

Summary
What causes critical periods (CPs) to open? For the best-studied case, ocular dominance plasticity in primary visual cortex in response to monocular deprivation (MD), the maturation of inhibition is necessary and sufficient. How does inhibition open the CP? We present a novel theory: the transition from pre-CP to CP plasticity arises because inhibition preferentially suppresses responses to spontaneous relative to visually-driven input activity, switching learning cues from internal to external sources. This differs from previous proposals in (1) arguing that the CP can open without changes in plasticity mechanisms when, through circuit development, activity patterns become more sensitive to sensory experience; (2) explaining not simply a transition from no plasticity to plasticity, but rather the change in outcome of MD-induced plasticity,from pre-CP to CP,. More broadly, hierarchical organization of sensory-motor pathways may develop through a cascade of CPs induced as circuit maturation progresses from “lower” to “higher” cortical areas.
Introduction
Many brain circuits and functions across species exhibit developmental critical periods (CPs) or sensitive periods, during which they become sensitive to particular changes in experience or activity patterns (Hensch, 2004; Knudsen, 2004). Critical periods can occur as a sequence of sensitivities to increasingly more complex aspects of sensory or sensorimotor experience (Brainard and Doupe, 2000; Werker et al., 2009; Hernandez and Li, 2007; Scott et al., 2007), suggesting a corresponding developmental sequence of CPs from lower to higher brain areas. The question of the neural mechanisms underlying CPs is a fundamental one for more generally understanding both cortical development and the development of perceptual and motor abilities.
A paradigmatic and best-studied example of a CP is ocular dominance (OD) plasticity in primary visual cortex (V1) in response to visual deprivation of one eye (Wiesel and Hubel, 1963; Espinosa and Stryker, 2012). During the CP, but not before, monocular deprivation (MD) yields a strong shift in cortical responsiveness toward the non-deprived eye (OD plasticity) and an associated decrease in acuity of deprived-eye vision (Fagiolini et al., 1994; Prusky and Douglas, 2003). This shift involves an initial decrease of deprived-eye and later homeostatic increase of non-deprived-eye response (Mrsic-Flogel et al., 2007; Kaneko et al., 2008; Cho et al., 2009).
The opening of this CP is triggered by the maturation of cortical inhibitory circuitry (Hensch et al., 1998; Hanover et al., 1999; Fagiolini and Hensch, 2000; Iwai et al., 2003; Hensch, 2005; Di Cristo et al., 2007; Katagiri et al., 2007; Sugiyama et al., 2008; Kanold et al., 2009; Southwell et al., 2010). Mice in which inhibition remains immature, either due to a lack of the synaptic isoform of glutamic acid decarboxylase (GAD65-KO) or to dark-rearing from birth, fail to enter the CP (Fagiolini and Hensch, 2000; Morales et al., 2002; Iwai et al., 2003; Katagiri et al., 2007). However, focal administration in V1 of diazepam, a benzodiazepine agonist that enhances inhibitory transmission, initiates OD plasticity in these animals (Hensch et al., 1998; Fagiolini and Hensch, 2000; Iwai et al., 2003; Kanold et al., 2009). Similarly, wild-type (WT) mice enter a CP prematurely upon early diazepam treatment (Fagiolini and Hensch, 2000; Fagiolini et al., 2004) or molecular interventions that promote early GABA circuit development (Hanover et al., 1999; Di Cristo et al., 2007; Sugiyama et al., 2008).
How does maturation of inhibition initiate a CP? One possibility is that inhibition specifically “turns on” some form of plasticity. Inhibition has been suggested to alter plasticity rules by changing the composition of NMDA receptors (Kanold et al., 2009) and/or by enabling LTD (Choi et al., 2002). However, this issue remains controversial because activity-dependent plasticity like that seen in CP animals is also observed in juvenile animals before the CP (Fox, 1995; Kuhlman et al., 2010; Chun et al., 2013) and in GAD65-KO mice (Hensch et al., 1998; and see further evidence presented in Discussion). Alternatively, without sufficient inhibition, many input patterns might activate the postsynaptic cell equally well so that there is no activity-dependent competition between them. Inhibitory maturation might then intensify competition among input patterns to initiate the CP (Hensch 2005, Kuhlman et al., 2010). These proposals are all challenged by the fact that MD before the CP also induces plasticity. During the week before the CP, MD of the contralateral eye retards the ongoing development of precision in both eyes' retinotopic maps and decreases the magnitude of responses to each eye without inducing a significant OD shift, that is, without appreciable change in the relative response to the two eyes (Smith and Trachtenberg, 2007). Thus, it is the outcome rather than the existence of experience-dependent plasticity that changes at CP onset. The CP specifically reflects a time when OD and visual acuity through a deprived eye becomes sensitive to MD, whereas other aspects of visual development are sensitive to MD prior to the CP.
In this study, we use the CP for OD plasticity in V1 as a model system to propose the theory that CPs open when there is a switch in the predominant learning cues from internally driven spontaneous activity to externally driven evoked activity without changes in plasticity rules. Unlike all previous models of CP opening of which we are aware (Kuhlman et al., 2010; Kanold and Shatz, 2006; Hensch, 2005; Kanold et al., 2009; Choi et al., 2002; Yazaki-Sugiyama et al., 2009; Sugiyama et al., 2008), our theory explains not simply an opening of experience-dependent plasticity but the change in this plasticity that is associated with the transition from pre-CP to CP in V1. Furthermore, this transition can occur strictly through alterations in activity patterns, with no requirement for changes in the rules governing synaptic modification. We examine this theory using a computational model and test key predictions in freely behaving mice.
The basic workings of our theory can be understood purely conceptually:
Spontaneous activity is always equal between the two eyes, whereas visually-evoked activity is unequal if one eye is closed. Thus, if spontaneous input sufficiently predominates over visual in driving cortical activity, MD will not induce OD plasticity; while if visual input contributes sufficiently, OD plasticity can occur.
The maturation of inhibition can suppress the contribution of spontaneous relative to visually-evoked activity and thus open the CP for OD plasticity. Our experiments confirm that inhibitory maturation relatively suppresses spontaneous activity specifically when it causes CP onset.
Closing one eye greatly broadens the spatial correlations between inputs from that eye (Faguet et al., 2009). Broader correlations yield less retinotopic refinement of receptive fields (RFs) under Hebbian (correlation-based) mechanisms of synaptic plasticity. This slowing of retinotopic refinement also decreases the peak synaptic strengths of both eyes' relative to normal development under homeostatic plasticity. Thus, broadening of deprived-eye spatial correlations can explain the effects of MD during the pre-CP.
This conceptual theory leaves open the quantitative issue of whether (1) and (3) can occur simultaneously: that is, whether visual activity can contribute sufficiently during the pre-CP to slow retinotopic refinement and weaken both eyes' inputs, but not sufficiently to allow OD plasticity. We use simple computational and mathematical models to demonstrate that (1) and (3) can occur simultaneously and more generally that the theory can reproduce multiple aspects of both pre-CP and CP behavior. We examine the models to, determine the elements responsible for each aspect of the outcome, arriving at a robust conceptual picture of the key elements required to explain pre-CP and CP behavior independent of model details. The main behaviors predicted by our theory arise cohesively at three very different levels of description: the conceptual level; in a computational biological model; and in a simpler model analyzed mathematically (Supplemental Analysis, S1a). This supports the robustness of our findings, which we argue in the Discussion may apply to cortical development more generally.
Results
Modeling framework
Here we describe the main features of the model; full details and further discussion of parameter dependencies are in Supplemental Methods, S2a–c. We model a single pyramidal neuron in V1 that receives input from lateral geniculate nucleus (LGN) neurons driven by each eye (c.f. Fig. 1A). We neglect interactions between V1 cells because a simpler model suffices to understand how our theory can explain experimental observations. Each eye's inputs are uniformly spaced in a two-dimensional retinotopic space. The density of contralateral-eye input neurons is about twice that for ipsilateral-eye inputs, as in mice (e.g. Hensch et al., 1998; Hofer et al., 2006). We assume synapses from LGN are modified by Hebbian plasticity, which is driven by correlations between postsynaptic and presynaptic activity (Hebb, 1949; Markram et al., 1997; Bi and Poo, 1998) and therefore is sensitive to the correlation structure of the input (e.g., Miller 1990; Dayan and Abbott 2001; Gerstner and Kistler 2002), and by homeostatic plasticity, which regulates the overall activity level and stabilizes learning (Turrigiano et al., 1998; Maffei et al., 2004; Mrsic-Flogel et al., 2007; Ibata et al., 2008; Kaneko et al., 2008).
Fig. 1.

(A) Model architecture: a cortical neuron receives excitatory input from 28×28 contralateral inputs and 20×20 ipsilateral inputs It also receives inhibitory input from nearby cortical neurons, which are not explicitly modeled. The excitatory synaptic strengths are subject to activity-dependent plasticity. (B) Hypothesis of how the maturation of inhibition initiates the CP for OD. Visual input is twice as strong as, but only 1/10 as frequent as, spontaneous input. During the pre-CP, the spontaneous input drives the cortical cell, so the contribution of the rare visual input to plasticity is relatively small. At CP onset, maturation of inhibition subtracts equally from all responses, causing greater proportional weakening of spontaneous than visual input and shifting many responses to spontaneous input below the threshold for Hebbian plasticity. This makes visual input the primary driver of plasticity. (C) Input correlations (covariance) in simulations are modeled as Gaussian functions of the distance between input retinotopic positions. At a given distance, inputs from the same eye (“contra” or “ipsi”) are more correlated than those from opposite eyes (“between-eye”). Under normal rearing (NR), the visually-evoked covariance (gray curves) is four times as strong as, but retinotopically as precise as, the spontaneous input (blue curves). During MD to the contralateral eye, the closed-eye and between-eye visual covariances (red curves) are reduced in amplitude and are retinotopically broadened because of the blurring of the visual stimulus through the closed eyelid.
