

Summary
Correct organ size is determined by the balance between cell death and proliferation. Perturbation of this delicate balance leads to cancer formation [1]. Hippo (Hpo), the Drosophila ortholog of MST1 and MST2 (Mammalian Sterile 20-like 1 and 2) is a key regulator of a signaling pathway that controls both cell death and proliferation [2, 3]. This pathway is so far composed of two Band 4.1 proteins, Expanded (Ex) and Merlin (Mer), two serine/threonine kinases, Hpo and Warts (Wts), the scaffold proteins Salvador (Sav) and Mats, and the transcriptional coactivator Yorkie (Yki). It has been proposed that Ex and Mer act upstream of Hpo, which in turn phosphorylates and activates Wts. Wts phosphorylates Yki and thus inhibits its activity and reduces expression of Yki target genes such as the caspase inhibitor DIAP1 and the micro RNA bantam [4-6]. However, the mechanisms leading to Hpo activation are still poorly understood. In mammalian cells, members of the Ras association family (RASSF) of tumor suppressors have been shown to bind to MST1 and modulate its activity [7]. In this study, we show that the Drosophila RASSF ortholog (dRASSF) restricts Hpo activity by competing with Sav for binding to Hpo. In addition, we observe that dRASSF also possesses a tumor-suppressor function.
Results and Discussion
The mammalian RASSF family comprises six different loci encoding a variety of splice variants. Most transcripts encode proteins that contain a Ras association domain (RA), an N-terminal C1-type zinc finger, and a C-terminal SARAH (Sav RASSF Hippo) domain ([8-13] and Figure S1A). RASSF family members, most notably RASSF1A, are frequently silenced in a variety of solid tumors [14], mainly by promoter methylation [15]. Thus, it has been proposed that RASSF genes act as tumor suppressors.
The biological function of these genes is not well understood. RASSF1A and Nore1A have both been shown to interact with MST1 via its SARAH domain [7]. Overexpression of RASSF1A or Nore1A inhibits MST1 activation, but coexpression of these RASSF proteins with Ras enhanced MST1 activity [16]. RASSF1A knockout mice have mildly increased tumor susceptibility [17], confirming that RASSF genes can act as tumor suppressors. The weakness of the mouse phenotype, which is at odds with the frequency of RASSF1A inactivation in human tumors, can be ascribed to redundancy with other family members.
By contrast, Drosophila melanogaster has a single RASSF family member, which is encoded by the CG4656 gene and which we will refer to as dRASSF. Like its vertebrate counterparts, dRASSF encodes a protein bearing an RA and SARAH domain at its C terminus (Figure S1A in the Supplemental Data available online). It also possesses a LIM domain that shares some similarities with C1 zinc fingers at its N terminus.
We generated mutant alleles of dRASSF by imprecise excision of two nearby transposons, GE23517 and EY2800 (see Supplemental Experimental Procedures). We obtained multiple alleles, which delete up to the fourth intron, including the initiating ATG (Figure S1B). Some transcript was still detected in dRASSFX16, dRASSFX36, but a strong reduction was found in dRASSF44.2, which lacks the transcription start (Figure S1C). However, antibodies raised against the C terminus (amino acids 792–806) and a nonconserved region (amino acids 294–308) of dRASSF showed that full-length dRASSF is absent in lysates from all mutant lines, suggesting our dRASSF mutants are indeed loss-of-function mutations for the locus (Figure S1D and data not shown). All of these alleles were viable and behaved identically in subsequent assays. In addition, dRASSF staining was severely reduced in FLP/FRT-generated dRASSF mutant clones in the eye-imaginal disc, the larval precursor to the adult eye (Figure S1E).
Although the dRASSF mutant flies are viable, they present a clear growth defect in comparison to wild-type animals when reared in carefully controlled conditions (Figure 1A). dRASSF mutant flies were 15% lighter than their wild-type counterparts (Figure 1D), a phenotype which was significantly rescued by introduction of a single copy of a dRASSF rescue construct, although wild-type levels of dRASSF were not fully restored (see Figure S1D). dRASSF mutant flies were fully fertile and normally proportioned (not shown) but sensitive to γ-irradiation (Figure S1F). Wing surface area was reduced by 8% in dRASSF mutant flies, whereas wing hair density was unaffected (Figures 1B, 1C, 1E, and 1F). This suggests that the growth defect of dRASSF mutant flies is due to a reduction in cell number and not a defect in cell size.
