factor synthesis 65, 66 and increases inflammatory cytokines, such as IL-6 and IL-8 67, 68.
Conclusions
The clotting process involves molecules, such as the selectins, that were originally thought to be involved primarily in inflammation (Box 1). Natural anticoagulant proteins exhibit a spectrum of anti-inflammatory activities, including inhibiting NF-κB signaling, inhibiting complement C5a and bradykinin and preventing apoptosis. Severe acute inflammatory challenges can downregulate the natural anticoagulants, thereby diminishing their ability to dampen the coagulant and inflammatory responses. When this happens, a progressive cycle can evolve in which the coagulation increases the inflammatory response, thereby increasing the coagulant response until severe vascular and organ injury occurs. Modulation of this complex series of interactive events promises to offer an attractive method for intervening clinically in diseases involving acute inflammatory challenges, such as severe sepsis. The recent successful demonstration that recombinant activated protein C can reduce the 28-day all cause mortality in patients with severe sepsis [69] provides support for this proposal.
Box 1. Questions for future research.
The exact signaling mechanisms responsible for the protective effects of natural anticoagulants need to be characterized more fully. At the preclinical level, differences in the protective effects of natural anticoagulants need to be explored more fully, particularly in situations where disease has already progressed, as would almost always be the case in patients. Future research will also need to address the impact of common disease processes, such as diabetes, on both the innate immune response and the ability of natural anticoagulant pathways to modulate the innate immune response. Combinations of microarray and proteomics approaches might aid in these studies.
In addition, although our understanding of the regulation of blood coagulation in systems with artificial membrane surfaces is relatively sophisticated, cellular control of coagulation is still an emerging area. For instance, activated protein C is a potent anticoagulant involving liposomes. By contrast, it is a relatively poor anticoagulant on platelet surfaces. In model systems of blood coagulation, in which platelets are present over endothelium, it appears that activated protein C anticoagulant functions occur on the endothelial cell rather than the platelet surface [76]. The nature of the membrane surface responsible for these differences remains to be elucidated.
Of interest, antithrombin [70] and TFPI both failed in large randomized clinical trials [71], despite, similar to activated protein C, having proven efficacious in non-human primate models of severe sepsis 72, 73. Several possibilities could explain the failure of the natural anticoagulants TFPI and antithrombin in the trials. First, in the clinical situation, bleeding complications limit dosage to a greater extent than in the animal studies. For instance, the anticoagulant doses in the baboon sepsis model were much higher than in the human trials [74]. Therefore, the doses of the anticoagulants might not have been adequate. In the antithrombin trial, heparin was used in many of the patients [70]. As noted earlier, heparin blocks several cellular effects of antithrombin. Although there was no overall effect on survival with antithromin, the patients with heparin alone or antithrombin alone tended to have a better survival rate than the patients who received both [70].
Alternatively, the trial result differences lie in some different functions among the natural anticoagulants. Antithrombin and activated protein C share several anti-inflammatory functions in vitro and in animal models. Few studies have explored differences between the natural anticoagulants with respect to the cellular responses and particular vascular beds that are impacted when an ongoing inflammatory challenge is in place at the time of inhibitor administration. Furthermore, in the patient populations, many co-morbidities might decrease efficacy of one of the inhibitors selectively. Co-morbidities, such as atherosclerosis, diabetes and hypertension, can exacerbate the innate inflammatory response by a variety of mechanisms, including downregulation of the natural anticoagulants [75].