We model the firing rate of each input neuron as the sum of two Gaussian random variables: the spontaneous component and the visually-evoked component. The spontaneous component represents baseline input statistics in the absence of a visual stimulus, which by definition are unchanged by visual stimuli. We separately model these two components to demonstrate that acute enhancement of inhibition is sufficient to suppress the contribution to Hebbian plasticity of the spontaneous component relative to that of the visually-evoked component.
We assume that the spontaneous component is always active but relatively weak. The visually-evoked component contributes only 10% of the time, but when activated is twice as strong as the spontaneous component under normal-rearing (NR) conditions (c.f. Fig. 1B). The infrequent visual contribution instantiates the idea that visual stimuli with orientation, position, and other features appropriate to visually drive a given neuron are present only infrequently in natural stimuli (Vinje, 2000). For each component, pairs of inputs are correlated with a strength that decreases with their retinotopic separation and, for a given separation, is stronger for inputs from the same eye than from opposite eyes: (Ohshiro and Weliky, 2006; Weliky and Katz, 1999)(c.f. Fig. 1C). Under NR, spontaneous- and visually-evoked components have the same spatial and inter-ocular structure, with within-eye correlations twice as strong as between-eye. LGN spontaneous activity has strong between-eye correlations due to drive to LGN by cortical feedback (Weliky and Katz, 1999; Huberman et al., 2008). The identical spatial structure of the two components, assumed for simplicity, is supported by experiments finding almost identical shape (though not necessarily amplitude) of spatial covariance with or without visual stimulation in LGN (Ohshiro and Weliky, 2006) and V1 (Fiser et al., 2004). The structure of between-eye LGN correlations in visually-evoked activity has not been characterized and so was taken for simplicity to be the same as in spontaneous activity.
To model MD, in which the visual stimulus reaches the contralateral eye only after penetration through the closed eyelid, we assume the visual component of the closed-eye is blurred (Faguet et al., 2009) and weakened by half. Such blurring is known to influence V1 development (White et al., 2001; Akerman et al., 2002), and has been suggested to underlie the slowing of retinotopic development by MD but not monocular inactivation (MI) during the pre-CP (Smith and Trachtenberg, 2007). To model MI induced by pharmacologically inactivating retinal activity or enucleating one eye, we set the visual component from the inactivated eye to zero. We ignore MI-induced changes in spontaneous activity for simplicity, although MI alters burstiness of responses and increases correlations in LGN spontaneous activity (Weliky and Katz, 1999; Linden et al., 2009).
The experience-dependent refinement of the retinotopic map during the pre-CP was measured by intrinsic imaging as a decrease in map scatter. Scatter was defined as the average over pixels of the difference between a pixel and its near neighbors in their preferred retinotopic positions (Smith and Trachtenberg, 2007). To model this, we assume that decreased scatter corresponds to decreased RF sizes in single neurons. A larger RF will be flatter near its peak. In the presence of measurement noise, flatter RFs will produce larger map scatter. Therefore, we simply equated map scatter with RF size in the model. Of course, aligning RF peak locations among nearby cortical neurons may also reduce scatter. We ignore this factor because addressing it requires modeling multiple, recurrently connected cortical cells and perhaps plasticity of intracortical synapses. This introduces complexities and arbitrary modeling choices that are not needed to illustrate the workings of our theory. However, we note that, if intracortical interactions lead nearby RFs to receive correlated inputs, then broader spatial correlations in LGN inputs should yield more widely scattered RF peaks of nearby cells as well as larger RFs. Thus, if we were to model recurrent interactions and the movements of RF peaks, we would expect qualitatively the same effects on map scatter as we find in the simpler model examined here.
Modeling of a cortical neuron and plasticity rule
The firing rate y of the output neuron is taken to be a threshold-linear function of the inputs: . Here, []+ is a linear rectification function, [x]+=x if x≥0, =0 otherwise; wi≧0 is the synaptic strength from, and xi the firing rate of, the ith presynaptic neuron; and m is the strength of inhibition driven by a nonspecific pooling of neighboring neurons. The instantaneous activity averaged over neighboring neurons is approximated by the long-term averaged firing rate, , of the postsynaptic neuron, assuming spatially homogeneous average firing rates. For simplicity, we model the maturation of cortical inhibition as an immediate increase of the inhibitory strength m at the onset of the CP for OD (Di Cristo et al., 2007; Fagiolini et al., 2004; Katagiri et al., 2007; Kuhlman et al., 2010; Sugiyama et al., 2008). Instead, if a gradual increase of inhibition (Morales et al., 2002) were modeled, it would progressively open the CP by increasing the magnitude of the OD shift under MD with increasing inhibitory strength (e.g., see Fig. 3E for the steady-state OD shift vs. inhibition level).
Fig. 3.
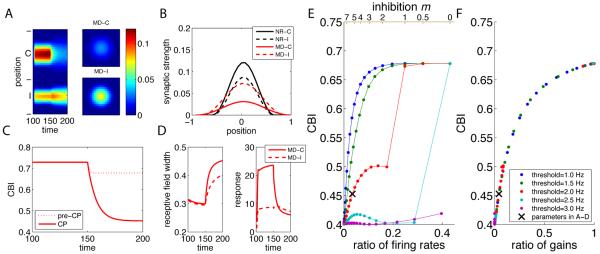
Model activity-dependent plasticity during the CP: activity-dependent retinotopic refinement and OD plasticity. The simulation was continued from the final state of Fig. 2 under NR and CP was modeled as mature inhibition (increase of m from 0 to 5 at time 100). (A) Development under NR to time 150 and MD, initiated at time 150. Now, MD causes a strong shift to the open (I) eye. Panels are as in Fig. 2A. Note, homeostatic plasticity rapidly increased synaptic strengths after inhibitory strengthening. (B) One-dimensional sections of the synaptic strengths at time 150 under NR (black) and at time 200 under MD (red) (solid: contra; dashed: ipsi). Conventions as in Fig. 2B. (C) Comparison of the time courses of the contralateral bias index (CBI) under MD for CP (m=5, solid-line) and pre-CP (m=0, dotted-line) conditions. Maturation of inhibition yields about a five-fold greater change in CBI. Note that the final CBI for the CP under MD indicates only slight ipsilateral advantage (CBI=0.45): the summed synaptic strength for each eye is similar, although I has stronger strengths, because of denser input from C. (D) The RF width and the response magnitude vs. time as in Fig. 2D–E (solid:C; dashed I). (E–F) Final CBI v.s. the spontaneous-to-visual ratio of firing rates (E) and the ratio of gains (F) under various levels of the inhibitory strength (varied from 0 to 10) and the Hebbian threshold (varied from 1 to 3 Hz; shown with different colors). Black crosses indicates parameters used in Panel A–D.
The instantaneous change in synaptic strengths, wi, is modeled as the sum of a Hebbian term and a homeostatic term. The Hebbian term is proportional to a measure of correlation of pre- and postsynaptic firing, ⟨xif(y)⟩ − a⟨xi⟩⟨f(y)⟩, where ⟨⟩ is a short-term average, a=1.001 is a constant slightly larger than 1 to force a competitive learning outcome by suppressing synapses with small input-output correlation, and f(y)= y-θ for y>θ, f(y)=0 otherwise. θ is a threshold level of postsynaptic activity required for Hebbian plasticity. We generally take θ=2 Hz but also consider varying θ. The homeostatic term is proportional to , with y0=1.2 Hz. Here, is the long-term average of the postsynaptic firing rate. This term scales synaptic strengths to bring towards the set-point firing rate y0. The proportionality to wi causes the homeostatic term to multiplicatively scale synaptic strengths. Such homeostatic scaling has been experimentally observed (Turrigiano et al., 1998; Stellwagen and Malenka, 2006) including in V1 of developing animals in vivo (Desai et al., 2002; Mrsic-Flogel et al., 2007; Kaneko et al., 2008).
Our choices of a thresholded input/output function (the []+ function in the expression for y) and subtractive inhibition ensure that inhibition suppresses spontaneous activity relative to visually-evoked activity, as illustrated in Fig. 1B. Subtraction of a fixed amount removes a greater proportion of the smaller spontaneous input than of the larger visual input and will also cause a greater fraction of spontaneous than of visual input to lie below the spiking threshold. Alternatively, inhibition might reduce all responses by an equal factor (e.g. Rothman et al., 2009). We choose a neuronal model in which inhibition changes the relative response strengths in order to explore our theory that such a change underlies CP opening. We will experimentally test the idea that inhibition acts in this way at CP opening. The function f in the plasticity rule further suppresses the contribution of weak spontaneous activity to Hebbian plasticity. We will explore how varying the threshold θ influences developmental outcome. The exact form of f is not important: e.g., f could be a smooth function that enhances Hebbian modifications at high postsynaptic firing rates instead of suppressing such modifications at low rates (see Supplemental Methods S2c).
Simulations of visual cortical development
We simulated pre-CP and CP plasticity under this model. Figure 2 shows activity-dependent development of retinotopy (as measured by RF size), OD, and response strength during the pre-CP, beginning at eye opening (time zero). The initial RF profile was chosen relatively broad. Under the NR condition (Fig. 2A Top; black lines in Figs. 2B–E), the neuron's RF narrowed (synaptic strength became more concentrated near the RF center, Fig. 2A,D) due to the spatial correlations of both the spontaneous and the visually-evoked activity components. This refinement is a typical outcome of competitive Hebbian plasticity, which tends to wire together a closely correlated group of neurons onto a given postsynaptic cell. Despite the retinotopic competition, there was relatively little between-eye competition, as reflected in the comparable strengths of the two eyes' final synaptic strengths (Fig. 2B). This was controlled by the relative magnitude of between-eye correlations vs. within-eye correlations for both the spontaneous and visually-evoked activity, with relatively weaker between-eye correlations yielding stronger inter-ocular competition. The contralateral bias index (CBI), which measures the bias toward the contralateral eye, reached slightly above 0.7 (Fig. 2C), primarily reflecting the 2X denser input from the contralateral eye (Gordon and Stryker, 2006).