Figure 1.
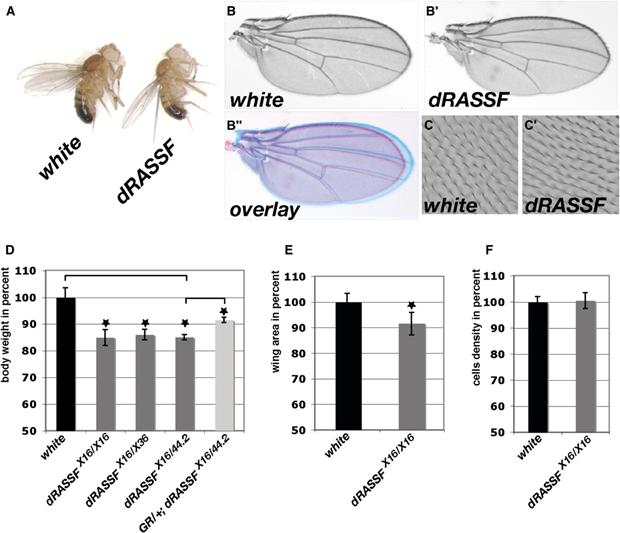
- (A) white and dRASSFX16/X16 adult flies showing that dRASSF flies present a size defect.
- (B–B″) dRASSFX16/X16 wings (B′) are smaller than white wings (B). (B″) Overlay of B and B′.
- (C and C′) Cell density is not affected in dRASSF wings. Phase-contrast image of wing hairs on the wing surface of white and dRASSFX16/X16 flies. Note identical hair densities, indicating normal cell size in mutant wings.
- (D) Histogram representing fly weights as percent of white control animals. dRASSF flies are 15% smaller than white flies. This weight defect is partially rescued in the presence of the genomic rescue construct (GR). *p < 0.05 (white n = 120, dRASSFX16/X16 n = 120, dRASSFX16/X36 n = 90, dRASSFX16/44.2 n = 90, GR;dRASSFX16/44.2 n = 90).
- (E) Histogram representing wing areas as percent of control (white) wings. dRASSFX16/X16 wings are 8% smaller than control wings. *p < 0.05 (white n = 14, dRASSFX16/X16 n = 11).
- (F) Histogram representing the number of trichomes in a defined wing area (see Supplemental Experimental Procedures) as a percent of control (white). No significant difference was found between control and dRASSFX16/X16 wings (p > 0.05). white, n = 430; dRASSF, n = 432. Error bars correspond to standard deviations.
In mammals, members of the RASSF family are known to interact with MST1 and thus to modulate its pro-apoptotic activity [7]. We therefore tested whether dRASSF can interact with Hpo. We performed coimmunoprecipitation (Co-IP) experiments in Drosophila Kc cells with dRASSF antibodies to immunoprecipitate endogenous protein. As expected, dRASSF robustly coimmunoprecipitated with Hpo (Figure 2A). The association between Hpo and Sav is mediated by these proteins' shared SARAH domains. Likewise, Hpo's SARAH domain is required for its association with dRASSF, as shown by the fact that a truncated form of Hpo (HpoΔC [18] lacking this domain fails to bring down dRASSF (compare Figures 2B and 2C). Thus, the Hpo SARAH domain can associate with both Sav and dRASSF.
Figure 2.
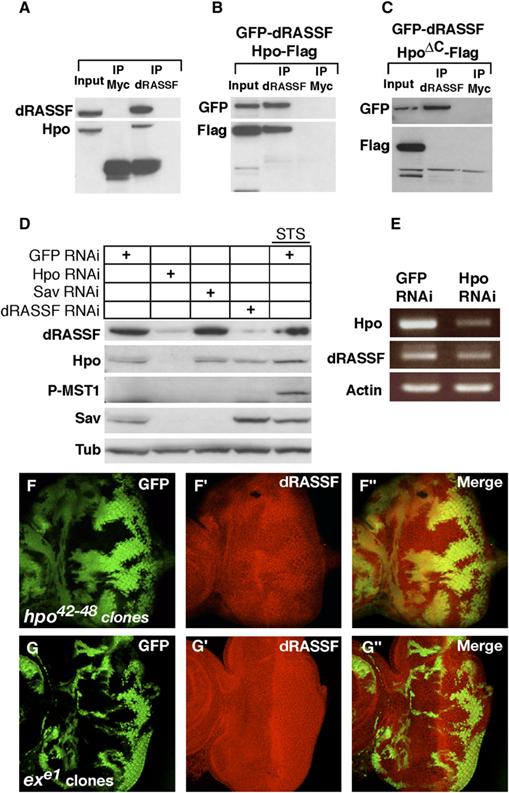
- (A) Hpo coimmunoprecipitates with dRASSF. Endogenous dRASSF was immunoprecipitated from Kc cells lysates with dRASSF antibodies or control (Myc antibodies). The Membrane was blotted with Hpo66 and dRASSF antibodies.