References
- 1.Opal S.M. Systemic host responses in severe sepsis analyzed by causative microorganism and treatment effects of drotrecogin alfa (activated) Clin. Infect. Dis. 2003;37:50–58. doi: 10.1086/375593. [DOI] [PubMed] [Google Scholar]
- 2.Lee N. A major outbreak of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 2003;348:1986–1994. doi: 10.1056/NEJMoa030685. [DOI] [PubMed] [Google Scholar]
- 3.Rauch U. Transfer of tissue factor from leukocytes to platelets is mediated by CD15 and tissue factor. Blood. 2000;96:170–175. [PubMed] [Google Scholar]
- 4.Bevers E.M. Transmembrane phospholipid distribution in blood cells: control mechanisms and pathophysiological significance. Biol. Chem. 1998;379:973–986. [PubMed] [Google Scholar]
- 5.Sims P.J. Assembly of the platelet prothrombinase complex is linked to vesiculation of the platelet plasma membrane. Studies in Scott Syndrome: An isolated defect in platelet procoagulant activity. J. Biol. Chem. 1989;264:17049–17057. [PubMed] [Google Scholar]
- 6.Sturn D.H. Expression and function of the endothelial protein C receptor in human neutrophils. Blood. 2003;102:1499–1505. doi: 10.1182/blood-2002-12-3880. [DOI] [PubMed] [Google Scholar]
- 7.Isobe H. Activated protein C prevents endotoxin-induced hypotension in rats by inhibiting excessive production of nitric oxide. Circulation. 2001;104:1171–1175. doi: 10.1161/hc3501.093799. [DOI] [PubMed] [Google Scholar]
- 8.Cheng T. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat. Med. 2003;9:338–342. doi: 10.1038/nm826. [DOI] [PubMed] [Google Scholar]
- 9.Joyce D.E. Gene expression profile of antithrombotic protein C defines new mechanisms modulating inflammation and apoptosis. J. Biol. Chem. 2001;276:11199–11203. doi: 10.1074/jbc.C100017200. [DOI] [PubMed] [Google Scholar]
- 10.Oelschläger C. Antithrombin III inhibits nuclear factor κB activation in human monocytes and vascular endothelial cells. Blood. 2002;99:4015–4020. doi: 10.1182/blood.v99.11.4015. [DOI] [PubMed] [Google Scholar]
- 11.Yasui H. Intratracheal administration of activated protein C inhibits bleomycin-induced lung fibrosis in the mouse. Am. J. Respir. Crit. Care Med. 2001;163:1660–1668. doi: 10.1164/ajrccm.163.7.9911068. [DOI] [PubMed] [Google Scholar]
- 12.Okajima K. Regulation of inflammatory responses by natural anticoagulants. Immunol. Rev. 2001;184:258–274. doi: 10.1034/j.1600-065x.2001.1840123.x. [DOI] [PubMed] [Google Scholar]
- 13.Opal S.M., Esmon C.T. Bench-to-bedside review: Functional relationships between coagulation and the innate immune response and their respective roles in the pathogenesis of sepsis. Crit. Care. 2003;7:23–38. doi: 10.1186/cc1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Furie B., Furie B.C. The molecular basis of blood coagulation. Cell. 1988;53:505–518. doi: 10.1016/0092-8674(88)90567-3. [DOI] [PubMed] [Google Scholar]
- 15.Esmon C.T. Blood coagulation. In: Nathan D.G., editor. Nathan and Oski's Hematology of Infancy and Childhood. W.B. Saunders Company; 2003. pp. 1475–1496. [Google Scholar]
- 16.Drake T.A. Selective cellular expression of tissue factor in human tissues: Implications for disorders of hemostasis and thrombosis. Am. J. Pathol. 1989;134:1087–1097. [PMC free article] [PubMed] [Google Scholar]
- 17.Celi A. Thrombus formation: direct real-time observation and digital analysis of thrombus assembly in a living mouse by confocal and widefield intravital microscopy. J. Thromb. Haemost. 2003;1:60–68. doi: 10.1046/j.1538-7836.2003.t01-1-00033.x. [DOI] [PubMed] [Google Scholar]
- 18.Giesen P.L.A. Blood-borne tissue factor: another view of thrombosis. Proc. Natl. Acad. Sci. U. S. A. 1999;96:2311–2315. doi: 10.1073/pnas.96.5.2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bouchard B.A., Tracy P.B. The participation of leukocytes in coagulant reactions. J. Thromb. Haemost. 2003;1:464–469. doi: 10.1046/j.1538-7836.2003.00089.x. [DOI] [PubMed] [Google Scholar]
- 20.Zwaal R.F.A., Schroit A.J. Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood. 1997;89:1121–1132. [PubMed] [Google Scholar]