Fig. 2.

Model activity-dependent plasticity during the pre-CP: activity-dependent retinotopic refinement without OD plasticity. In Figs. 2–3, C and I stand for contralateral and ipsilateral eye, respectively. (A) Left panels: Development of synaptic strengths (color) over time during the pre-CP under NR (Top) and MD (Bottom; C closed from time 0). Final strengths are insensitive to initial conditions, which are identical in the two cases. Upper and lower halves show C and I strengths, respectively, over a one-dimensional (1-D) section of retinotopic positions through the center of the 2-D RFs (note, there are more C than I input neurons). Right panels: final 2-D RFs of both eyes. Under NR, retinotopic refinement occurs with little OD competition. Under MD, there is less retinotopic refinement and both eyes' strengths weaken relative to NR, but again there is little OD competition. (B) 1-D sections of the final synaptic strengths at time 100 under NR (black) and MD (red) (solid: C; dashed: I). (C) Contralateral bias index (CBI: see Supplemental Methods, S2d), which ranges from 0 to 1 for responses varying from purely driven by I to purely driven by C; the CBI settles to 0.75 under NR (black) and 0.68, only slightly lower, under MD (red), reflecting denser input from C than I. (D–E) RF width (D) and the response magnitude (E) (defined in Supplemental Methods, S2d) vs. time. Solid lines: C; dashed lines: I; black: NR; red: MD; green, monocular inactivation (MI) of C.
Homeostatic plasticity stabilized the Hebbian plasticity, which otherwise was unstable. The multiplicative homeostatic scaling yielded a bell-shaped synaptic strength profile, because it more strongly suppressed stronger synapses. In contrast, stabilization by a hard upper bound on synaptic strengths pushes all synapses to near the maximal or minimal allowed values (e.g., Song et al., 2000; Miller and MacKay, 1994).
Under MD of the contralateral eye initiated at time zero during the pre-CP, the refinement of the RF was decreased compared to the NR condition (Fig. 2A–B,D) but with little change in OD (Fig. 2C). The refinement decreased due to the broadly correlated residual visual stimulus through the deprived eyelid (Faguet et al., 2009). Despite a dramatic change in the distance over which visual inputs were correlated, the RF widened only by about 30% (Fig. 2D), because the contribution of the frequent spontaneous input to plasticity was greater than that of the rare visually evoked input during the pre-CP.
The widening of the RF, in turn, reduced the maximum synaptic strength of each eye under the approximate conservation of total synaptic strength enforced by the homeostatic term (Fig. 2E). The total synaptic strength was roughly preserved because the reduction in input activity induced by MD was small given the dominant spontaneous input. MD did not induce much change in OD because the frequent input from spontaneous activity produced almost equal overall levels of input from both the deprived and open eyes (Fig. 2C). The CBI under MD was again about 0.7, despite similar synaptic strengths from both eyes, because of the denser input from the contralateral eye. Under MI (blockade of all visually-evoked activity) of the contralateral eye, the ipsilateral RF did not show the full widening and peak reduction seen under MD (Fig. 2D–E), qualitatively in agreement with experiments (Smith and Trachtenberg, 2007). This was due to the absence of the broadly-correlated residual visual activity that remains during MD, as suggested by (Smith and Trachtenberg, 2007).
Initiation of MD after the opening of the CP has a very different effect (Fig. 3). We continued the NR condition from Fig. 2, changing the inhibitory strength m from 0 to 5 at time 100 to represent CP opening. As discussed above (c.f., Fig. 1B), the increased inhibition suppressed the spontaneous input component more strongly than the visually-evoked component. Homeostatic plasticity compensated for the inhibition-induced loss of activity by scaling up synaptic strengths to approximately preserve the long-term average of the postsynaptic firing rate, largely maintaining the reduced spontaneous-to-visual activity ratio. The reduced ratio, together with the threshold that suppresses Hebbian modifications under small postsynaptic activity, reduced the contribution to plasticity of spontaneous activity relative to visually-evoked activity, rendering the neuron more sensitive to changes in visual statistics. As a result, MD, initiated at time 150, caused a strong OD shift toward the open (ipsilateral) eye (Fig. 3A–C), as well as greater broadening of the ipsilateral synaptic strength profile than was seen during the pre-CP (deprived-eye RF width decreased by about 50%, Fig. 3B,D; see Discussion).
The amount of OD shift depended on parameters – the strength of inhibition and the Hebbian threshold – in a seemingly complex way, but underlying this is a simple dependence of the OD shift on the degree to which visually-evoked vs. spontaneous activity contributed to Hebbian plasticity. Figure. 3E plots the final CBI after MD as a function of the spontaneous-to-visual ratio of the mean firing rate (the rate without visual input divided by the rate with visual input) at various levels of inhibition and of the Hebbian threshold. For any given threshold, a lower spontaneous-to-visual ratio of firing rates, (due to stronger inhibition) increased the OD shift. As threshold increased, the ratio of firing rates that could produce a strong OD shift increased. This is because the spontaneous component did not contribute to plasticity when its rate fell below the Hebbian threshold (c.f. Fig. 1B).
The many curves for different thresholds in Fig. 3E all collapse onto a single curve if the CBI is plotted against the spontaneous-to-visual ratio of the gains for Hebbian plasticity (Fig. 3F; see Supplemental Methods, S2c,d for a mathematical definition of the gain). Under the current set of parameters, the gain is equivalent to the frequency of events that drive Hebbian plasticity, i.e., the percentage of time the output firing rate exceeds the Hebbian threshold. That is, across parameters, the CBI is simply determined by the relative contributions of spontaneous and visual activity to Hebbian plasticity. This is the key, robust prediction of the model. A mathematical analysis of the Hebbian component also confirms that this is the key quantity for predicting the magnitude of the OD shift (see Supplemental Methods, S2c).
In Supplemental Analysis, Section S1 and Fig. S1, we present a simplified mathematical model for which we can obtain an analytic expression for the weights that develop. In the simplified model, postsynaptic activity is a simple linear sum of the weights times the input rates, and Hebbian plasticity is proportional to the covariance of pre- and postsynaptic activity without the nonlinearity used above and with a=1. The homeostatic plasticity rule and the statistics of excitatory input to the cortical neuron are unchanged. Rather than modeling subtractive inhibition, spike threshold, and Hebbian threshold as above, we simply assume that, in the absence of visual drive, postsynaptic activity is suppressed a certain fraction of the time by inhibition, where this fraction increases with inhibitory strength. This simpler model reproduces the main results of our theory: in the pre-CP, MD yields broader RFs and decreases both eyes' inputs strengths with minimal change in OD; while in the CP, MD yields strong OD shifts (and also broader RFs). The analytical expression for the weights shows that these are all robust outcomes, with one exception. The decrease in both eyes' weights induced by MD in the pre-CP involves a tradeoff between the decreased input activity, which reduces the weights overall; homeostatic plasticity, which increases the weights; and the spatially broader input activity, which spreads the weights and thus reduces the peak weight under homeostatic plasticity. Thus, relatively strong reduction in input activity, relatively weak homeostasis, and relatively broad correlations all favor reduction in the two eyes' weights, while with different sets of these three choices the open eye's weights or both eye's weights can instead be increased by MD in the pre-CP. The model also predicts that the final steady-state of the synaptic strengths is insensitive to the initial synaptic strengths, which was generally true in the previous simulations. The ability of the simple model to reproduce the results demonstrates their overall robustness, showing that the results depend on the overall framework of Hebbian plus homeostatic plasticity and an inhibition-induced reduction in the spontaneous-to-visual activity ratio, but not on the details of how inhibition causes this reduction.
Spontaneous-to-visual activity ratio in freely behaving mice
We have shown that a suppression of the spontaneous-to-visual activity ratio induced by the maturation of inhibition is sufficient to explain the transition from pre-CP to CP plasticity. To test the prediction that such suppression occurs, we recorded extracellularly with head-mounted tetrodes from putative pyramidal cells in V1 of freely behaving adult mice (Supplemental Methods, S2e). We studied both WT and GAD65-KO animals. The GAD65-KO mouse models a pre-CP state and will enter a CP even in adulthood upon chronic strengthening of inhibition by administration of diazepam (Fagiolini and Hensch, 2000). We also studied adult GAD65-KO mice that had previously been exposed to chronic diazepam for six days around P28 (KOc), which had caused them to enter and then exit the CP. These mice, like adult WT mice, are in a post-CP state in which plasticity is no longer triggered by administration of diazepam (Fagiolini and Hensch, 2000; Iwai et al., 2003).
Acute strengthening of inhibition by diazepam lowered the spontaneous-to-visual activity ratio specifically in those animals in which this treatment, when maintained over days, opens the CP. We measured both sensory-evoked responses driven by LED flashes and baseline firing rates (Fig. 4A, Fig. S2). Average firing responses to LED flashes of typical multi-unit activity from fully mature WT and GAD65-KO mice are shown in Fig. 4B before and after acute administration of diazepam (see Fig. S2 for firing rates of all isolated recorded cells under all treatment and stimulus conditions). We then computed the spontaneous-to-visual ratio of firing rates (baseline firing rate divided by LED-evoked firing rate; Fig. 4C). In agreement with the model's prediction, acute enhancement of inhibitory transmission by diazepam suppressed the spontaneous-to-visual ratio in GAD65-KO mice (p=0.002 Mann-Whitney test; 9 mice, n=68 cells), in which diazepam opens the CP (Fagiolini and Hensch, 2000). Surprisingly, diazepam did not alter the spontaneous-to-visual ratio in animals in which diazepam no longer opens plasticity (Iwai et al., 2003), neither in adult WT mice (p=0.6 Mann-Whitney test; 3 mice, n=21 cells) nor in KOc mice (p=0.1 Mann-Whitney test; 2 mice, n=18 cells).