- (B) Hpo coimmunoprecipitates with dRASSF in Kc cells. Hpo-Flag and GFP-dRASSF were cotransfected in Kc cells. dRASSF or control (Myc antibodies) immunoprecipitates were blotted for GFP-dRASSF and Hpo-Flag.
- (C) Hpo lacking its SARAH domain (HpoΔC) does not interact with dRASSF. HpoΔC-Flag and GFP-dRASSF were cotransfected in Kc cells. Anti-dRASSF or control (anti-Myc) immunoprecipitates were blotted for GFP-dRASSF and Hpo-Flag.
- (D) Hpo controls dRASSF and Sav protein levels. Kc cells were treated with GFP, Hippo, Sav, or dRASSF dsRNAs. Lane 5 cells were treated with GFP dsRNA and 3 hr of Staurosporine (STS). Protein extracts were blotted with dRASSF, Hpo66, Sav, P-MST1, and tubulin antibodies. Hpo RNAi strongly reduces both Sav and dRASSF protein levels. dRASSF RNAi stabilizes Sav but is not sufficient to induce Hpo phosphorylation.
- (E) Hpo loss of function had no effect on dRASSF mRNA expression. RT-PCRs performed on Kc cell lysates treated with GFP or Hpo RNAi. Hpo, dRASSF, and Actin mRNA levels are shown.
- (F–F″) Hpo controls dRASSF protein levels in vivo. hpo mutant clones (marked by a lack of GFP) were generated in eye discs via the hpo42–48 allele. A robust reduction of dRASSF staining (in red [F′]) is observed in hpo clones.
- (G–G″) Ex does not affect dRASSF protein levels. Clones of ex mutant cells (marked by a lack of GFP) were generated in eye discs via the exe1 allele. dRASSF (in red [G′]) staining is unaffected in the clones.
Sav is stabilized by the presence of Hpo ([18, 19] and Figure 2D, lane 2). We therefore tested whether dRASSF levels are modulated by Hpo. dRASSF immunostaining was reduced in clones mutant for a hpo allele that lacks the SARAH domain (Figure 2F). In addition, RNAi-mediated depletion of Hpo from Drosophila Kc cells resulted in a reduction of endogenous dRASSF expression (Figure 2D), whereas dRASSF transcripts were unaffected (Figure 2E). By contrast, dRASSF levels were unaffected in clones mutant for other Hpo-pathway members, such as ex (Figure 2G), sav, and wts (Figures S2A and S2B). These results suggest that direct binding to Hpo through its SARAH domain, rather than signaling through the Hpo pathway, is necessary for dRASSF stability. This is analogous to the situation for Sav, which is also stabilized by a kinase-dead form of Hpo [18].
Because Hpo, Sav, and dRASSF all contain a SARAH domain, we speculated that dRASSF might also bind Sav. To test this, we investigated whether dRASSF interacts with Sav by co-IP but repeatedly failed to detect such an interaction (Figure S2C and data not shown). Because the possibility of a ternary complex had been raised by Scheel and Hofmann [13], we then tested whether the three proteins could be found in the same complex. We coexpressed Hpo, Sav, and dRASSF in cultured Kc cells. As expected, Hpo was able to bind Sav and dRASSF (Figure 3A). However, Sav immunoprecipitates only contained Hpo and not dRASSF, and dRASSF immunoprecipitates contained Hpo but not Sav (Figure 3A). We obtained identical results with endogenous IPs by using dRASSF and Sav antibodies (Figure S2C). These data support the notion that Sav and dRASSF are not present in the same complex but are in two different Hpo complexes.