- 21.Rezaie A.R. Protein C inhibitor is a potent inhibitor of the thrombin-thrombomodulin complex. J. Biol. Chem. 1995;270:25336–25339. doi: 10.1074/jbc.270.43.25336. [DOI] [PubMed] [Google Scholar]
- 22.Lawson J.H. Complex-dependent inhibition of factor VIIa by antithrombin III and heparin. J. Biol. Chem. 1993;268:767–770. [PubMed] [Google Scholar]
- 23.Rosenberg R.D. Regulation of the hemostatic mechanism. In: Stamatoyannopoulos G., editor. The Molecular Basis of Blood Diseases. W.B. Saunders Company; 1987. pp. 534–574. [Google Scholar]
- 24.Marcum J.A., Rosenberg R.D. Anticoagulantly active heparin-like molecules from the vascular tissue. Biochemistry. 1984;23:1730–1737. doi: 10.1021/bi00303a023. [DOI] [PubMed] [Google Scholar]
- 25.Ishiguro K. Complete antithrombin deficiency in mice results in embryonic lethality. J. Clin. Invest. 2000;106:873–878. doi: 10.1172/JCI10489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gu J-M. Disruption of the endothelial cell protein C receptor gene in mice causes placental thrombosis and early embryonic lethality. J. Biol. Chem. 2002;277:43335–43343. doi: 10.1074/jbc.M207538200. [DOI] [PubMed] [Google Scholar]
- 27.Huang Z-F. Tissue factor pathway inhibitor gene disruption produces intrauterine lethality in mice. Blood. 1997;90:944–951. [PubMed] [Google Scholar]
- 28.Weiler-Guettler H. A targeted point mutation in thrombomodulin generates viable mice with a prethrombotic state. J. Clin. Invest. 1998;101:1983–1991. doi: 10.1172/JCI2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jalbert L.R. Inactivation of the gene for anticoagulant protein C causes lethal perinatal consumptive coagulopathy in mice. J. Clin. Invest. 1998;102:1481–1488. doi: 10.1172/JCI3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dreyfus M. Treatment of homozygous protein C deficiency and neonatal purpura fulminans with a purified protein C concentrate. N. Engl. J. Med. 1991;325:1565–1568. doi: 10.1056/NEJM199111283252207. [DOI] [PubMed] [Google Scholar]
- 31.Levi M., ten Cate H. Disseminated intravascular coagulation. N. Engl. J. Med. 1999;341:586–592. doi: 10.1056/NEJM199908193410807. [DOI] [PubMed] [Google Scholar]
- 32.Klein N.J. Alteration in glycosaminoglycan metabolism and surface charge on human umbilical vein endothelial cells induced by cytokines, endotoxin and neutrophils. J. Cell Sci. 1992;102:821–832. doi: 10.1242/jcs.102.4.821. [DOI] [PubMed] [Google Scholar]
- 33.Klein N.J. The influence of capsulation and lipooligosaccharide structure on neutrophil adhesion molecule expression and endothelial injury by Neisseria meningitidis. J. Infect. Dis. 1996;173:172–179. doi: 10.1093/infdis/173.1.172. [DOI] [PubMed] [Google Scholar]
- 34.Conway E.M., Rosenberg R.D. Tumor necrosis factor suppresses transcription of the thrombomodulin gene in endothelial cells. Mol. Cell. Biol. 1988;8:5588–5592. doi: 10.1128/mcb.8.12.5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fukudome K., Esmon C.T. Identification, cloning and regulation of a novel endothelial cell protein C/activated protein C receptor. J. Biol. Chem. 1994;269:26486–26491. [PubMed] [Google Scholar]
- 36.Takano S. Plasma thrombomodulin in health and diseases. Blood. 1990;76:2024–2029. [PubMed] [Google Scholar]
- 37.Faust S.N. Dysfunction of endothelial protein C activation in severe meningococcal sepsis. N. Engl. J. Med. 2001;345:408–416. doi: 10.1056/NEJM200108093450603. [DOI] [PubMed] [Google Scholar]
- 38.Liaw P.C.Y. A monoclonal antibody against activated protein C allows rapid detection of activated protein C in plasma and reveals a calcium ion dependent epitope involved in factor Va inactivation. J. Thromb. Haemost. 2003;1:662–670. doi: 10.1046/j.1538-7836.2003.00153.x. [DOI] [PubMed] [Google Scholar]
- 39.Fisher C.J., Yan S.B. Protein C levels as a prognostic indicator of outcome in sepsis and related diseases. Crit. Care Med. 2000;28:S49–S56. doi: 10.1097/00003246-200009001-00011. [DOI] [PubMed] [Google Scholar]