Fig. 4.

Tetrode recording from freely behaving adult mice. Spontaneous-to-visual activity ratio is a physiological correlate of OD plasticity as predicted by the model. (A) Experimental setup: periodic LED flashes (1 sec) were presented to freely behaving adult mice while putative pyramidal cells were recorded from V1. (B) Average firing response to LED flashes of typical multi-unit activity from WT (top) and GAD65-KO mice (bottom) before (left) and after (right) acute administration of diazepam. Green bars mark stimulus duration. (C) Summary of the spontaneous-to-visual ratio of firing rates before and after acute administration of diazepam (+Dz) in different mouse groups. Diazepam decreased the spontaneous-to-visual ratio of GAD65-KO mice but did not significantly change the ratios of adult WT mice or GAD65-KO mice chronically treated with diazepam earlier in life (KOc). Box-and-whisker plots show 25th to 75th percentiles (box), median (red line in box), the range of data falling outside the 25th to 75th percentiles by up to 1.5 the distance from 25th to 75th percentile (whiskers) and outliers outside this range (red crosses). (D) Cumulative distribution of spontaneous-to-visual ratios of firing rates of various mouse groups. Ratios of WT mice were smaller than those of GAD65-KO mice. Acute diazepam decreased the ratio of GAD65-KO mice far beyond adult WT levels. In KOc mice, the visual response ratio had returned to a level similar to naïve GAD65-KO mice, but no longer responded acutely to the drug as in adult WT mice (C).
To compare activity ratios across different animal groups, additional cohorts were recorded without acute diazepam treatment (c.f., Fig. S2, showing all firing rates for all animals in all conditions). In Fig. 4D, the cumulative distributions of the spontaneous-to-visual ratios of firing rates are shown for different mouse groups. WT mice (11 mice, n=66 cells) exhibited significantly smaller ratios than the GAD65-KO mice (p=0.003, Mann-Whitney test; KO, 21 mice, n=112 cells), consistent with the pre-CP state of the GAD65-KO mice. Acute administration of diazepam in the GAD65-KO mice (9 mice, n=68 cells) reduced their ratios to levels smaller than in adult WT mice (p=0.03, Mann-Whitney test). WT mice also showed significantly smaller ratios than in KOc mice (p=0.02, Mann-Whitney test; KOc, 21 mice, n=127 cells), whose ratios were not significantly different from those of naïve GAD65-KO mice (p=0.6, Mann-Whitney test). Thus, diazepam only transiently suppresses ratios in GAD65-KO mice to open the CP, after which the ratios in KOc animals return to their pre-CP levels. This finding was unexpected, but could reflect the enduring weak inhibition due to the persistent lack of GAD-65.
One caveat to these results is that we have somewhat overestimated the spontaneous-to-visual ratio. We over-estimated spontaneous activity, which we took as the baseline activity in the absence of LED stimulation. Animals experience natural vision under these conditions, so infrequently effective visual drive was added to the spontaneous. Moreover, an LED flash is not an optimal visual stimulus for visual cortical cells, i.e. we underestimated visually-evoked activity. However, assuming linear addition of LED-evoked and other visual responses to spontaneous firing rates, the spontaneous-to-visual ratio estimated here shows a monotonic relation to the underlying true ratio (see Supplemental Analysis, S1b). Our primary aim is to make qualitative rather than quantitative predictions, due to the simplicity of our models. Nonetheless, the measured ratio changes, corrected for overestimation using reasonable parameter values, and applied to the simple plasticity model we have presented here, account quantitatively for the changes in plasticity in the majority of cells (Fig. S3).
Discussion
In this study, we have presented a novel hypothesis for the origin of CPs that explains the diverse effects of visual plasticity in a unified manner. The transition from developmental plasticity seen prior to and during the CP in V1 arises through inhibition-induced shifts in cortical activity, with no additional requirement for changes in plasticity rules. The key idea is that the maturation of inhibition that initiates the CP reduces the ratio of spontaneous-to-visual activity. In simulations and analysis, we showed that changes in the ratio suffice to account for defining features of pre-CP and CP plasticity: MD during the pre-CP retards retinotopic refinement of RFs and weakens both eyes' inputs with little OD change (Smith and Trachtenberg, 2007); MD during the CP produces a large OD change (Gordon and Stryker, 1996). The work supports the more general idea that CPs may open when the dominant learning cue switches from internal to external sources, without requiring changes in plasticity rules.
In our extracellular recordings from mature, awake behaving mice, we directly tested the hypothesis that the maturation of inhibition that opens the CP decreases the spontaneous-to-visual activity ratio. Consistent with our theory, diazepam potently decreased the spontaneous-to-visual activity ratio in naïve GAD65-KO mice, in which it opens the CP (Fagiolini and Hensch, 2000). Diazepam did not alter this ratio in WT or KOc animals, in which the CP had already opened and closed (Iwai et al., 2003). Furthermore, animals entering the CP -- GAD65-KO mice treated with diazepam -- exhibited significantly lower spontaneous-to-visual activity ratios than either pre-CP mice (untreated GAD65-KO mice) or post-CP mice (WT or KOc).
We are proposing that maturation of inhibition induces a transition in the dominant input driving cortical activity, from spontaneous to visually-driven. This fits with a more general picture that brain circuits self-organize based on innate activity and molecular cues during an early stage and more finely tune later in development according to stimuli from the external world (Katz and Shatz, 1996; Feller and Scanziani, 2005), but with the caveat that experience also contributes during the pre-CP. Before the CP, spontaneous activity in V1 and its inputs plays fundamental roles in the cortical acquisition of retinotopic organization (Cang et al., 2005; Cang et al., 2008), synaptic strengthening and pruning (Hooks and Chen, 2006), OD segregation (Huberman et al., 2006), and orientation selectivity (Fregnac and Imbert, 1984; Chapman and Stryker, 1993; Crair et al., 1998; Chapman and Gödecke, 2000; White et al., 2001, Fagiolini et al., 2003). After CP opening, the heightened sensitivity of visual development to sensory experience is manifest not only in the opening of OD plasticity, but also in the dependence on visual experience of (1) the maintenance and further development of orientation selectivity, which in the pre-CP are independent of experience (Fregnac and Imbert, 1984; Crair et al., 1998; White et al., 2001) and (2) at least in rodents, the binocular matching of orientation preference, which occurs during the CP (Wang et al., 2010). Spontaneous activity plays a similar role in the organization of tonotopic maps in the auditory system before the onset of hearing (Leao et al., 2006; Moody and Bosma, 2005; Tritsch et al., 2007).
Mechanisms of CP initiation
No previous model of CP induction has addressed the fact that MD-induced plasticity is present before the CP, but differs in quality from CP plasticity. The alternative models that bear the closest resemblance to ours also suggest that inhibition opens the CP by changing activity patterns, but focus on changes that could open inter-ocular competition under spike-timing dependent plasticity (STDP). Kanold and Shatz (2006) argued that excess spontaneous activity of cortical origin (as opposed to activity driven by the spontaneous component of LGN input, as we consider) will decorrelate postsynaptic spiking from presynaptic inputs, causing STDP to strengthen less active inputs relative to more active inputs. Reduction of cortical spontaneous activity by inhibition causes STDP to instead strengthen more active relative to less active inputs, allowing the OD shift caused by MD. The model requires fine tuning of the level of pre-CP cortical spontaneous activity so that pre-CP MD causes no change in OD. Kuhlman et al (2010) argued that IPSPs follow EPSPs with a short time delay, so that stronger inhibition tightens the temporal window within which inputs must fire together to drive postsynaptic spikes and yield potentiation under STDP. The more temporally coherent, visually-driven open-eye inputs would therefore have an advantage over more temporally dispersed deprived-eye inputs. Inhibitory maturation would increase this advantage sufficiently that the initially weaker open eye could out-compete the deprived eye. Hensch (2005) suggested that weak inhibition could prevent interocular competition by allowing weaker as well as stronger inputs to drive backpropagating action potentials and so be strengthened by STDP.
There have been many suggestions that molecular changes in plasticity mechanisms open the CP. These proposals again would not account for the transition from pre-CP to CP plasticity, but this does not preclude contributions of molecular mechanisms. Gene and protein expression patterns develop in a manner dramatically dependent upon visual input (Tropea et al., 2006; Majdan and Shatz, 2006; Lyckman et al., 2008). Several studies suggest that plasticity rules do not change during the pre-CP to CP transition (Fox, 1995; Hensch et al., 1998; Kuhlman et al., 2010; Chun et al., 2013), as we have assumed, but others report changes in plasticity rules (Jiang et al., 2007).
One molecular proposal focuses on the switch in the predominant subunit composition of NMDA receptors from NR2B to NR2A, which occurs over the first five postnatal weeks and is boosted within hours by visual experience and attenuated or reversed by visual deprivation (Flint et al., 1997; Quinlan et al., 1999a, 1999b; Roberts and Ramoa, 1999). It has been argued that inhibitory maturation might open the CP by triggering this change in NMDA receptors: GAD65-KO mice show reduced NMDA receptor subunit NR2A levels and correspondingly slower NMDA currents, which are rescued by diazepam application (Kanold et al., 2009). However, while NR2A may modulate OD plasticity (Fagiolini et al., 2003; Cho et al., 2009), it is not required for opening or normal closing of the CP (Fagiolini et al., 2003).
There have been conflicting reports about cortical LTD in GAD65-KO mice. One study reported a defect of cortical LTD in these animals that could be rescued by chronic diazepam (Choi et al., 2002). However, our earlier studies (Hensch et al., 1998) found clear synaptic depression following low-frequency stimulation (LFS) in the same GAD65-KO mouse line of Kash et al. (1997) studied by Choi et al. 2002. Failure to observe LTD in these fragile animals could be an artifact of poor general nutrition or stress due to housing or slice recording conditions (Artola et al., 2006; Kash et al., 1997; Nishie et al., 2012).