Figure 3.
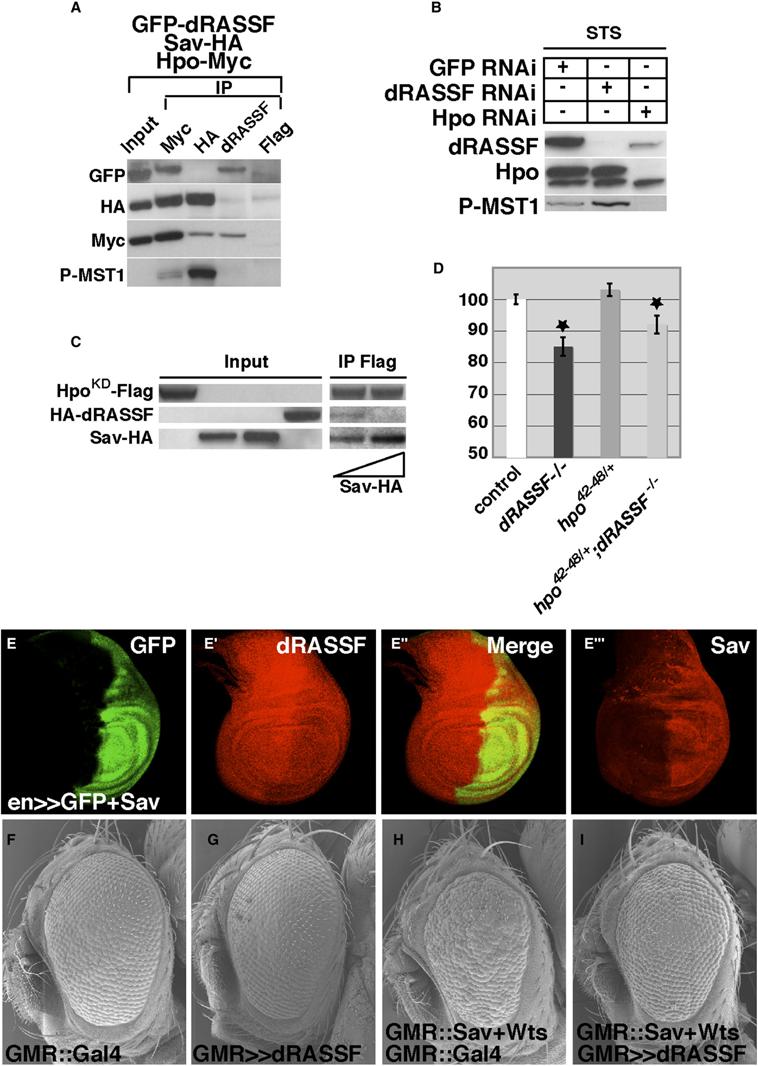
- (A) Hpo/Sav and Hpo/dRASSF are two distinct complexes. Hpo-Myc, Sav-HA, and GFP-dRASSF were cotransfected in Kc cells. Anti-Myc, anti-HA, anti-dRASSF, or control (anti-Flag) immunoprecipitates were blotted for GFP-dRASSF, Sav-HA, Hpo-Myc, and phospho-MST1. Hpo interacts with Sav and dRASSF (lane 2). Sav and dRASSF interact only with Hpo and not with each other (lanes 3 and 4). The Hpo fraction bound to Sav is highly phosphorylated (lane 3), and that bound to dRASSF is not (lane 4).
- (B) dRASSF inhibits Hpo phosphorylation. Kc cells were treated for 4 days with eGFP, dRASSF, or Hpo dsRNAi. The addition of STS 3 hr prior to lysis induced Hpo phosphorylation (lane 1). Western blots were probed with dRASSF, Hpo34, and phospho-MST1 antibodies. In the presence of dRASSF dsRNAi, Hpo phosphorylation increased (lane 2). As expected, the Hpo band disappeared upon Hpo dsRNAi treatment (lane 3).
- (C) Sav competes with dRASSF to bind Hpo. Kc cell lysates expressing, respectively, HpoKD-Flag, HA-dRASSF, Sav-HA (200 ng), and Sav-HA (400 ng) were mixed, incubated overnight, and immunoprecipitated with Flag antibody. Blots were probed with HA and Flag antibodies. Increasing the amount of Sav displaced dRASSF from HpoKD (compare lanes 5 and 6).