- 40.Souter P.J. Antithrombin inhibits lipopolysaccharide-induced tissue factor and interleukin-6 production by mononuclear cells, human umbilical vein endothelial cells, and whole blood. Crit. Care Med. 2001;29:134–139. doi: 10.1097/00003246-200101000-00027. [DOI] [PubMed] [Google Scholar]
- 41.Altieri D.C. Structurally homologous ligand binding of integrin Mac-1 and viral glycoprotein C receptors. Science. 1991;254:1200–1202. doi: 10.1126/science.1957171. [DOI] [PubMed] [Google Scholar]
- 42.Altieri D.C. Adhesive receptor Mac-1 coordinates the activation of factor X on stimulated cells of monocytic and myeloid differentiation: An alternative initiation of the coagulation protease cascade. Proc. Natl. Acad. Sci. U. S. A. 1988;85:7462–7466. doi: 10.1073/pnas.85.20.7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamauchi T. Antithrombin III stimulates prostacyclin production by cultured aortic endothelial cells. Biochem. Biophys. Res. Commun. 1989;163:1404–1411. doi: 10.1016/0006-291x(89)91135-2. [DOI] [PubMed] [Google Scholar]
- 44.Kaneider N.C. Syndecan-4 as antithrombin receptor of human neutrophils. Biochem. Biophys. Res. Commun. 2001;287:42–46. doi: 10.1006/bbrc.2001.5534. [DOI] [PubMed] [Google Scholar]
- 45.Ostrovsky L. Antithrombin III prevents and rapidly reverses leukocyte recruitment in ischemia/reperfusion. Circulation. 1997;96:2302–2310. doi: 10.1161/01.cir.96.7.2302. [DOI] [PubMed] [Google Scholar]
- 46.Ye J. The fifth and sixth growth factor-like domains of thrombomodulin bind to the anion exosite of thrombin and alter its specificity. J. Biol. Chem. 1992;267:11023–11028. [PubMed] [Google Scholar]
- 47.Liu L-W. The region of the thrombin receptor resembling hirudin binds to thrombin and alters enzyme specificity. J. Biol. Chem. 1991;266:16977–16980. [PubMed] [Google Scholar]
- 48.Wang W. Elements of the primary structure of thrombomodulin required for efficient thrombin-activable fibrinolysis inhibitor activation. J. Biol. Chem. 2000;275:22942–22947. doi: 10.1074/jbc.M001760200. [DOI] [PubMed] [Google Scholar]
- 49.Campbell W. Carboxypeptidase R is an inactivator of complement-derived inflammatory peptides and an inhibitor of fibrinolysis. Immunol. Rev. 2001;180:162–167. doi: 10.1034/j.1600-065x.2001.1800114.x. [DOI] [PubMed] [Google Scholar]
- 50.Myles T. Thrombin activatable fibrinolysis inhibitor: a potential regulator of vascular inflammation. J. Biol. Chem. 2003;278:51059–51067. doi: 10.1074/jbc.M306977200. [DOI] [PubMed] [Google Scholar]
- 51.Esmon C.T. The roles of protein C and thrombomodulin in the regulation of blood coagulation. J. Biol. Chem. 1989;264:4743–4746. [PubMed] [Google Scholar]
- 52.Conway E.M. The lectin-like domain of thrombomodulin confers protection from neutrophil-mediated tissue damage by suppressing adhesion molecule expression via nuclear factor κB and mitogen-activated protein kinase pathways. J. Exp. Med. 2002;196:565–577. doi: 10.1084/jem.20020077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hancock W.W. Binding of activated protein C to a specific receptor on human mononuclear phagocytes inhibits intracellular calcium signaling and monocyte-dependent proliferative responses. Transplantation. 1995;60:1525–1532. doi: 10.1097/00007890-199560120-00026. [DOI] [PubMed] [Google Scholar]
- 54.White B. Activated protein C inhibits lipopolysaccharide-induced nuclear translocation of nuclear factor κB (NF-κB) and tumour necrosis factor α (TNF-α) production in the THP-1 monocytic cell line. Br. J. Haematol. 2000;110:130–134. doi: 10.1046/j.1365-2141.2000.02128.x. [DOI] [PubMed] [Google Scholar]
- 55.Yuksel M. Activated protein C inhibits lipopolysaccharide-induced tumor necrosis factor-α production by inhibiting activation of both nuclear factor-κB and activator protein-1 in human monocytes. Thromb. Haemost. 2002;88:267–273. [PubMed] [Google Scholar]
- 56.Murphy, C. et al. (2001) Activated protein C inhibits upregulation of both tissue factor and TNFα in endotoxin stimulated monocytes. Thromb. Haemost. Suppl. P509 (Abstr.)