To resolve this issue, we have revisited it in a second GAD65-KO mouse line (of Asada et al., 1996) using a two-pathway paradigm (Kirkwood and Bear, 1994; Renger et al., 2002) in which standard V1 coronal slices from GAD65-KO mice were cut vertically from white matter to layer 4 (Supplemental Methods, S2f). One `test' input is first determined to be independent of a `control' input across the cut by linearly additive responses and a lack of paired-pulse interactions when stimuli are applied at short intervals (< 50 msec) to each side. The test pathway showed clear LTD in response to LFS, while the unconditioned control input remained stable (Fig. S4A). This confirms both the independence of the two pathways and the good health of our slice preparations for the duration of the recording. In a separate series of experiments, repeated LFS to one pathway saturated LTD at comparable levels in WT and GAD65-KO mice (Fig. S4B). Thus, our data now confirm robust LTD in two lines of GAD65-KO mice.
Other factors might play a role in CP initiation. In this study we focus on plasticity of synapses from excitatory feed-forward inputs onto excitatory cells, but several recent reports also suggest a role for rapid plasticity of excitatory synapses onto inhibitory neurons (Gandhi et al., 2008; Yazaki-Sugiyama et al., 2009; Kameyama et al., 2010) and inhibitory output plasticity (Maffei et al., 2006). Further studies are required to determine the contributions made by each of these factors and the dependencies between them. While multiple mechanisms may potentially contribute to the range of phenomena in pre-CP and CP plasticity, we find it striking that the model we propose can account for both stages in a simple, unified way.
Experimental Tests
The most direct test of the mechanism we propose would involve directly manipulating the spontaneous-to-visual activity ratio. If this can switch plasticity between pre-CP and CP regimes without altering other hypothesized plasticity factors, it would demonstrate both that our proposed mechanism is sufficient and that alternative factors are not necessary to explain this shift. This might be achieved by optogenetic techniques (Knöpfel et al., 2010; Wyatt et al., 2012) or by genetically engineered reduction / increase in potassium currents to increase / decrease spontaneous activity, respectively (Mizuno et al., 2007).
Correlative tests could be performed by measuring activity patterns across experimental manipulations that change plasticity regime and / or measuring plasticity regime across manipulations that alter activity patterns. This would most directly relate to our theory if conducted before or during the animal's initial CP. While reopening of the CP in adult animals might occur by different mechanisms than initial CP opening, as further discussed below, it would also be interesting to examine spontaneous-to-visual activity ratio in a variety of paradigms of adult CP reactivation (Morishita and Hensch, 2008; Bavelier et al., 2010). These include inhibitory precursor cell transplants (Southwell et al., 2010), peri-neuronal net removal or Otx2 reduction (Beurdeley et al., 2012), Nogo receptor deletion (McGee et al., 2005), caloric restriction (Spolidoro et al., 2011), dark exposure, fluoxetine administration, and environmental enrichment (Morishita and Hensch, 2008; Bavelier et al., 2010). A particularly tractable example is the impact of cholinergic enhancement, which has long been appreciated to improve neuronal signal-to-noise (Sato et al., 1987; Parkinson et al., 1988), and which rescues amblyopia in adulthood (Morishita et al., 2010).
Our model predicts that OD shifts should be observed during the pre-CP if LGN input from one eye were mostly blocked, as this would eliminate both visually-evoked and spontaneous input from that eye. (Note that blockade of activity in one eye, rather than in LGN, is unlikely to suffice, as substantial spontaneous activity remains in the inactivated eye's LGN layers; Weliky and Katz 1999). Such OD plasticity in the pre-CP would demonstrate that spontaneous activity in the LGN is preventing an OD shift during the pre-CP and that developmental changes in other factors from pre-CP to CP are not necessary for OD plasticity. Another prediction is that dark-reared animals, in which CP onset (Fagiolini et al., 1994) and the maturation of inhibition (Morales et al., 2002; Katagiri et al., 2007) are delayed until light exposure, should have larger, pre-CP-like spontaneous-to-visual ratios that should rapidly reduce to CP-like levels upon visual experience. Consistent with this, previous experiments (Benevento et al., 1992; Gianfranceschi et al., 2003) reported larger spontaneous-to-visual activity ratios in dark-reared animals than in age-matched controls.
Points the Theory Does Not Address
We do not address the mechanisms through which inhibition suppresses the spontaneous-to-visual activity ratio in some circumstances but not others. In GAD65-KO mice, in which many neurons showed low baseline firing rates after acute diazepam administration (Fig. S2), our suggestion that subtractive inhibition and spike threshold cause suppression seems likely. It is possible that the operating mode of parvalbumin-positive basket cells that initiate the CP (Hensch, 2005) might subsequently change so that inhibition mainly divides input after the CP (Atallah et al., 2012). In any case, our theory only requires that the maturation of inhibition that starts the CP suppress the ratio, as we observed experimentally, regardless of mechanism.
Due to suppression of spontaneous input during the CP, our model predicts that the increase of RF size in MD animals relative to NR animals is much stronger during the CP than in the pre-CP. Such relative expansions of RF size are experimentally observed after long-term (Pizzorusso et al., 2006) but not brief (Gordon and Stryker, 1996) MD. The simple learning rule we used in this paper may not capture the time-course of changes in RF size, which may involve mechanisms beyond those we modeled such as anatomical changes. These may have much longer time courses than our simple model predicts.
Implications for Adult Plasticity and Disease
Our theory addresses only the opening of the CP. It does not address its ending, to which many factors may contribute (Bavelier et al., 2010; Hensch, 2005; Morishita et al., 2010; Harauzov et al., 2010; Pizzorusso et al., 2006; Maya Vetencourt et al., 2008; Southwell et al., 2010). As such, it does not make predictions about the evolution of the spontaneous-to-visual ratio after CP opening. Nonetheless, the fact that this ratio was increased in post-CP animals relative to CP animals raises the possibility that and increased ratio could be one factor in CP closure. The fact that acute inhibitory strengthening did not lower the ratio in post-CP animals, in which inhibitory strengthening does not open a CP, is also consistent with this idea.
Evidence that inhibition can alter the spontaneous-to-visual activity ratio, which in turn can alter plasticity, comes from neurological disorders. Mice experiencing early hypoxia-ischemia display reduced parvalbumin expression, increased spontaneous-to-visual ratio (due to smaller visual responses), and less OD plasticity (Failor et al., 2010). In senescent monkeys, the ratio decreases under administration of GABA or muscimol and increases under bicuculline (Leventhal et al., 2003). Rett syndrome may provide a particularly informative case study. In the MeCP2-KO mouse model, there is virtually no spontaneous activity in V1 (Dani et al., 2005) and correspondingly low spontaneous-to-visual activity ratios, relative to WT animals, as well as parvalbumin cell hyper-connectivity and a loss of visual acuity (Durand et al., 2012). These results are suggestive that hyper-inhibition and too low a spontaneous-to-visual ratio can also be deleterious to normal developmental plasticity. Notably, this ratio, parvalbumin cell connectivity and visual acuity can all be restored to CP levels by dark-rearing or by genetic deletion of NR2A receptors (Durand et al., 2012). Thus, optimization of spontaneous-to-visual activity ratio may be essential for defining different CP stages. Artificially adjusting spontaneous firing rates might then offer potential therapeutic or learning strategies when specific types or properties of developmental plasticity are desirable.
General Implications for Cortical Development
Many sensory and sensorimotor systems exhibit sequences of critical or sensitive periods that progress from simpler to increasingly more complex aspects of experience (Cang et al., 2005; Hensch, 2004; Brainard and Doupe, 2000; Werker et al., 2009; Hernandez and Li, 2007; Scott et al., 2007). In the auditory system, for example, a cascade of CPs induces plasticity for tonotopy, bandwidth tuning, binaural integration, directionality, phoneme discrimination, audio-visual matching, and semantic and syntactic development (Barkat et al., 2011; Insanally et al., 2009; Popescu and Polley, 2010; Werker et al., 2009; Robinson, 1998; Lenneberg, 1967). An appealing hypothesis is that these sequences of CPs may correspond to sequences of reductions in the spontaneous-to-evoked activity ratio along hierarchies of cortical areas (Felleman and Van Essen, 1991; Sharpee et al., 2011), switching the factors most strongly driving learning in each area from internally to externally generated (Tritsch et al., 2007; Moody and Bosma, 2005; Katz and Shatz, 1996; Hooks and Chen, 2006). Sequences of reductions in activity ratio may in turn involve sequential maturation of key GABA circuits (Condé et al., 1996), which might arise through sequential activity-dependent maturation of peri-neuronal nets. These nets induce maturation of inhibition upon binding Otx2 (Beurdeley et al. 2012), which is delivered both from retina to LGN to V1 (Sugiyama et al., 2008) and across cortex from the choroid plexus (Spatazza et al., 2013). It will be interesting to test whether a cascade of reduced spontaneous-to-evoked ratios across areas coincides with cascades of CPs, whether maturation of inhibition is implicated both in these CP onsets and in corresponding ratio changes, and whether postponement of these ratio changes postpones CPs.
In sum, the co-occurrence of a CP with the switching of the dominant learning cue from internal to external may be a general principle governing brain development.