- (D) The dRASSF phenotype is sensitive to hpo loss of function. The histogram represents the total body weight as a percent of control flies (white). The reduction in body size in dRASSF flies can be partially rescued by removal of one copy of hpo (hpo42–48 allele). *p < 0.05 (white n = 80, dRASSFX16/X16 n = 80, FRT42D, hpo42–48/+ n = 80, and FRT42D, hpo42–48/+; dRASSFX16/X36 n = 80).
- (E–E″′) Sav controls dRASSF protein level. GFP (in green) and Sav were expressed in the posterior half of the wing disc by the engrailed-GAL4 (en-GAL4) driver. A robust reduction of dRASSF staining (in red [E′]) was observed in the en domain. [E″′] shows Sav overexpression in a separate disc.
- (F–I) dRASSF reduces apoptosis induced by sav and wts coexpression. Shown are scanning electron micrographs of Drosophila heads from GMR::Gal4 control animals (F) or from GMR::Gal4/UAS::dRASSF (G), GMR::Gal4;GMR::sav+wts (H), or (I) GMR::Gal4/UAS::dRASSF;GMR:: sav+wts (I). Overexpression of dRASSF inhibits the rough-eye phenotype generated by coexpression of Sav and Wts.
See Supplemental Experimental Procedures for exact genotypes. Error bars correspond to standard deviations.
Sav has been shown to be a positive regulator of the Hpo pathway, whereas our genetic results suggest that dRASSF might antagonize Hpo function. We were therefore interested in determining whether complexing with Sav or dRASSF might influence Hpo activity. We probed our immunoprecipitates with an phospho-MST1 antibody that recognizes phosphorylated (active) Hpo [20]. Interestingly, although Hpo that was coimmunoprecipitated with dRASSF showed barely detectable levels of phosphorylation, the Sav-associated fraction was highly phosphorylated (Figure 3A). Thus, Hpo can exist as two pools, a highly active Sav-associated pool and an inactive dRASSF-associated pool. This correlates with data showing that Nore1 can repress MST1 activity in mammalian cells [16]. This also suggests that Sav can promote Hpo activation and provides the first direct evidence of a function for the Hpo/Sav interaction.
Next, we wanted to test our prediction that dRASSF depletion would promote Hpo activation. Like that of Hpo's mammalian counterparts, phosphorylation of endogenous Hpo can be potently stimulated by the drug Staurosporine (STS) in Kc cells ([16, 20, 21] and Figure 3B, lane 1). Although RNAi depletion of dRASSF alone was not able to induce Hpo phosphorylation (Figure 2D, compare lanes 4 and 5), dRASSF depletion markedly potentiated STS-induced Hpo activation (Figure 3B, compare lanes 1 and 2). Thus, dRASSF restricts Hpo activation in cultured cells.
Given their opposing effects on Hpo activation, we investigated the relationship between Sav and dRASSF. Depletion of dRASSF in Kc cells gives rise to an increase in Sav protein levels (Figure 2D lines 1 and 4). Although dRASSF levels were unaltered in sav mutant clones (Figure S2A), overexpression of Sav in the wing disc results in a robust decrease of dRASSF staining (Figure 3E). We then tested whether dRASSF and Sav compete to bind Hpo. To address this question, because Sav and dRASSF repress each other's expression and dRASSF has reduced affinity for phosphorylated Hpo, we mixed separate Kc cell lysates expressing a kinase-dead form of Hpo (HpoKD-Flag), Sav-HA, and HA-dRASSF and performed IPs after the proteins were allowed to bind overnight. Both Sav and dRASSF were able to interact with Hpo (Figure 3C). In these conditions, increasing the amount of Sav was able to displace the dRASSF fraction bound to Hpo, showing that Sav and dRASSF are competing to bind Hpo. The outcome of the competition probably determines the stability of Sav and dRASSF; both proteins are downregulated when Hpo is depleted by RNAi (Figure 2D). Thus, we suggest that interplay between the inhibitor dRASSF and the activator Sav determines the level of Hpo activation and therefore affects body size.