- 57.Shu F. Activated protein C suppresses tissue factor expression on U937 cells in the endothelial protein C receptor dependent manner. FEBS Lett. 2000;477:208–212. doi: 10.1016/s0014-5793(00)01740-3. [DOI] [PubMed] [Google Scholar]
- 58.Riewald M. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;296:1880–1882. doi: 10.1126/science.1071699. [DOI] [PubMed] [Google Scholar]
- 59.Shibata M. Anti-inflammatory, antithrombotic, and neuroprotective effects of activated protein C in a murine model of focal ischemic stroke. Circulation. 2001;103:1799–1805. doi: 10.1161/01.cir.103.13.1799. [DOI] [PubMed] [Google Scholar]
- 60.Oganesyan V. The crystal structure of the endothelial protein C receptor and a bound phospholipid. J. Biol. Chem. 2002;277:24851–24854. doi: 10.1074/jbc.C200163200. [DOI] [PubMed] [Google Scholar]
- 61.Moody D.B. CD1c-mediated T-cell recognition of isoprenoid glycolipids in Mycobacterium tuberculosis infection. Nature. 2000;404:884–888. doi: 10.1038/35009119. [DOI] [PubMed] [Google Scholar]
- 62.Hong S. Lipid antigen presentation in the immune system learned from CD1d knockout mice. Immunol. Rev. 1999;169:31–44. doi: 10.1111/j.1600-065x.1999.tb01304.x. [DOI] [PubMed] [Google Scholar]
- 63.Hurtado, V. et al. Autoantibodies against EPCR are found in antiphospholipid syndrome and are a risk factor for foetal death. Blood (in press) [DOI] [PubMed]
- 64.Burstein S.A. Cytokines, platelet production and hemostasis. Platelets. 1997;8:93–104. doi: 10.1080/09537109709169324. [DOI] [PubMed] [Google Scholar]
- 65.Pendurthi U.R. Binding of factor VIIa to tissue factor induces alterations in gene expression in human fibroblast cells: up-regulation of poly(A) polymerase. Proc. Natl. Acad. Sci. U. S. A. 1997;94:12598–12603. doi: 10.1073/pnas.94.23.12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miller D.L. CD40L–CD40 interactions regulate endothelial cell surface tissue factor and thrombomodulin expression. J. Leukoc. Biol. 1998;63:373–379. doi: 10.1002/jlb.63.3.373. [DOI] [PubMed] [Google Scholar]
- 67.André P. CD40L stabilizes arterial thrombi by a β3 integrin-dependent mechanism. Nat. Med. 2002;8:247–252. doi: 10.1038/nm0302-247. [DOI] [PubMed] [Google Scholar]
- 68.Henn V. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. 1998;391:591–594. doi: 10.1038/35393. [DOI] [PubMed] [Google Scholar]
- 69.Bernard G.R. Efficacy and safety of recombinant human activated protein C for severe sepsis. N. Engl. J. Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 70.Warren B.L. Caring for the critically ill patient. High-dose antithrombin III in severe sepsis: a randomized controlled trial. J. Am. Med. Assoc. 2001;286:1869–1878. doi: 10.1001/jama.286.15.1869. [DOI] [PubMed] [Google Scholar]
- 71.Abraham E. Efficacy and safety of tifacogin (recombinant tissue factor pathway inhibitor) in severe sepsis: a randomized controlled trial. J. Am. Med. Assoc. 2003;290:238–247. doi: 10.1001/jama.290.2.238. [DOI] [PubMed] [Google Scholar]
- 72.Minnema M.C. Recombinant human antithrombin III improves survival and attenuates inflammatory responses in baboons lethally challenged with Escherichia coli. Blood. 2000;95:1117–1123. [PubMed] [Google Scholar]
- 73.Creasey A.A. Tissue factor pathway inhibitor (TFPI) reduces mortality from Escherichia coli septic shock. J. Clin. Invest. 1993;91:2850–2860. doi: 10.1172/JCI116529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Esmon C.T. Introduction: are natural anticoagulants candidates for modulating the inflammatory response to endotoxin? Blood. 2000;95:1113–1116. [PubMed] [Google Scholar]
- 75.Esmon C.T. Why do animal models (sometimes) fail to mimic human sepsis? Crit. Care Med. 2004;32(Suppl):S219–S222. doi: 10.1097/01.ccm.0000127036.27343.48. [DOI] [PubMed] [Google Scholar]
- 76.Oliver J.A. Activated protein C cleaves factor Va more efficiently on endothelium than on platelet surfaces. Blood. 2002;100:539–546. doi: 10.1182/blood.v100.2.539. [DOI] [PubMed] [Google Scholar]
- 77.Esmon C.T. Cell mediated events that control blood coagulation and vascular injury. Annu. Rev. Cell Biol. 1993;9:1–26. doi: 10.1146/annurev.cb.09.110193.000245. [DOI] [PubMed] [Google Scholar]
- 78.Baglin T.P. Crystal structures of native and thrombin-complexed heparin cofactor II reveal a multistep allosteric mechanism. Proc. Natl. Acad. Sci. U. S. A. 2002;99:11079–11084. doi: 10.1073/pnas.162232399. [DOI] [PMC free article] [PubMed] [Google Scholar]