Supplementary Material
Acknowledgments
We would like to thank M. Eisele and X. Pitkow for helpful discussions and C. Yokoyama for comments on the manuscript. T. T. was supported by The Japan Society for the Promotion of Science (JSPS), Grant-in-Aid 1806772 for JSPS fellows, The Robert Leet and Clara Guthrie Patterson Trust Postdoctoral Fellowship, Bank of America, Trustee, The Special Postdoctoral Researchers Program of RIKEN, and RIKEN Brain Science Institute. The Japan Science and Technology Agency supported H.M. (PRESTO), Y.Y-S. and T.K.H. (CREST). N.A. and T.K.H. were further supported by RIKEN Brain Science Institute. K.D.M. was supported by R01-EY11001 from the NEI and by the Gatsby Charitable Foundation through the Gatsby Initiative in Brain Circuitry at Columbia University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
T.T. and K.D.M. developed the theory in discussion with Y.Y-S. T.T. simulated the model. H.M. and T.K.H. designed the tetrode experiment. H.M. recorded the data. T.T. and H.M. analyzed the data. N.A. provided the two pathway LTD experiment. T.T., T.K.H., and K.D.M. wrote the manuscript.
References
- Akerman CJ, Smyth D, Thompson ID. Visual experience before eye-opening and the development of the retinogeniculate pathway. Neuron. 2002;36:869–879. doi: 10.1016/s0896-6273(02)01010-3. [DOI] [PubMed] [Google Scholar]
- Artola A, von Frijtag JC, Fermont PC, Gispen WH, Schrama LH, Kamal A, Spruijt BM. Long-lasting modulation of the induction of LTD and LTP in rat hippocampal CA1 by behavioural stress and environmental enrichment. Eur J Neurosci. 2006;23:261–272. doi: 10.1111/j.1460-9568.2005.04552.x. [DOI] [PubMed] [Google Scholar]
- Asada H, Kawamura Y, Maruyama K, Kume H, Ding R, Ji FY, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K. Mice lacking the 65 kDa isoform of glutamic acid decarboxylase (GAD65) maintain normal levels of GAD67 and GABA in their brains but are susceptible to seizures. Biochem Biophys Res Commun. 1996;229:891–895. doi: 10.1006/bbrc.1996.1898. [DOI] [PubMed] [Google Scholar]
- Atallah BV, Bruns W, Carandini M, Scanziani M. Parvalbumin-expressing interneurons linearly transform cortical responses to visual stimuli. Neuron. 2012;73:159–170. doi: 10.1016/j.neuron.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkat TR, Polley DB, Hensch TK. A critical period for auditory thalamocortical connectivity. Nat Neurosci. 2011;14:1189–1194. doi: 10.1038/nn.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavelier D, Levi DM, Li RW, Dan Y, Hensch TK. Removing brakes on adult brain plasticity: from molecular to behavioral interventions. J Neurosci. 2010;30:14964–14971. doi: 10.1523/JNEUROSCI.4812-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurdeley M, Spatazza J, Lee HH, Sugiyama S, Bernard C, Di Nardo AA, Hensch TK, Prochiantz A. Otx2 binding to perineuronal nets persistently regulates plasticity in the mature visual cortex. J Neurosci. 2012;32:9429–9437. doi: 10.1523/JNEUROSCI.0394-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi GQ, Poo MM. Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci. 1998;18:10464–10472. doi: 10.1523/JNEUROSCI.18-24-10464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brainard MS, Doupe AJ. Auditory feedback in learning and maintenance of vocal behaviour. Nat Rev Neurosci. 2000;1:31–40. doi: 10.1038/35036205. [DOI] [PubMed] [Google Scholar]
- Cang J, Niell CM, Liu X, Pfeiffenberger C, Feldheim DA, Stryker MP. Selective disruption of one Cartesian axis of cortical maps and receptive fields by deficiency in ephrin-As and structured activity. Neuron. 2008;57:511–523. doi: 10.1016/j.neuron.2007.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cang J, Rentería RC, Kaneko M, Liu X, Copenhagen DR, Stryker MP. Development of precise maps in visual cortex requires patterned spontaneous activity in the retina. Neuron. 2005;48:797–809. doi: 10.1016/j.neuron.2005.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman B, Gödecke I. Cortical cell orientation selectivity fails to develop in the absence of ON-center retinal ganglion cell activity. J Neurosci. 2000;20:1922–1930. doi: 10.1523/JNEUROSCI.20-05-01922.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman B, Stryker MP. Development of orientation selectivity in ferret visual cortex and effects of deprivation. J Neurosci. 1993;13:5251–5262. doi: 10.1523/JNEUROSCI.13-12-05251.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho KK, Khibnik L, Philpot BD, Bear MF. The ratio of NR2A/B NMDA receptor subunits determines the qualities of ocular dominance plasticity in visual cortex. Proc Natl Acad Sci U S A. 2009;106:5377–5382. doi: 10.1073/pnas.0808104106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SY, Morales B, Lee HK, Kirkwood A. Absence of long-term depression in the visual cortex of glutamic Acid decarboxylase-65 knock-out mice. J Neurosci. 2002;22:5271–5276. doi: 10.1523/JNEUROSCI.22-13-05271.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condé F, Lund JS, Lewis DA. The hierarchical development of monkey visual cortical regions as revealed by the maturation of parvalbumin-immunoreactive neurons. Brain Res Dev Brain Res. 1996;96:261–276. doi: 10.1016/0165-3806(96)00126-5. [DOI] [PubMed] [Google Scholar]
- Crair MC, Gillespie DC, Stryker MP. The role of visual experience in the development of columns in cat visual cortex. Science. 1998;279:566–570. doi: 10.1126/science.279.5350.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayan P, Abbott LF. Theoretical neuroscience computational and mathematical modeling of neural systems. Massachusetts Institute of Technology Press; Cambridge, Mass.: 2001. [Google Scholar]
- Dani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R, Nelson SB. Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2005;102:12560–12565. doi: 10.1073/pnas.0506071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai NS, Cudmore RH, Nelson SB, Turrigiano GG. Critical periods for experience-dependent synaptic scaling in visual cortex. Nat Neurosci. 2002;5:783–789. doi: 10.1038/nn878. [DOI] [PubMed] [Google Scholar]
- Di Cristo G, Chattopadhyaya B, Kuhlman SJ, Fu Y, Bélanger MC, Wu CZ, Rutishauser U, Maffei L, Huang ZJ. Activity-dependent PSA expression regulates inhibitory maturation and onset of critical period plasticity. Nat Neurosci. 2007;10:1569–1577. doi: 10.1038/nn2008. [DOI] [PubMed] [Google Scholar]
- Dorrn AL, Yuan K, Barker AJ, Schreiner CE, Froemke RC. Developmental sensory experience balances cortical excitation and inhibition. Nature. 2010;465:932–936. doi: 10.1038/nature09119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand S, Patrizi A, Quast KB, Hachigian L, Pavlyuk R, Saxena A, Carninci P, Hensch TK, Fagiolini M. NMDA receptor regulation prevents regression of visual cortical function in the absence of Mecp2. Neuron. 2012;76:1078–90. doi: 10.1016/j.neuron.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa JS, Stryker MP. Development and plasticity of the primary visual cortex. Neuron. 2012;75:230–49. doi: 10.1016/j.neuron.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagiolini M, Hensch TK. Inhibitory threshold for critical-period activation in primary visual cortex. Nature. 2000;404:183–186. doi: 10.1038/35004582. [DOI] [PubMed] [Google Scholar]
- Fagiolini M, Fritschy JM, Löw K, Möhler H, Rudolph U, Hensch TK. Specific GABAA circuits for visual cortical plasticity. Science. 2004;303:1681–1683. doi: 10.1126/science.1091032. [DOI] [PubMed] [Google Scholar]
- Fagiolini M, Katagiri H, Miyamoto H, Mori H, Grant SG, Mishina M, Hensch TK. Separable features of visual cortical plasticity revealed by N-methyl-D-aspartate receptor 2A signaling. Proc Natl Acad Sci U S A. 2003;100:2854–2859. doi: 10.1073/pnas.0536089100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagiolini M, Pizzorusso T, Berardi N, Domenici L, Maffei L. Functional postnatal development of the rat primary visual cortex and the role of visual experience: dark rearing and monocular deprivation. Vision Res. 1994;34:709–720. doi: 10.1016/0042-6989(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Faguet J, Maranhao B, Smith SL, Trachtenberg JT. Ipsilateral eye cortical maps are uniquely sensitive to binocular plasticity. J Neurophysiol. 2009;101:855–861. doi: 10.1152/jn.90893.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Failor S, Nguyen V, Darcy DP, Cang J, Wendland MF, Stryker MP, McQuillen PS. Neonatal cerebral hypoxia-ischemia impairs plasticity in rat visual cortex. J Neurosci. 2010;30:81–92. doi: 10.1523/JNEUROSCI.5656-08.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman DE. Timing-based LTP and LTD at vertical inputs to layer II/III pyramidal cells in rat barrel cortex. Neuron. 2000;27:45–56. doi: 10.1016/s0896-6273(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Felleman DJ, Van Essen DC. Distributed hierarchical processing in the primate cerebral cortex. Cereb Cortex. 1991;1:1–47. doi: 10.1093/cercor/1.1.1-a. [DOI] [PubMed] [Google Scholar]
- Feller MB, Scanziani M. A precritical period for plasticity in visual cortex. Curr Opin Neurobiol. 2005;15:94–100. doi: 10.1016/j.conb.2005.01.012. [DOI] [PubMed] [Google Scholar]
- Fiser J, Chiu C, Weliky M. Small modulation of ongoing cortical dynamics by sensory input during natural vision. Nature. 2004;431:573–578. doi: 10.1038/nature02907. [DOI] [PubMed] [Google Scholar]