We tested this model by performing genetic-interaction experiments. We crossed a mutant allele of hpo into the dRASSF mutant background and measured the adult body mass (Figure 3D). The body-mass reduction of dRASSF mutant flies (15% reduction) was substantially rescued by removal of just one copy of Hpo (8% reduction). Flies overexpressing Sav showed a reduction of 10% in weight and 5% in wing area, mimicking dRASSF loss of function (Figure S3). This wing defect was significantly increased in a dRASSF mutant background (Figures S3B–S3D). In addition, misexpression of dRASSF was able to robustly rescue the rough-eye phenotype elicited by coexpression of Sav and Wts (Figures 3F-3I). These data support the notion that dRASSF can antagonize Sav-mediated Hpo activation in vivo.
Though our results are consistent with biochemical data on mammalian RASSF family members [7, 16], they are at odds with the fact that RASSF genes are commonly silenced in tumor cells. Avruch and colleagues have proposed that one RASSF protein, Nore1, possesses a tumor-suppressor function that is independent of MST1 and MST2 [22]. We found two lines of evidence to support this notion. First, we made clones that are mutant for two hpo hypomorphic alleles, hpo42–48 ([19] compare Figures 4C and 4F), hpoKC203 ([23] not shown), that remove the SARAH domain in a dRASSF mutant background in the head by using the eyeless FLP system [24]. Interestingly, the overgrowth phenotype elicited by these hpo alleles was strongly enhanced by loss of dRASSF. By contrast, a hpo allele (hpo42–47 [19]) bearing an inactivating deletion in the kinase domain but an intact SARAH domain was barely if at all enhanced by dRASSF loss of function (Figures 4B and 4E). This suggests that dRASSF may possess a tumor-suppressor function, which may be uncovered when the Hpo function is compromised.
Figure 4.

- (A–C, E, and F) Scanning Electron Micrographs of Drosophila heads from control animals (A), animals bearing hpo42–47 clones (B), hpo42–48 clones (C), hpo42–47 clones in a dRASSF loss-of-function background (E), or hpo42–48 clones in a dRASSF loss-of-function background (F). The overgrowth phenotype elicited by the loss of hpo is enhanced by the removal of dRASSF. See Supplemental Experimental Procedures for genotypes.
- (D) Schematic representation of Hpo protein showing the different mutations used. The hpo42–47 allele causes a deletion of six amino acids in the kinase domain, and this deletion probably inhibits Hpo-ATP binding. The hpo42–48 allele is a deletion of 20 bp and gives rise to a premature stop codon. hpoKC203 changes G to A at the 5′ splicing site and the translation run into a stop codon in the intron.
- (G–H″) dRASSF rescues Ras1 loss of function. (G–G″) Ras1c40b clones (marked by a lack of GFP) are small. (H–H″) Rasc40b dRASSFX36 clones (marked by a lack of GFP) are larger than Rasc40b clones. dRASSF staining is in red (G′ and H′).
In addition, we examined the relationship between Ras1 and dRASSF because the mammalian RASSF proteins have all been shown to bind Ras proteins [8-10, 25].In Drosophila imaginal tissues, Ras1 mutant clones grow poorly and are eliminated by apoptosis ([26, 27] and Figure 4G). When we made double-mutant clones for Ras1 and dRASSF in the developing eye, we observed a substantial rescue of the growth defect observed in clones mutant for Ras1 alone (Figure 4H). This rescue of Ras loss of function was the result of both increased proliferation (Figure S4) quantified with phosphorylated Histone 3 staining and a reduction of apoptosis visualized with a cleaved-Caspase 3 antibody (Figure S5). Thus, dRASSF appears to antagonize Ras1 signaling in growth control, which is again suggestive of a “tumour-suppressing” effect distinct from its “oncogenic” role in opposing the Hpo pathway. However, Aoyama et al. suggest that NORE1 may also have both Ras- and MST-independent functions [22]. Future experiments will therefore be aimed at gaining a better understanding of the RASSFs' growth-restricting functions. The fact that the dRASSF mutations are viable might therefore reflect the facts that its ability to regulate the Hpo pathway may be redundant with other modes of regulation and that loss of dRASSF's tumor-suppressive activity is balanced by loss of its growth-promoting activity. O'Neill et al. have proposed that MST2 may be inactivated by binding to Raf-1. It will be interesting to determine whether this mode of regulation is redundant with RASSF [28].
In summary, we have generated mutant alleles of the sole Drosophila ortholog of the RASSF family of tumor suppressors. Surprisingly, dRASSF mutant flies are smaller than control flies. This growth defect can probably be ascribed in part to dRASSF's ability to antagonize Hpo signaling by competing with Sav for binding to Hpo. In addition, we have shown that dRASSF also possesses a tumor-suppressor activity, which is uncovered when hpo or Ras1 function is compromised. It will be interesting to investigate whether some mammalian RASSF proteins share these properties.
Supplementary Material
Acknowledgments
We thank S. Leevers, I. Hariharan, D. Pan, G. Halder, and the Bloomington Drosophila Stock Center for fly stocks. We are grateful to T. Gilbank, S. Murray, and F. Earl for transgenic generation, A. Weston at the London Research Institute EM facility for SEMs, and D. Andersen, J. Colombani, and F. Josué for comments on the manuscript. We thank J. Overton, B. Aerne, F. Josué, and J. Batut for technical help. Work in N.H.B's laboratory was supported by the Wellcome Trust grant 69943. Work in N.T.'s laboratory was supported by Cancer Research UK. C.P. is supported by a FEBS long-term fellowship.
Footnotes
Supplemental Data
Supplemental Data include Experimental Procedures and Five Figures and are available online at http://www.current-biology.com/full/16/24/2459/DC1/.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Edgar BA. From cell structure to transcription: Hippo forges a new path. Cell. 2006;124:267–273. doi: 10.1016/j.cell.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 3.Hergovich A, Stegert MR, Schmitz D, Hemmings BA. NDR kinases regulate essential cell processes from yeast to humans. Nat. Rev. Mol. Cell Biol. 2006;7:253–264. doi: 10.1038/nrm1891. [DOI] [PubMed] [Google Scholar]
- 4.Thompson BJ, Cohen SM. The Hippo pathway regulates the bantam microRNA to control cell proliferation and apoptosis in Drosophila. Cell. 2006;126:767–774. doi: 10.1016/j.cell.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 5.Nolo R, Morrison CM, Tao C, Zhang X, Halder G. The bantam MicroRNA is a target of the Hippo tumor-suppressor pathway. Curr. Biol. 2006;16:1895–1904. doi: 10.1016/j.cub.2006.08.057. [DOI] [PubMed] [Google Scholar]
- 6.Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122:421–434. doi: 10.1016/j.cell.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 7.Khokhlatchev A, Rabizadeh S, Xavier R, Nedwidek M, Chen T, Zhang XF, Seed B, Avruch J. Identification of a novel Ras-regulated proapoptotic pathway. Curr. Biol. 2002;12:253–265. doi: 10.1016/s0960-9822(02)00683-8. [DOI] [PubMed] [Google Scholar]
- 8.Vavvas D, Li X, Avruch J, Zhang XF. Identification of Nore1 as a potential Ras effector. J. Biol. Chem. 1998;273:5439–5442. doi: 10.1074/jbc.273.10.5439. [DOI] [PubMed] [Google Scholar]
- 9.Eckfeld K, Hesson L, Vos MD, Bieche I, Latif F, Clark GJ. RASSF4/AD037 is a potential ras effector/tumor suppressor of the RASSF family. Cancer Res. 2004;64:8688–8693. doi: 10.1158/0008-5472.CAN-04-2065. [DOI] [PubMed] [Google Scholar]
- 10.Vos MD, Ellis CA, Elam C, Ulku AS, Taylor BJ, Clark GJ. RASSF2 is a novel K-Ras-specific effector and potential tumor suppressor. J. Biol. Chem. 2003;278:28045–28051. doi: 10.1074/jbc.M300554200. [DOI] [PubMed] [Google Scholar]
- 11.Tommasi S, Dammann R, Jin SG, Zhang Xf XF, Avruch J, Pfeifer GP. RASSF3 and NORE1: Identification and cloning of two human homologues of the putative tumor suppressor gene RASSF1. Oncogene. 2002;21:2713–2720. doi: 10.1038/sj.onc.1205365. [DOI] [PubMed] [Google Scholar]