- Flint AC, Maisch US, Weishaupt JH, Kriegstein AR, Monyer H. NR2A subunit expression shortens NMDA receptor synaptic currents in developing neocortex. J Neurosci. 1997;17:2469–2476. doi: 10.1523/JNEUROSCI.17-07-02469.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox MD, Raichle ME. Spontaneous fluctuations in brain activity observed with functional magnetic resonance imaging. Nat Rev Neurosci. 2007;8:700–711. doi: 10.1038/nrn2201. [DOI] [PubMed] [Google Scholar]
- Gandhi SP, Yanagawa Y, Stryker MP. Delayed plasticity of inhibitory neurons in developing visual cortex. Proc Natl Acad Sci U S A. 2008;105:16797–16802. doi: 10.1073/pnas.0806159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstner W, Kistler WM. Spiking neuron models : single neurons, populations, plasticity. Cambridge University Press; Cambridge, U.K.; New York: 2002. [Google Scholar]
- Gianfranceschi L, Siciliano R, Walls J, Morales B, Kirkwood A, Huang ZJ, Tonegawa S, Maffei L. Visual cortex is rescued from the effects of dark rearing by overexpression of BDNF. Proc Natl Acad Sci U S A. 2003;100:12486–12491. doi: 10.1073/pnas.1934836100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JA, Stryker MP. Experience-dependent plasticity of binocular responses in the primary visual cortex of the mouse. J Neurosci. 1996;16:3274–3286. doi: 10.1523/JNEUROSCI.16-10-03274.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanover JL, Huang ZJ, Tonegawa S, Stryker MP. Brain-derived neurotrophic factor overexpression induces precocious critical period in mouse visual cortex. J Neurosci. 1999;19:RC40. doi: 10.1523/JNEUROSCI.19-22-j0003.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harauzov A, Spolidoro M, DiCristo G, De Pasquale R, Cancedda L, Pizzorusso T, Viegi A, Berardi N, Maffei L. Reducing intracortical inhibition in the adult visual cortex promotes ocular dominance plasticity. J Neurosci. 2010;30:361–371. doi: 10.1523/JNEUROSCI.2233-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebb DO. The organization of behavior. Wiley & Sons; New York: 1949. [Google Scholar]
- Hensch TK. Critical period regulation. Annu Rev Neurosci. 2004;27:549–579. doi: 10.1146/annurev.neuro.27.070203.144327. [DOI] [PubMed] [Google Scholar]
- Hensch TK. Critical period plasticity in local cortical circuits. Nat Rev Neurosci. 2005;6:877–888. doi: 10.1038/nrn1787. [DOI] [PubMed] [Google Scholar]
- Hensch TK, Fagiolini M, Mataga N, Stryker MP, Baekkeskov S, Kash SF. Local GABA circuit control of experience-dependent plasticity in developing visual cortex. Science. 1998;282:1504–1508. doi: 10.1126/science.282.5393.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez AE, Li P. Age of acquisition: its neural and computational mechanisms. Psychol Bull. 2007;133:638–650. doi: 10.1037/0033-2909.133.4.638. [DOI] [PubMed] [Google Scholar]
- Hofer SB, Mrsic-Flogel TD, Bonhoeffer T, Hübener M. Prior experience enhances plasticity in adult visual cortex. Nat Neurosci. 2006;9:127–132. doi: 10.1038/nn1610. [DOI] [PubMed] [Google Scholar]
- Hooks BM, Chen C. Distinct roles for spontaneous and visual activity in remodeling of the retinogeniculate synapse. Neuron. 2006;52:281–291. doi: 10.1016/j.neuron.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Huberman AD, Speer CM, Chapman B. Spontaneous retinal activity mediates development of ocular dominance columns and binocular receptive fields in V1. Neuron. 2006;52:247–254. doi: 10.1016/j.neuron.2006.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huberman AD, Feller MB, Chapman B. Mechanisms underlying development of visual maps and receptive fields. Annu Rev Neurosci. 2008;31:479–509. doi: 10.1146/annurev.neuro.31.060407.125533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibata K, Sun Q, Turrigiano GG. Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron. 2008;57:819–826. doi: 10.1016/j.neuron.2008.02.031. [DOI] [PubMed] [Google Scholar]
- Insanally MN, Köver H, Kim H, Bao S. Feature-dependent sensitive periods in the development of complex sound representation. J Neurosci. 2009;29:5456–5462. doi: 10.1523/JNEUROSCI.5311-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai Y, Fagiolini M, Obata K, Hensch TK. Rapid critical period induction by tonic inhibition in visual cortex. J Neurosci. 2003;23:6695–6702. doi: 10.1523/JNEUROSCI.23-17-06695.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B, Treviño M, Kirkwood A. Sequential development of long-term potentiation and depression in different layers of the mouse visual cortex. J Neurosci. 2007;27:9648–9652. doi: 10.1523/JNEUROSCI.2655-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameyama K, Sohya K, Ebina T, Fukuda A, Yanagawa Y, Tsumoto T. Difference in binocularity and ocular dominance plasticity between GABAergic and excitatory cortical neurons. J Neurosci. 2010;30:1551–1559. doi: 10.1523/JNEUROSCI.5025-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M, Stellwagen D, Malenka RC, Stryker MP. Tumor necrosis factor-alpha mediates one component of competitive, experience-dependent plasticity in developing visual cortex. Neuron. 2008;58:673–680. doi: 10.1016/j.neuron.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanold PO, Shatz CJ. Subplate neurons regulate maturation of cortical inhibition and outcome of ocular dominance plasticity. Neuron. 2006;51:627–638. doi: 10.1016/j.neuron.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Kanold PO, Kim YA, GrandPre T, Shatz CJ. Co-regulation of ocular dominance plasticity and NMDA receptor subunit expression in glutamic acid decarboxylase-65 knock-out mice. J Physiol. 2009;587:2857–2867. doi: 10.1113/jphysiol.2009.171215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kash SF, Johnson RS, Tecott LH, Noebels JL, Mayfield RD, Hanahan D, Baekkeskov S. Epilepsy in mice deficient in the 65-kDa isoform of glutamic acid decarboxylase. Proc Natl Acad Sci U S A. 1997;94:14060–14065. doi: 10.1073/pnas.94.25.14060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katagiri H, Fagiolini M, Hensch TK. Optimization of somatic inhibition at critical period onset in mouse visual cortex. Neuron. 2007;53:805–812. doi: 10.1016/j.neuron.2007.02.026. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Kenet T, Bibitchkov D, Tsodyks M, Grinvald A, Arieli A. Spontaneously emerging cortical representations of visual attributes. Nature. 2003;425:954–956. doi: 10.1038/nature02078. [DOI] [PubMed] [Google Scholar]
- Kirkwood A, Bear MF. Homosynaptic long-term depression in the visual cortex. J Neurosci. 1994;14:3404–3412. doi: 10.1523/JNEUROSCI.14-05-03404.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knöpfel T, Lin MZ, Levskaya A, Tian L, Lin JY, Boyden ES. Toward the second generation of optogenetic tools. J Neurosci. 2010;30:14998–15004. doi: 10.1523/JNEUROSCI.4190-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen EI. Sensitive periods in the development of the brain and behavior. J Cogn Neurosci. 2004;16:1412–1425. doi: 10.1162/0898929042304796. [DOI] [PubMed] [Google Scholar]
- Kuhlman SJ, Lu J, Lazarus MS, Huang ZJ. Maturation of GABAergic inhibition promotes strengthening of temporally coherent inputs among convergent pathways. PLoS Comput Biol. 2010;6:e1000797. doi: 10.1371/journal.pcbi.1000797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leao RN, Sun H, Svahn K, Berntson A, Youssoufian M, Paolini AG, Fyffe RE, Walmsley B. Topographic organization in the auditory brainstem of juvenile mice is disrupted in congenital deafness. J Physiol. 2006;571:563–578. doi: 10.1113/jphysiol.2005.098780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenneberg EH. Biological Foundations of Language. Wiley; New York: 1967. [Google Scholar]
- Leventhal AG, Wang Y, Pu M, Zhou Y, Ma Y. GABA and its agonists improved visual cortical function in senescent monkeys. Science. 2003;300:812–815. doi: 10.1126/science.1082874. [DOI] [PubMed] [Google Scholar]
- Lyckman AW, Horng S, Leamey CA, Tropea D, Watakabe A, Van Wart A, McCurry C, Yamamori T, Sur M. Gene expression patterns in visual cortex during the critical period: synaptic stabilization and reversal by visual deprivation. Proc Natl Acad Sci U S A. 2008;105:9409–9414. doi: 10.1073/pnas.0710172105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei A, Nataraj K, Nelson SB, Turrigiano GG. Potentiation of cortical inhibition by visual deprivation. Nature. 2006;443:81–84. doi: 10.1038/nature05079. [DOI] [PubMed] [Google Scholar]
- Maffei A, Nelson SB, Turrigiano GG. Selective reconfiguration of layer 4 visual cortical circuitry by visual deprivation. Nat Neurosci. 2004;7:1353–1359. doi: 10.1038/nn1351. [DOI] [PubMed] [Google Scholar]
- Majdan M, Shatz CJ. Effects of visual experience on activity-dependent gene regulation in cortex. Nat Neurosci. 2006;9:650–659. doi: 10.1038/nn1674. [DOI] [PubMed] [Google Scholar]
- Markram H, Lübke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- Maya Vetencourt JF, Sale A, Viegi A, Baroncelli L, De Pasquale R, O'Leary OF, Castrén E, Maffei L. The antidepressant fluoxetine restores plasticity in the adult visual cortex. Science. 2008;320:385–388. doi: 10.1126/science.1150516. [DOI] [PubMed] [Google Scholar]