- 12.Dammann R, Li C, Yoon JH, Chin PL, Bates S, Pfeifer GP. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nat. Genet. 2000;25:315–319. doi: 10.1038/77083. [DOI] [PubMed] [Google Scholar]
- 13.Scheel H, Hofmann K. A novel interaction motif, SARAH, connects three classes of tumor suppressor. Curr. Biol. 2003;13:R899–R900. doi: 10.1016/j.cub.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 14.Agathanggelou A, Cooper WN, Latif F. Role of the Ras-association domain family 1 tumor suppressor gene in human cancers. Cancer Res. 2005;65:3497–3508. doi: 10.1158/0008-5472.CAN-04-4088. [DOI] [PubMed] [Google Scholar]
- 15.Pfeifer GP, Yoon JH, Liu L, Tommasi S, Wilczynski SP, Dammann R. Methylation of the RASSF1A gene in human cancers. Biol. Chem. 2002;383:907–914. doi: 10.1515/BC.2002.097. [DOI] [PubMed] [Google Scholar]
- 16.Praskova M, Khoklatchev A, Ortiz-Vega S, Avruch J. Regulation of the MST1 kinase by autophosphorylation, by the growth inhibitory proteins, RASSF1 and NORE1, and by Ras. Biochem. J. 2004;381:453–462. doi: 10.1042/BJ20040025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tommasi S, Dammann R, Zhang Z, Wang Y, Liu L, Tsark WM, Wilczynski SP, Li J, You M, Pfeifer GP. Tumor susceptibility of Rassf1a knockout mice. Cancer Res. 2005;65:92–98. [PubMed] [Google Scholar]
- 18.Pantalacci S, Tapon N, Leopold P. The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat. Cell Biol. 2003;5:921–927. doi: 10.1038/ncb1051. [DOI] [PubMed] [Google Scholar]
- 19.Wu S, Huang J, Dong J, Pan D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell. 2003;114:445–456. doi: 10.1016/s0092-8674(03)00549-x. [DOI] [PubMed] [Google Scholar]
- 20.Colombani J, Polesello C, Josue F, Tapon N. Dmp53 activates the Hippo pathway to promote cell death in response to DNA damage. Curr. Biol. 2006;16:1453–1458. doi: 10.1016/j.cub.2006.05.059. [DOI] [PubMed] [Google Scholar]
- 21.Glantschnig H, Rodan GA, Reszka AA. Mapping of MST1 kinase sites of phosphorylation. Activation and auto-phosphorylation. J. Biol. Chem. 2002;277:42987–42996. doi: 10.1074/jbc.M208538200. [DOI] [PubMed] [Google Scholar]
- 22.Aoyama Y, Avruch J, Zhang XF. Nore1 inhibits tumor cell growth independent of Ras or the MST1/2 kinases. Oncogene. 2004;23:3426–3433. doi: 10.1038/sj.onc.1207486. [DOI] [PubMed] [Google Scholar]
- 23.Udan RS, Kango-Singh M, Nolo R, Tao C, Halder G. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat. Cell Biol. 2003;5:914–920. doi: 10.1038/ncb1050. [DOI] [PubMed] [Google Scholar]
- 24.Newsome TP, Asling B, Dickson BJ. Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaics. Development. 2000;127:851–860. doi: 10.1242/dev.127.4.851. [DOI] [PubMed] [Google Scholar]
- 25.Vos MD, Martinez A, Ellis CA, Vallecorsa T, Clark GJ. The pro-apoptotic Ras effector Nore1 may serve as a Ras-regulated tumor suppressor in the lung. J. Biol. Chem. 2003;278:21938–21943. doi: 10.1074/jbc.M211019200. [DOI] [PubMed] [Google Scholar]
- 26.Diaz-Benjumea FJ, Hafen E. The sevenless signalling cassette mediates Drosophila EGF receptor function during epidermal development. Development. 1994;120:569–578. doi: 10.1242/dev.120.3.569. [DOI] [PubMed] [Google Scholar]
- 27.Prober DA, Edgar BA. Ras1 promotes cellular growth in the Drosophila wing. Cell. 2000;100:435–446. doi: 10.1016/s0092-8674(00)80679-0. [DOI] [PubMed] [Google Scholar]
- 28.O'Neill E, Rushworth L, Baccarini M, Kolch W. Role of the kinase MST2 in suppression of apoptosis by the proto-oncogene product Raf-1. Science. 2004;306:2267–2270. doi: 10.1126/science.1103233. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.