- McGee AW, Yang Y, Fischer QS, Daw NW, Strittmatter SM. Experience-driven plasticity of visual cortex limited by myelin and Nogo receptor. Science. 2005;309:2222–2226. doi: 10.1126/science.1114362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KD. Correlation-based models of neural development. In: Gluck MA, Rumelhart DE, editors. Neuroscience and connectionist theory. Lawrence Erlbaum Associates; Hillsdale, NJ: 1990. pp. 267–353. [Google Scholar]
- Miller KD, MacKay DJC. The role of constraints in Hebbian learning. Neural Comput. 1994;6:100–126. [Google Scholar]
- Mizuno H, Hirano T, Tagawa Y. Evidence for activity-dependent cortical wiring: formation of interhemispheric connections in neonatal mouse visual cortex requires projection neuron activity. J Neurosci. 2007;27:6760–6770. doi: 10.1523/JNEUROSCI.1215-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody WJ, Bosma MM. Ion channel development, spontaneous activity, and activity-dependent development in nerve and muscle cells. Physiol Rev. 2005;85:883–941. doi: 10.1152/physrev.00017.2004. [DOI] [PubMed] [Google Scholar]
- Morales B, Choi SY, Kirkwood A. Dark rearing alters the development of GABAergic transmission in visual cortex. J Neurosci. 2002;22:8084–8090. doi: 10.1523/JNEUROSCI.22-18-08084.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita H, Hensch TK. Critical period revisited: impact on vision. Curr Opin Neurobiol. 2008;18:101–107. doi: 10.1016/j.conb.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Morishita H, Miwa JM, Heintz N, Hensch TK. Lynx1, a cholinergic brake, limits plasticity in adult visual cortex. Science. 2010;330:1238–1240. doi: 10.1126/science.1195320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrsic-Flogel TD, Hofer SB, Ohki K, Reid RC, Bonhoeffer T, Hübener M. Homeostatic regulation of eye-specific responses in visual cortex during ocular dominance plasticity. Neuron. 2007;54:961–972. doi: 10.1016/j.neuron.2007.05.028. [DOI] [PubMed] [Google Scholar]
- Nishie H, Miyata R, Fujikawa R, Kinoshita K, Muroi Y, Ishii T. Post-weaning mice fed exclusively milk have deficits in induction of long-term depression in the CA1 hippocampal region and spatial learning and memory. Neurosci Res. 2012;73:292–301. doi: 10.1016/j.neures.2012.05.006. [DOI] [PubMed] [Google Scholar]
- Ohshiro T, Weliky M. Simple fall-off pattern of correlated neural activity in the developing lateral geniculate nucleus. Nat Neurosci. 2006;9:1541–1548. doi: 10.1038/nn1799. [DOI] [PubMed] [Google Scholar]
- Oja E. Neural networks, principal components, and subspaces. International journal of neural systems. 1989;1:61–68. [Google Scholar]
- Parkinson D, Kratz KE, Daw NW. Evidence for a nicotinic component to the actions of acetylcholine in cat visual cortex. Exp Brain Res. 1988;73:553–568. doi: 10.1007/BF00406614. [DOI] [PubMed] [Google Scholar]
- Pizzorusso T, Medini P, Landi S, Baldini S, Berardi N, Maffei L. Structural and functional recovery from early monocular deprivation in adult rats. Proc Natl Acad Sci U S A. 2006;103:8517–8522. doi: 10.1073/pnas.0602657103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusky GT, Douglas RM. Developmental plasticity of mouse visual acuity. Eur J Neurosci. 2003;17:167–173. doi: 10.1046/j.1460-9568.2003.02420.x. [DOI] [PubMed] [Google Scholar]
- Quinlan EM, Olstein DH, Bear MF. Bidirectional, experience-dependent regulation of N-methyl-D-aspartate receptor subunit composition in the rat visual cortex during postnatal development. Proc Natl Acad Sci U S A. 1999a;96:12876–12880. doi: 10.1073/pnas.96.22.12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan EM, Philpot BD, Huganir RL, Bear MF. Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat Neurosci. 1999b;2:352–357. doi: 10.1038/7263. [DOI] [PubMed] [Google Scholar]
- Renger JJ, Hartman KN, Tsuchimoto Y, Yokoi M, Nakanishi S, Hensch TK. Experience-dependent plasticity without long-term depression by type 2 metabotropic glutamate receptors in developing visual cortex. Proc Natl Acad Sci U S A. 2002;99:1041–1046. doi: 10.1073/pnas.022618799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts EB, Ramoa AS. Enhanced NR2A subunit expression and decreased NMDA receptor decay time at the onset of ocular dominance plasticity in the ferret. J Neurophysiol. 1999;81:2587–2591. doi: 10.1152/jn.1999.81.5.2587. [DOI] [PubMed] [Google Scholar]
- Robinson K. Implications of developmental plasticity for the language acquisition of deaf children with cochlear implants. Int J Pediatr Otorhinolaryngol. 1998;46:71–80. doi: 10.1016/s0165-5876(98)00125-6. [DOI] [PubMed] [Google Scholar]
- Rothman JS, Cathala L, Steuber V, Silver RA. Synaptic depression enables neuronal gain control. Nature. 2009;457:1015–1018. doi: 10.1038/nature07604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Hata Y, Masui H, Tsumoto T. A functional role of cholinergic innervation to neurons in the cat visual cortex. J Neurophysiol. 1987;58:765–780. doi: 10.1152/jn.1987.58.4.765. [DOI] [PubMed] [Google Scholar]
- Scott LS, Pascalis O, Nelson CA. A Domain-General Theory of the Development of Perceptual Discrimination. Curr Dir Psychol Sci. 2007;16:197–201. doi: 10.1111/j.1467-8721.2007.00503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharpee TO, Atencio CA, Schreiner CE. Hierarchical representations in the auditory cortex. Curr Opin Neurobiol. 2011;21:761–767. doi: 10.1016/j.conb.2011.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SL, Trachtenberg JT. Experience-dependent binocular competition in the visual cortex begins at eye opening. Nat Neurosci. 2007;10:370–375. doi: 10.1038/nn1844. [DOI] [PubMed] [Google Scholar]
- Song S, Miller KD, Abbott LF. Competitive Hebbian learning through spike-timing-dependent synaptic plasticity. Nat Neurosci. 2000;3:919–926. doi: 10.1038/78829. [DOI] [PubMed] [Google Scholar]
- Southwell DG, Froemke RC, Alvarez-Buylla A, Stryker MP, Gandhi SP. Cortical plasticity induced by inhibitory neuron transplantation. Science. 2010;327:1145–1148. doi: 10.1126/science.1183962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spatazza J, Lee HHC, Di Nardo AA, Tibaldi L, Joliot A, Hensch TK, Prochiantz A. Choroid-plexus-derived Otx2 homeoprotein constrains adult cortical plasticity. Cell Reports. 2013;3:1–9. doi: 10.1016/j.celrep.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spolidoro M, Baroncelli L, Putignano E, Maya-Vetencourt JF, Viegi A, Maffei L. Food restriction enhances visual cortex plasticity in adulthood. Nat Commun. 2011;2:320. doi: 10.1038/ncomms1323. [DOI] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC. Synaptic scaling mediated by glial TNF-alpha. Nature. 2006;440:1054–1059. doi: 10.1038/nature04671. [DOI] [PubMed] [Google Scholar]
- Sugiyama S, Di Nardo AA, Aizawa S, Matsuo I, Volovitch M, Prochiantz A, Hensch TK. Experience-dependent transfer of Otx2 homeoprotein into the visual cortex activates postnatal plasticity. Cell. 2008;134:508–520. doi: 10.1016/j.cell.2008.05.054. [DOI] [PubMed] [Google Scholar]
- Sun YJ, Wu GK, Liu BH, Li P, Zhou M, Xiao Z, Tao HW, Zhang LI. Fine-tuning of pre-balanced excitation and inhibition during auditory cortical development. Nature. 2010;465:927–931. doi: 10.1038/nature09079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Yi E, Gale JE, Glowatzki E, Bergles DE. The origin of spontaneous activity in the developing auditory system. Nature. 2007;450:50–55. doi: 10.1038/nature06233. [DOI] [PubMed] [Google Scholar]
- Tropea D, Kreiman G, Lyckman A, Mukherjee S, Yu H, Horng S, Sur M. Gene expression changes and molecular pathways mediating activity-dependent plasticity in visual cortex. Nat Neurosci. 2006;9:660–668. doi: 10.1038/nn1689. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature. 1998;391:892–896. doi: 10.1038/36103. [DOI] [PubMed] [Google Scholar]
- Vinje WE. Sparse Coding and Decorrelation in Primary Visual Cortex During Natural Vision. Science. 2000;287:1273–1276. doi: 10.1126/science.287.5456.1273. [DOI] [PubMed] [Google Scholar]
- Wang BS, Sarnaik R, Cang J. Critical period plasticity matches binocular orientation preference in the visual cortex. Neuron. 2010;65:246–256. doi: 10.1016/j.neuron.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weliky M, Katz LC. Correlational Structure of Spontaneous Neuronal Activity in the Developing Lateral Geniculate Nucleus in Vivo. Science. 1999;285:599–604. doi: 10.1126/science.285.5427.599. [DOI] [PubMed] [Google Scholar]
- Werker JF, Byers-Heinlein K, Fennell CT. Bilingual beginnings to learning words. Philos Trans R Soc Lond B Biol Sci. 2009;364:3649–3663. doi: 10.1098/rstb.2009.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White LE, Coppola DM, Fitzpatrick D. The contribution of sensory experience to the maturation of orientation selectivity in ferret visual cortex. Nature. 2001;411:1049–1052. doi: 10.1038/35082568. [DOI] [PubMed] [Google Scholar]
- Wiesel TN, Hubel DH. Effects of visual deprivation on morphology and physiology of cells in the cats lateral geniculate body. J Neurophysiol. 1963;26:978–993. doi: 10.1152/jn.1963.26.6.978. [DOI] [PubMed] [Google Scholar]
- Wyatt RM, Tring E, Trachtenberg JT. Pattern and not magnitude of neural activity determines dendritic spine stability in awake mice. Nat Neurosci. 2012 doi: 10.1038/nn.3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazaki-Sugiyama Y, Kang S, Câteau H, Fukai T, Hensch TK. Bidirectional plasticity in fast-spiking GABA circuits by visual experience. Nature. 2009;462:218–221. doi: 10.1038/nature08485. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.